

SUMMARY
The Src-homology 2 (SH2) domain is a protein interaction domain that directs myriad phosphotyrosine (pY) signaling pathways. Genome-wide screening of human SH2 domains reveals that ≈90% of SH2 domains bind plasma membrane lipids and many have high phosphoinositide specificity. They bind lipids using surface cationic patches separate from pY-binding pockets, thus binding lipids and the pY motif independently. The patches form grooves for specific lipid headgroup recognition or flat surfaces for non-specific membrane binding and both types of interaction is important for cellular function and regulation of SH2 domain-containing proteins. Cellular studies with ZAP70 showed that multiple lipids bind its C-terminal SH2 domain in a spatiotemporally specific manner and thereby exert exquisite spatiotemporal control over its protein binding and signaling activities in T cells. Collectively, these studies reveal how lipids control SH2 domain-mediated cellular protein-protein interaction networks and suggest a new strategy for therapeutic modulation of pY signaling pathways.
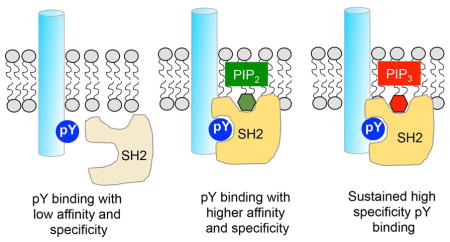
Graphical abstract
INTRODUCTION
The Src-homology 2 (SH2) domain is a prototypal modular protein interaction domain (PID) and has long served as an excellent model system for studying protein-protein interactions (Pawson, 2004). Since its discovery as the first PID in v-fps/fes oncogene in Fujinami sarcoma virus (Sadowski et al., 1986), the SH2 domain has been identified in diverse cellular proteins in a wide variety of eukaryotes, but primarily in metazoans. The human genome encodes 121 SH2 domains in 111 different proteins, including kinases, adaptors, phosphatases, and other signaling molecules (Pawson, 2004). As a major reader of phosphotyrosine (pY) signaling information, SH2 domain-containing proteins play crucial roles in linking various protein tyrosine kinases to downstream molecules, thereby controlling the specificity of pY signaling (Lim and Pawson, 2010).
Structural analysis of a wide range of SH2 domains and their complexes with pY-peptides has revealed that SH2 domains have a common architecture made of two α-helices flanking antiparallel β strands (Pawson, 2004; Waksman et al., 1993). They specifically recognize pY and a few residues immediately C-terminal to pY using a pY-binding pocket and a secondary binding site, respectively (Pawson, 2004). However, quantitative analysis has shown that SH2 domains bind pY-containing peptides with variable affinity and a significant degree of promiscuity (Ladbury and Arold, 2000; Machida and Mayer, 2005). Exquisite protein interaction specificity required for high-fidelity pY signaling thus has to be augmented by other mechanisms, such as protein compartmentalization (Good et al., 2011; Scott and Pawson, 2009), signaling complex formation (Bray, 1998; Cho, 2006), or pY-independent secondary protein interactions (Bae et al., 2009).
Recent studies have suggested that membrane lipids play a role in modulating cellular protein-protein interactions mediated by another PID, PDZ (PSD95, Dlg1, ZO-1) domain (Chen et al., 2012; Feng and Zhang, 2009; Sheng et al., 2012; Sheng et al., 2014; Wu et al., 2007; Zimmermann et al., 2002). It was also reported that some SH2 domains could bind lipids, which either inhibited (Rameh et al., 1995) or promoted (Bae et al., 1998) the activity of their host proteins; however, the mechanisms, the physiological significance, and the universality of these findings remain controversial (Surdo et al., 1999). In this study, we thoroughly and systematically investigated the potential roles of lipids in regulating SH2 domain-mediated protein-protein interactions and cellular signaling activities.
RESULTS
Membrane Binding Analysis of Human SH2 Domains
We quantitatively measured the lipid binding affinity and specificity of human SH2 domains on a genomic scale by surface plasmon resonance (SPR) analysis. Since many of these SH2 domains were not stably expressed in Escherichia coli, we expressed them as enhanced green fluorescence protein (EGFP)-fusion proteins, which significantly improved the protein expression yield for most SH2 domains. Our control experiments showed that a properly placed EGFP tag did not affect the membrane binding properties of SH2 domains (see Table S1). Out of 121 human SH2 domains, we were able to rigorously characterize 76 domains that can be prepared stably and in amounts sufficient for biophysical studies. A majority of SH2 domain proteins interact with pY-containing proteins that are either associated with or act near the cytofacial leaflet of the plasma membrane (PM). We thus used for our measurements the vesicles whose lipid composition recapitulates that of the cytofacial PM (referred to as PM-mimetic vesicles hereafter) (Cho and Stahelin, 2005).
Our initial strategy was to screen for SH2 domains that yielded a high response in rapid SPR screening with the PM-mimetic vesicles but since a majority of them gave high enough SPR signals, we determined the Kd values for all available SH2 domains. The Kd values of 76 human (and one yeast) SH2 domains (Table 1) revealed that 74% of them have submicromolar affinity for the PM-mimetic vesicles. This level of membrane affinity is comparable to that of other lipid binding proteins (Cho and Stahelin, 2005). Also, 13 additional SH2 domains have Kd’s in the range of 1–5 μM. Only 8 SH2 domains (≈10%) exhibited no detectable binding to the PM-mimetic vesicles.
Table 1.
Lipid Binding Properties of Human SH2 Domains
| SH2 domainsa | Residue Number | Kd (nM) for PMb | Lipid binding residuesc | PtdInsP Selectivityd |
|---|---|---|---|---|
| STAT6-SH2 | 517-632 | 20 ± 10 | ||
| GRB7-SH2 | 411-532 | 70 ± 12 | Low selectivityi | |
| FRK(PTK5)-SH2 | 109-213 | 80 ± 12 | ||
| YES1-SH2 | 151-260 | 110 ± 12 | R215, K216 | PI45P2 > PIP3 > others |
| BLNK-SH2 | 326-456 | 120 ± 19 | PIP3 > PI45P2 ≫ others | |
| APS(SH2B2)-SH2 | 394-514 | 140 ± 11 | Low selectivity | |
| NPLCγ2-cSH2e | 626-740 | 150 ± 13 | R727, K728 | PIP3 > PI45P2 ≫ others |
| BRK(PTK6)-SH2 | 71-175 | 150 ± 50 | R131, R136 | Low selectivity |
| Tensin3-SH2 | 1152-1287 | 180 ± 23 | ||
| SHIP1-SH2 | 1-106 | 190 ± 30 | PIP3 ≈ PI45P2 ≫ others | |
| Tensin2-SH2 | 1120-1250 | 200 ± 67 | ||
| NSYK-cSH2e | 148-264 | 210 ± 9 | PIP3 > PI45P2 ≫ others | |
| ITK-SH2 | 230-343 | 210 ± 372 | Low selectivity | |
| HCK-SH2 | 157-267 | 220 ± 20 | ||
| PI3K P85α-cSH2 | 604-718 | 220 ± 20 | ||
| NPTPN6(SHP1)-nSH2f | 1-99 | 240 ± 11 | PIP3 > PI45P2≫ others | |
| FYN-SH2 | 141-250 | 250 ± 70 | K182, R206, K207 | Low selectivity |
| ZAP70-cSH2e | 164-259 | 340 ± 35 | K176, K186, K206, K251 | PIP3 > PI45P2 > others |
| NPLCγ1-cSH2e | 648-761 | 290 ± 16 | R748, K749 | |
| GRB10-SH2 | 473-594 | 250 ± 50 | ||
| Tensin1-SH2 | 1443-1577 | 300 ± 30 | PIP3 ≫ others | |
| NVAV3-SH2e | 652-766 | 320 ± 49 | ||
| LYN-SH2 | 121-230 | 320 ± 56 | ||
| NPI3K p55γ-cSH2e | 338-452 | 330 ± 27 | ||
| STAP1-SH2 | 159-276 | 350 ± 46 | ||
| Tensin4-SH2 | 429-561 | 350 ± 35 | ||
| BLK-SH2 | 116-224 | 360 ± 29 | ||
| SPT6-cSH2e | 1424-1515 | 420 ± 30 | Low selectivity | |
| Yeast SPT6-cSH2e,h | 1351-1440 | 430 ± 60 | ||
| PI3K p85β-cSH2e | 602-716 | 420 ± 350 | ||
| SH2D2A-SH2 | 75-190 | 440 ± 35 | ||
| NRASA1-nSH2f | 161-276 | 440 ± 51 | R188, R207 | PIP3 ≈ PI45P2 > others |
| PI3K P85α-nSH2f | 312-427 | 440 ± 80 | ||
| NPI3K p55γ-nSH2f | 45-160 | 450 ± 31 | ||
| SHC3-SH2 | 479-590 | 460 ± 15 | ||
| PI3K p85β-nSH2f | 310-425 | 460 ± 67 | ||
| NPTPN6(SHP1)-cSH2e | 90-214 | 480 ± 47 | ||
| SOCS6-SH2 | 364-481 | 480 ± 24 | ||
| SRC-SH2 | 144-253 | 450 ± 60 | ||
| FGR(SRC2)-SH2 | 137-264 | 500 ± 41 | ||
| GRB2-SH2 | 57-155 | 520 ± 15 | ||
| BMX-SH2 | 276-397 | 550 ± 70 | K313, K315 | PI45P2 > PIP3 > others |
| SH3BP2-SH2 | 446-558 | 580 ± 95 | ||
| BTK-SH2 | 273-382 | 640 ± 55 | K311, K314 | Low selectivity |
| NCSK-SH2 | 71-191 | 670 ± 160 | ||
| SH2D4A-SH2 | 327-443 | 670 ± 60 | ||
| NPLCγ1-nSH2f | 530-659 | 710 ± 50 | ||
| SH2B1-SH2 | 507-624 | 710 ± 350 | ||
| NCK1-SH2 | 262-376 | 730 ± 30 | ||
| GRB14-SH2 | 418-540 | 730 ± 100 | PIP3≫ others | |
| STAT3-SH2 | 580-670 | 670±120 | ||
| MATK-SH2 | 102-216 | 860 ± 90 | ||
| CRK-SH2 | 1-124 | 960 ± 120 | ||
| NVAV2-SH2 | 653-767 | 970 ± 190 | ||
| SHF-SH2 | 303-419 | 970 ± 60 | ||
| SHE-SH2 | 375-491 | 1000 ± 100 | ||
| NPLCγ2-nSH2f | 512-637 | 1100 ± 110 | ||
| NGRAP2-SH2 | 55-152 | 1300 ± 170 | ||
| EAT2-SH2 | 1-106 | 1300 ± 280 | ||
| SH2D3A-SH2 | 1-115 | 1500 ± 130 | ||
| NGRAP-SH2 | 57-155 | 1600 ± 410 | ||
| NCRKL-SH2 | 1-108 | 1600 ± 550 | ||
| SH2D1A-SH2 | 1-107 | 1700 ± 260 | ||
| NRASA1-cSH2 | 340-446 | 1800 ± 60 | ||
| PTPN11(SHP2)-cSH2e | 92-217 | 2000 ± 400 | ||
| ABL1-SH2 | 120-222 | 2100 ± 400 | ||
| PTPN11(SHP2)-nSH2f | 1-102 | 2300 ± 570 | ||
| SHC2-SH2 | 467-578 | 5400 ± 1800 | ||
| SHC1-SH2 | 468-579 | NMg | ||
| SYK-nSH2f | 1-112 | NM | ||
| ZAP70-nSH2f | 1-107 | NM | ||
| LNK-SH2 | 344-461 | NM | ||
| SHD-SH2 | 220-336 | NM | ||
| FES-SH2 | 440-550 | NM | ||
| NTXK-SH2 | 141-266 | NM | ||
| Supt6h-nSH2f | 1331-1423 | NM |
All SH2 domains are expressed as C-terminal EGFP-fusion proteins unless specified otherwise (e.g., NPLCγ2-cSH2 indicates the N-terminal EGFP-fusion PLCγ2-cSH2). For most SH2 domains, N-terminal and C-terminal EGFP tags have essentially the same effect: i.e., they significantly improved the protein expression yield without affecting membrane binding properties of SH2 domains. The C-terminal EGFP tag was selected over the N-terminal one because the former tends to stabilize the SH2 domain better than the latter. For some SH2 domains, we employed the N-terminal EGFP tag because the C-terminal EGFP tag interferes with their lipid binding.
Mean ± S.D. from triplicate equilibrium SPR measurements using PM mimetic vesicles (POPC/POPE/POPS/PI/cholesterol/PtdIns(4,5)P2 (12:35:22:8:22:1)).
Determined by SPR analysis of mutants using PM mimetic vesicles.
Determined by SPR analysis using seven different POPC/POPS/PtdInsP (77:20:3) vesicles. PIP3, phosphatidylinositol-3,4,5-trisphosphate; PI34P2, phosphatidylinositol-3,4-bisphosphate; PI35P2, phosphatidylinositol-3,5-bisphosphate; PI45P2, phosphatidylinositol-4,5-bisphosphate; PI3P, phosphatidylinositol-3-monophosphate; PI4P, phosphatidylinositol-4-monophosphate; PI5P, phosphatidylinositol-5-monophosphate.
C-terminal SH2 domain of tandem SH2 domains
N-terminal SH2 domain of tandem SH2 domains
Not measurable with up to 10 μM proteins (i.e., Kd >10 μM)
SPT6-cSH2 domain from S. cerevisiae.
Those SH2 domains that do not clearly distinguish among PI-bisphosphates and among PI-monophosphate.
To assess the specific nature of SH2 domain-lipid binding, we randomly selected 18 SH2 domains with high PM affinity and measured their lipid headgroup selectivity. In particular, we determined selectivity for phosphoinositides (PtdInsP), which play key roles in cell signaling (Di Paolo and De Camilli, 2006). Among tested SH2 domains, 12 of them exhibited PtdInsP selectivity that is comparable to that of the Akt1 pleckstrin homology (PH) domain, a canonical PtdInsP-binding domain (Fig. S1 and Table 1). Many phosphatidylinositol-3,4,5-trisphosphate (PIP3)-binding PH domains, including Akt1-PH, bind phosphatidylinositol-3,4-bisphosphate (PI34P2) as well as PIP3 (Fig. S1) (Manna et al., 2007). Interestingly, most of PtdInsP-selective SH2 domains prefer PIP3 and phosphatidylinositol-4,5-bisphosphate (PI45P2) to other PtdInsPs. Their selectivity for PI45P2 over isoelectric PI34P2 and phosphatidylinositol-3,5-bisphosphate (PI35P2) shows that their PtdInsP binding is driven not by non-specific electrostatic binding but by specific PtdInsP headgroup recognition. This notion is further supported by the finding that some SH2 domains, including BMX-SH2 and YES1-SH2, prefer PI45P2 to more anionic PIP3 (Fig. S1). Overall, favorable binding of most SH2 domains to PI45P2 might be important for their PM binding under physiological condition given that PI45P2 is the most abundant PtdInsP in the PM of mammalian cells (McLaughlin et al., 2002). Six SH2 domains did not show clear PtdInsP selectivity: i.e., they did not clearly distinguish among PI45P2, PI34P2, and PI35P2 (e.g., see Btk-SH2 and PTK6-SH2 in Fig. S1). Even for these SH2 domains, however, addition of PIP3 and PI45P2 to vesicles significantly enhanced their membrane affinity, suggesting that PIP3 and PI45P2 might also facilitate their PM binding under physiological conditions.
To prove that PIP3 and PI45P2 play an important role in PM localization of SH2 domains, we measured the PM localization of PIP3-selective (ZAP70-cSH2: the C-terminal unit of the tandem SH2 domains), PI45P2-selective (BMX-SH2 and YES1-SH2), and non-selective (PTK6-SH2) SH2 domains in response to rapamycin-triggered PI45P2 depletion (Inoue et al., 2005; Varnai et al., 2006) and PIP3 enrichment (Inoue and Meyer, 2008) in the PM. Before rapamycin treatment, all these SH2 domains showed PM localization, albeit to different degrees (Fig. 1). When PI45P2 was depleted, PI45P2-selective BMX-SH2 (Fig, 1A) and YES1-SH2 domains (Fig. 1B) were rapidly (i.e., < 1 min) removed from the PM whereas PIP3-selective ZAP70-cSH2 (Fig. 1D) showed only a minor degree of PM displacement. In contrast, PIP3 enrichment significantly increased the PM localization of ZAP70-cSH2 (Fig. 1D) while showing little effect on that of BMX-SH2 (Fig. 1A) and YES1-SH2 (Fig. 1B). These results support the physiological significance of PIP3 and PI45P2 selectivity of these SH2 domains. Lastly, PM recruitment of non-selective PTK6-SH2 was also affected by both PI45P2 depletion and PIP3 enrichment (Fig. 1C), albeit to lesser degrees, supporting the notion that both PI45P2 and PIP3 also facilitate its PM recruitment under physiological conditions.
Fig. 1.

Effect of PI45P2 depletion and PIP3 enrichment on the subcellular localization of mCherry-tagged SH2 domains. (A) PI45P2 depletion greatly reduced PM localization of PI45P2-selective BMX-SH2 WT whereas PIP3 enrichment had little effect. R302A behaved similarly to WT whereas K313A/K315A showed dramatically reduced PM localization. (B) PI45P2 depletion greatly inhibited PM localization of YES1-SH2 WT whereas PIP3 enrichment had little effect. R185A behaved similarly to WT whereas R215A/K216A showed dramatically reduced PM localization. (C) Both PI45P2 depletion and PIP3 enrichment had only modest effects on PTK6-SH2 WT. R85A/R105A behaved similarly to WT whereas R131A/R136A showed significantly reduced PM localization. (D) PI45P2 depletion had a minor effect on PM localization of PIP3-selective ZAP70-cSH2 WT whereas PIP3 enrichment significantly enhanced its PM localization. R190A/R192A behaved similarly to WT. Between two lipid binding LOF mutants, K176E/K186E did not respond to PIP3 enrichment while K206E/K251E showed some degree of enhanced PM localization. Both LOF mutants showed much reduced PM localization than WT. (E) Control imaging of PI45P2 and PIP3 sensors demonstrates the robustness of our PI45P2 depletion and PIP3 enrichment systems. All images were taken before and 1 min after PI45P2 depletion and PIP3 enrichment. HeLa cells were used for all measurements. Scale bars indicate 5 μm.
Identification of Lipid Binding Sites of SH2 Domains
Electrostatic potential computation of human SH2 domains shows that all have highly cationic pY binding pockets (Fig. 2). Interestingly, many of them also contain alternate cationic patches (ACPs) (Fig. 2). In general, these ACPs are not as electropositive as pY pockets, but most of them contain neighboring hydrophobic and aromatic residues, which is reminiscent of lipid binding sites of membrane binding proteins (Cho and Stahelin, 2005). We thus suspected that either or both of these cationic sites might serve as a binding site for anionic lipids including PtdInsPs. Previous studies suggested that the pY pocket might be the main site for lipid binding (Hong et al., 2009; Rameh et al., 1995; Tokonzaba et al., 2006). We found, however, mutation of single or multiple cationic residues in the pY pockets of 5 randomly selected SH2 domains did not cause significant decreases in vesicle binding (Fig. S2A). In contrast, mutations of ACP residues of these SH2 domains plus three additional SH2 domains reduced vesicle affinity to a much greater extent (Fig. S2B). Furthermore, non-lipid-binding SH2 domains, such as Syk N-terminal SH2 domain (nSH2) and SHD-SH2, lack ACPs while possessing cationic pY pockets (Fig. 2). These results suggest that not the pY pockets but ACPs serve as primary lipid binding sites for most SH2 domains. This notion is further supported by the subcellular localization patterns of SH2 domain mutants in HeLa cells. For BMX-SH2, YES-SH2, and ZAP70-cSH2, mutations of ACP residues greatly reduced their PM localization whereas mutations of pY pocket residues had little effect on their PM recruitment (Fig. 1).
Fig. 2.

Surface electrostatic potential of selected SH2 domains. For each SH2 domain, the pY-binding pocket and the main alternate cationic patch (ACP) are indicated by red and green arrows, respectively. For ZAP70-cSH2 and BMX-SH2, structures are shown in two different orientations with 180° rotation because their main ACPs are located on the opposite side of the pY pockets. Non-lipid binding Syk-nSH2 and SHD-SH2 do not have ACP. The PIP3-specific Btk-PH domain (PDB ID: 1b55) is shown for comparison. A green arrow indicates its PIP3-binding pocket. All structures are shown in the same scale. In all cases, the minimal and maximal electrostatic potentials are −5kT/e (red) and +5kT/e (blue), respectively.
Molecular location of lipid binding ACPs is highly variable (Fig. 2 and Fig. 3A). For instance, they are located on the opposite side to their pY pockets for ZAP70-cSH2 and BMX-SH2 whereas they are on the same surface with or about perpendicular to the pY pockets for other SH2 domains, including Fyn-SH2, Syk-cSH2, phospholipase Cγ2 (PLCγ2)-cSH2, YES1-SH2, PLCγ1-cSH2, RASA1-nSH2, PTK-SH2, and Btk-SH2. Despite their positional variability, these sites do not overlap with any known protein binding sites, including the secondary binding site to the pY motif (Pawson, 2004), a pY-independent protein binding site reported for the PLCγ-nSH2 (Bae et al., 2009), and a second pY binding site reported for PLCγ1-cSH2 (Groesch et al., 2006). Indeed, molecular modeling of ZAP70-cSH2, Fyn-SH2, and PTK6-SH2 suggests that they can bind a lipid headgroup and a pY-motif simultaneously and independently (Fig. 3B). This notion was supported by their identical pY-peptide binding with and without PM vesicles (Fig. S3A) and unaltered vesicle binding in the presence of pY-peptides (Fig. S3B). We also performed the modeling of ZAP70-cSH2 interacting with the PM-mimetic bilayer and the phosphorylated T cell receptor (TCR) ζ chain (TCR-ζ), a key interaction partner of ZAP70 in the activated TCR signaling complex (Wang et al., 2010) (Fig. S3C). The modeling suggests that ZAP70-cSH2 can simultaneously bind a PI45P2 molecule in the lipid bilayer and a pY in its membrane-imbedded target protein. Further, the presence of PM-mimetic vesicles did not affect the binding of intact ZAP70 to the phosphorylated immunoreceptor tyrosine-based activation motif (ITAM) of TCR-ζ (Fig. S3D). These results indicate that ZAP70-cSH2 and other SH2 domains can simultaneously bind a pY and lipids under physiological conditions.
Fig. 3.

Variable location and morphology of lipid binding sites in SH2 domains. (A) The structures of Fyn-SH2 and PTK6-SH2 are shown in the same orientation with the pY-binding pocket (red arrows) pointing upward. ZAP70-cSH2 is rotated 180° to show the face opposite to the pY-binding pocket. Key cationic residues involved in lipid binding are shown in space filling representation and labeled. (B) Model structures of SH2-pY peptide-IP4 ternary complexes. SH2 domains are shown in surface representation and peptides (red arrows) and IP4 (green arrows) in stick representation. Notice that there is no steric overlap between the peptide and IP4 for all SH2 domains. See Movie S1–S3. (C) NMR chemical shift perturbations (CSPs) for backbone amide signals of 13C-labeled Fyn-SH2 in the presence of IP4. Residues with top 15% CSPs are labeled in the left panel and highlighted in red in the right panel. The residues whose NMR signals are missing after IP4 titration are colored in green. Notice that major CSPs are focused near K182 and R206 for Fyn-SH2.
Lipid binding ACPs are morphologically diverse. For example, they form a pocket or groove for ZAP70-cSH2, BMX-SH2, Fyn-SH2, Syk-cSH2, PLCγ2-cSH2, and YES1-SH2 whereas they are located on relatively flat surfaces for PTK6-SH2 and Btk-SH2 (Fig 2). This distinct morphology seems to affect the way they interact with lipids. In general, SH2 domains with an ACP groove show higher PtdInsP selectivity than those with a flat ACP. For instance, ZAP70-cSH2 has selectivity for PIP3 (Fig. S1) and mutation of residues in their cationic grooves (i.e., K176 and K186) abrogated the PIP3 selectivity (Fig. 4A). Also, our computational protein cavity analysis revealed a correlation between the ACP groove dimension and the lipid headgroup specificity (Table S2). The PIP3-selective ACP groove of ZAP70-cSH2 is ≈60% larger than that of the PI45P2-selective ACP groove of BMX-SH2. All these data support the notion that their ACP grooves are involved in specific PtdInsP headgroup recognition. However, it should be noted that ACP grooves of SH2 domains are considerably smaller than that of the PIP3-specific BTK PH domain (Fig. 2 and Table S2). In conjunction with the different PtdInsP selectivity displayed by SH2 and PH domains (Fig. S1), these structural differences suggest that SH2 and PH domains might recognize the PtdInsP headgroup by different mechanisms.
Fig. 4.

Membrane-binding properties and cellular activities of ZAP70 WT and mutants. (A) PIP3 dependence of ZAP70-cSH2 and its LOF mutants. ZAP70-cSH2 shows higher affinity when PI45P2 in the PM-mimetic vesicles (PM) is replaced by PIP3 (PM*) (blue curves). K176E/K186E (red curves) lost PIP3 selectivity whereas K206E/K251E (green curves) retained it, suggesting that K176 and K186 are directly involved in PIP3 headgroup recognition while K206 and K251 in non-specific binding to anionic membranes. (B) PIP3 dependence of a ZAP70-cSH2 GOF mutant. D184K shows enhanced membrane affinity while retaining PIP3 selectivity (orange). PM = PM mimetic vesicles. PM* = PM – PI45P2 + PIP3. (C) Time-course of Tyr phosphorylation of TCR signaling proteins in P116 cells expressing EGFP-tagged ZAP70 WT, lipid binding LOF mutants, and a pY binding site mutant after OKT3 stimulation. (D) Calcium mobilization in P116 cells expressing EGFP-tagged ZAP70 WT and mutants after OKT3 stimulation. Each circle represents a single cell. *** P < 0.001 (Student’s t-test) (E) IL-2 release from P116 cells expressing EGFP-tagged ZAP70 WT and mutants before and after OKT3 stimulation.
To corroborate that ACP grooves are involved in specific PtdInsP headgroup recognition, we monitored by solution NMR analysis the binding of 13C-labeled Fyn-SH2 to a soluble head group analog of PIP3, D-myo-inositol-(1,3,4,5)-tetrakisphosphate (IP4). Upon binding IP4, major chemical shift perturbations were detected primarily on the residues constituting (i.e., K182 and R206) or surrounding (e.g., Y185) the ACP groove; however, chemical shift perturbations were insignificant for the pY pocket residues (Fig. 3C). Collectively, these results support the notion that the ACP grooves serve as specific lipid binding sites for those SH2 domains with high lipid headgroup selectivity.
Role of Lipid Binding of ZAP70 c-SH2 Domain in T Cell Signaling
To investigate the physiological significance of lipid binding activities of SH2 domains, we selected ZAP70 for functional studies. ZAP70 is a Syk family tyrosine kinase that plays a central role in TCR signaling (Wang et al., 2010). It promotes the downstream T cell signaling by binding to double pY motifs of ITAMs of TCR-ζ via its tandem SH2 domains (tSH2) and subsequently phosphorylating LAT and SLP-76. This leads to further downstream signaling activities, including phosphorylation of PLCγ1 and ERK1/2 (Pollizzi and Powell, 2014).
Our modeling (Fig. 3B) suggested that K176 and K186 might be involved in PIP3 recognition whereas K206 and K251 might non-specifically interact with all anionic PM lipids. Consistent with this prediction, K176E/K186E lost PIP3 selectivity whereas K206E/K251E did not (Fig. 4A). Furthermore, PM localization of ZAP70-cSH2 wild type (WT) and K206E/K251E in HeLa cells was enhanced in response to rapamycin-triggered PIP3 enrichment whereas that of K176E/K186E was not altered (Fig. 1D). Both mutants had 4 to 5-fold lower membrane affinity than WT (Table S3), indicating that both specific PIP3 binding and non-specific anionic lipid binding contribute to membrane binding of ZAP70-cSH2.
To check if the lipid binding site of ZAP70-cSH2 is active in the context of the full-length protein, we measured the vesicle binding of ZAP70-tSH2 and intact ZAP70. SPR measurements showed that ZAP70-cSH2, ZAP70-tSH2 and the intact ZAP70 have similar vesicle affinity (Fig. S4A and Table S3). Also, K176E/K186E and K206E/K251E mutations reduced the vesicle binding affinity of ZAP70-cSH2, ZAP70-tSH2 and the intact ZAP70 to comparable degrees (Fig. 4A, Fig. S4C, and Table S3). These results, which are consistent with the crystal structure of ZAP70-tSH2 (Hatada et al., 1995) in which the ACP groove of ZAP70-cSH2 is fully exposed (Fig. S4D), show that the lipid binding site of ZAP70-cSH2 is fully functional in intact ZAP70 and that lipid binding activity of ZAP70 lies entirely within its cSH2 domain. Finally, neither mutation altered binding of ZAP70-cSH2 to a pY-containing peptide (Fig. S3E) or binding of intact ZAP70 to the dual phosphorylated ITAM of TCR-ζ (Fig. S3F).
To elucidate how differently the two types of lipid binding affect cellular activities of ZAP70, we compared the effects of introducing WT and the two lipid-binding loss-of-function (LOF) mutants of ZAP70 to ZAP70-deficient (P116) Jurkat cells on TCR signaling activities. Specifically, we monitored the time-dependent tyrosine phosphorylation of ZAP70 and its downstream proteins, LAT, PLCγ1 and ERK1/2 after stimulation with an anti-CD3 antibody, OKT3. Tyr phosphorylation of these proteins was completely suppressed in P116 cells; however, when ZAP70 WT was reconstituted in P116 cells, all proteins were rapidly phosphorylated after stimulation, which lasted for 10–15 min (Fig. 4C). Interestingly, two LOF mutants of ZAP70 showed distinctively different temporal phenotypes. K206E/K251E was much less effective than WT throughout the entire activation period whereas the PIP3-specific mutant, K176E/K186E, was almost as active as WT at an earlier stage but became much less effective at later stages (i.e., >5 min). These results suggest that PIP3-ZAP70 interaction is important not for initiating the TCR signaling activity of ZAP70 but for sustaining it. We also measured the cellular calcium flux and interleukin-2 release after OKT3 stimulation. P116 cells again showed no calcium flux and interleukin-2 release after OKT3 stimulation, but stable expression of ZAP70 WT dramatically enhanced these activities (Fig. 4D and 4E). In both assays, K176E/K186E and K206E/K251E were much less active than WT although the former was slightly more active than the latter.
We also prepared a gain-of-function (GOF) mutant for ZAP70, D184K, which had higher membrane affinity than WT (Table S3) but retained PIP3 specificity (Fig. 4B) and pY-binding affinity (Fig. S3E). Consistent with increased membrane affinity, D184K was considerably more active than the WT protein in both calcium flux and ERK1/2 phosphorylation assay (Fig. S5). As a control, we also measured the signaling activity of a pY binding-deficient mutant of ZAP70 (R190A/R192A) and it showed no detectable activity in all assays (Fig. 4C, 4D, and 4E).
To better understand the mechanisms underlying the distinct cellular phenotypes of the ZAP70 mutants, we monitored dynamic interaction of ZAP70 WT and mutants with TCR-ζ by two-color single molecule tracking (Koyama-Honda et al., 2005; Sheng et al., 2012). We first transfected EGFP-ZAP70 and the SNAP-tetramethylrhodamine (TMR)-labeled TCR-ζ into P116 cells and tracked these molecules simultaneously. Although ZAP70 is a cytosolic protein, it showed a high degree of PM localization before stimulation as previously reported (Huby et al., 1997). However, it showed a low degree of colocalization with TCR-ζ until cells were stimulated with OKT3 (Fig. 5A and Movie S1 and S2).
Fig. 5.

Single molecule tracking of ZAP70 and TCR-ζ in the TCR complex. (A)-(B) Representative images of EGFP-ZAP70 WT (A) or K206E/K251E (B) and SNAP-TMR-labeled TCR-ζ in a P116 cell before and after OKT3 stimulation are shown. Green, red, and yellow lines are trajectories of ZAP70, TCR-ζ, and co-localized molecules, respectively. Arrows indicate the starting points of tracking. See also Movie S4–S7. The scale bar indicates 1 μm. (C) Time-courses of the half-life of colocalization for EGFP-ZAP70 WT and lipid-binding mutants with SNAP-TMR-TCR-ζ. (D) Time-courses of the half-life of colocalization for EGFP-ZAP70 WT and a pY-site mutant with SNAP-TMR-TCR-ζ. The half-life of co-localization was calculated from the dissociation rate constant. The same size of PM surface was analyzed for each histogram. Error bars indicate S.D. (n = 50–100).
Detailed kinetic analysis of ZAP70-TCR-ζ colocalization showed how differently OKT3 stimulation modulates the dynamic colocalization of TCR-ζ with ZAP70 WT and various mutants. For ZAP70 WT, the OKT3 stimulation for 2 min caused about a ≈2-fold increase in ZAP70-TCR-ζ colocalization in terms of half-life of colocalization, which lasted for >10 min before it was reduced to the basal level (Fig. 5C and Fig. S6A). Under the same conditions, K206E/K251E displayed no significant increase in colocalization with TCR-ζ throughout the entire activation period (Fig. 5B, 5C, Movie S3–S4, and Fig. S6B). Interestingly, K176E/K186E showed a significant degree of co-localization with TCR-ζ at 2–5 min just as WT did, but the co-localization rapidly decreased thereafter (Fig. 5C and Fig. S6C). Pre-treatment of P116 cells with a phosphoinositide 3-kinase (PI3K) inhibitor, LY294002, exerted essentially the same effect as the K176E/K186E mutation (Fig. 5C and Fig. S6D). The GOF mutant (D184K) showed considerably enhanced PM recruitment even before OKT3 stimulation and a higher degree of co-localization with TCR-ζ than WT over the entire OKT3 stimulation period (Fig. 5C and Fig. S6E). In contrast, the pY binding-deficient mutant, R190A/R192A, consistently showed much lower co-localization with TCR-ζ than WT (Fig. 5D). Collectively, these results suggest that initial PM lipid (e.g., PI45P2) binding of ZAP70-cSH2 is essential for facilitating the interaction of ZAP70 with TCR-ζ and that its subsequent binding to PIP3 produced by PI3K activation is important for their sustained interaction. This notion is also consistent with the unique temporal effects of the PIP3-specific K176E/K186E mutation on the TCR signaling activity of ZAP70 (Fig. 4C).
To assess the physiological relevance of the lipid-mediated control of ZAP70-TCR-ζ interaction observed in single molecule imaging, we also performed co-immuoprecipitation of ZAP70 WT and mutants with TCR-ζ. As expected, OKT stimulation dramatically enhanced the co-immuoprecipitation of ZAP70 WT with TCR-ζ (Fig. S6F). Under the same conditions, however, OKT stimulation caused ≈50% less co-immuoprecipitation between ZAP70-K206E/K251E and TCR-ζ, which was comparable to the degree of co-immuoprecipitation between ZAP70-R190A/R192A and TCR-ζ. Since both mutants still possess an intact nSH2, they are expected to show some degree of interaction with TCR-ζ under the conditions of co-immuoprecipitation involving overexpressed proteins. It should be noted, however, that even under these conditions compromised lipid binding by the K206E/K251E mutation caused essentially the same negative effect on ZAP70-TCR-ζ interaction as loss of pY binding by the R190A/R192A mutation.
Role of Non-Specific Lipid Binding of SH2 Domains in Cell Signaling Pathways
PTK6 is a non-myristoylated, non-receptor tyrosine kinase that is aberrantly expressed in several types of human cancer (Brauer and Tyner, 2009). To check if non-specific lipid binding of SH2 domains is also important for the cellular function and regulation of their host proteins, we measured the effect of the altering lipid binding activity of PTK6-SH2 on the cellular activity of PTK6. It was recently reported that in a prostate cancer cell line (PC3) the pool of endogenous activated PTK6, marked by auto-phosphorylation at Y342, was localized at the PM and promoted the phosphorylation and activation of ERK5 and Akt (Zheng et al., 2012). Consistent with the report, activation of HEK293 cells overexpressing PTK6 WT led to PTK6 Y342 phosphorylation (Fig. S7B), PM recruitment (Fig. S7E), and phosphorylation of ERK5 (Fig. S7C) and Akt1 (Fig. S7D). However, a LOF mutant (R131A/R136A) with ≈5-fold lower PM affinity than WT (Fig. S7A) showed a dramatically reduced degree of Y342 autophosphorylation (Fig. S7B) and PM localization (Fig. S7E), and was much less effective than WT in phosphorylating ERK5 and Akt1 (Fig. S7C and S7D). As a control, we also measured the effects of the pY binding site mutation (R85A/R105A) and it was much less active than WT in all assays. These results lend further credence to the notion that lipid binding activity of SH2 domains, whether it is through specific headgroup recognition or non-specific binding, is important for cell signaling.
DISCUSSION
The present study demonstrates that most SH2 domains bind PM lipids with high affinity via ACPs. Many ACP-containing SH2 domains also bind PtdInsPs as specifically as PH domains. All SH2 domains characterized herein have lipid-binding ACPs that are topologically and functionally separate from the pY pockets, allowing them to bind the pY peptide and lipids independently. This salient feature enables them to serve as dual-specificity lipid- and protein-binding modules whose protein interaction may be directly and specifically modulated by various lipids.
Although the location of lipid binding ACPs is highly variable, they are expected to be surface-exposed and fully functional in the full-length proteins, as evidenced by comparable membrane affinity of ZAP70-cSH2, ZAP70-tSH2, and the intact ZAP70 (Table S3). This would allow constitutive PM recruitment of SH2 domain-containing proteins (see Fig. 1), priming them for agonist-induced binding to pY-containing proteins. Lipids can recruit and compartmentalize a wide range of effector proteins to confined membrane locations (Cho and Stahelin, 2005; Lemmon, 2008) and also allosterically regulate their structures and orientation (Cho, 2006). These activities should directly and specifically facilitate binding of SH2 domains to pY-containing protein partners. Consequently, relative location of the ACP and the pY pocket would dictate how PM-associated SH2 domain proteins interact with their interaction partners. Highly variable location of lipid binding ACPs in SH2 domains would thus allow for flexible lipid-mediated regulatory mechanisms for pY signaling pathways.
Variable morphology of lipid-binding ACPs also enables SH2 domains to bind lipids by different mechanisms. Groove-forming ACPs can specifically recognize lipid headgroups whereas flat ACPs tend to non-specifically interact with anionic lipids. This correlation between the ACP morphology and the lipid binding mechanism is corroborated by our PtdInsP selectivity measurements and computational cavity analysis. Our SPR analysis shows that ≈60% of tested SH2 domains have definite PtdInsP specificity. This in turn indicates that a significant number of SH2 domains non-specifically bind PM lipids. Physiological significance of specific PtdInsP binding by lipid binding domains has been well documented (Di Paolo and De Camilli, 2006; Lemmon, 2008) but importance of non-specific lipid binding in cellular function and regulation has only recently started to be appreciated (Cho, 2006; Heo et al., 2006; McLaughlin and Murray, 2005; Winters et al., 2005; Wu et al., 2007). Our cell studies on ZAP70 and PTK6 show that both specific headgroup recognition and non-specific lipid binding by SH2 domains are physiologically important. It is thus expected that lipids will play important roles in modulating cellular activities of most, if not all, proteins with high-affinity lipid-binding SH2 domains, whether they bind lipids specifically or non-specifically. Functional studies on ZAP70 and PTK6 with different cellular functions also suggest that the lipid-SH2 interaction may be a common critical regulatory step for most pY signaling pathways involving lipid-binding SH2 domains.
Functional and single molecule studies of ZAP70 demonstrate how multiple lipids work in concert to achieve exquisite spatiotemporal control over SH2 domain-mediated protein-protein interactions and cell signaling activities. ZAP70-cSH2 has PIP3 selectivity but it can also tightly bind PI45P2-containing PM-mimetic vesicles (Fig. S1 and Table 1). As a result, ZAP70-cSH2 (Fig. 1) and intact ZAP70 (Fig. 5) exhibit a high degree of PM recruitment before TCR stimulation. Judging from the inability of ZAP70 K206E/K251, which has much reduced affinity for the PM but intact affinity for pY, to effectively interact with TCR-ζ in response to OKT3 stimulation, this constitutive PM binding seems to be essential for facilitating binding of ZAP70 to target proteins in the activated TCR signaling complex. The subsequent PI3K-mediated conversion of PI45P2 into PIP3, which would specifically bind to the ACP groove of ZAP70-cSH2 and thus enhance the PM binding of ZAP70, appears to be crucial for elongated interaction of ZAP70 with TCR-ζ in the activated TCR complex. Differential temporal regulation of ZAP70 activities by these two distinct types of lipid binding epitomizes how a concerted action of multiple lipids enables exquisite spatiotemporal coordination of protein-protein interactions and cell signaling activities. Given that other proteins involved in B and T cell signaling also have high PM affinity and PIP3 selectivity (Table 1), PM lipids and PIP3 might spatiotemporally modulate and coordinate the actions of multiple SH2 domain proteins during T or B cell activation, leading to high-fidelity pY signaling.
Strong lipid binding activity of two primordial SH2 domains (Liu and Nash, 2012), SPT6-cSH2 and STAT6-SH2, suggests that this activity was adopted at least as early as their pY binding activity in evolution (Table 1). This point is further supported by the finding that human and Saccharomyces cerevisiae SPT6-cSH2 domains have essentially identical affinity for PM-mimetic vesicles. Since both SPT6 and STAT are nuclear proteins involved in gene regulation, the lipid binding activity of SH2 domains must have been acquired even before the PM became the central stage for pY signaling. Normally, functionally important residues are highly conserved through evolution. As described above, however, variability of lipid binding residues among different SH2 domains is evolutionarily more advantageous than conversation because the former allows SH2 domains to adopt different modes of lipid and pY binding that ideally suit their cellular functions. It should be stressed, however, that for each SH2 domain, the conservation of lipid binding residues among species is extremely high. For instance, multiple sequence alignment of ZAP70-cSH2 and Fyn-SH2 orthologs shows that essential lipid binding residues are absolutely or highly conserved through evolution.
Since many SH2 domain proteins are implicated in human diseases, SH2 domains have been targeted for drug development (Kraskouskaya et al., 2013); however, extensive efforts to modulate their pY binding have met with limited success, due to structural similarity and ligand promiscuity of the pY-binding pockets. Our study showing the ligand specificity and structural diversity of lipid binding sites of SH2 domains, in conjunction with the finding that many disease-causing mutations of SH2 domain proteins are found in the lipid-binding sites of SH2 domains (Table 2), could point a way toward a new alternate strategy for controlling pY signaling activities with improved specificity and potency.
Table 2.
Disease-Causing Mutations in the Lipid Binding Sites of SH2 Domainsa
| SH2 domains | Lipid binding residuesb | Mutationsc |
|---|---|---|
| BRK(PTK6)-SH2 | R131, R136 | COSMIC: R131P (1), R131L (1) |
| YES1-SH2 | R215, K216 | COSMIC: R215M (1) |
| BLNK-SH2 | K380, R399, R411, K412 | ClinVar: R399N (malignant melanoma) COSMIC: R399Q (1) |
| PLCγ2-cSH2 | K687, R727, K728 | COSMIC: R727Q (2) |
| FYN-SH2 | K182, R206, K207 | COSMIC: R206C (5) |
| BMX-SH2 | K313, K315 | COSMIC: K313Q (1) |
Obtained from three public-domain databases: HGMD (Human Gene Mutation Database), ClinVar, and COSMIC (Catalogue Of Somatic Mutations In Cancer). Mutations in the Professional version of HGMD are not included.
The Identified from our mutational and NMR analyses.
The numbers in the parenthesis mean the number of reported occurrence in the COSMIC database.
Lastly, it should be noted that our results on lipid binding sites of SH2 domains are at odds with previous studies suggesting that some SH2 domains bind lipids in their pY pockets (Hong et al., 2009; Rameh et al., 1995; Tokonzaba et al., 2006). Although the origin of this discrepancy is not fully understood, one can speculate that some SH2 domains without ACPs may be able to bind a lipid(s) in their pY pockets given that the shape and electrostatic properties of pY pockets vary significantly among SH2 domains. For such SH2 domains lipid and protein binding could be mutually exclusive or interfere with each other. Obviously, further studies on other SH2 domain-containing proteins are necessary to fully elucidate different regulatory mechanisms for lipid-mediated protein interactions in pY signaling.
EXPERIMENTAL PROCEDURES
Full protocols can be found in Extended Experimental Procedures.
Protein Expression and Purification
All EGFP-tagged SH2 domains and full-length proteins were expressed as His6-tagged proteins in E. coli BL21 (DE3) pLysS (Novagen) and purified using the Ni-NTA-Agarose resin (Qiagen).
Lipid Vesicles Preparation and SPR Analysis
PM-mimetic vesicles were prepared by mixing POPC, POPE, POPS, cholesterol, PI, and PI45P2 in a molar ratio of 12:35:22:22:8:1. All SPR measurements were performed at 23°C using the lipid-coated L1 chip in the BIACORE X or T100 system as described (Stahelin and Cho, 2001). 20 mM Tris-HCl, pH 7.4, containing 0.16 M NaCl was used as the running buffer while PM-mimetic vesicles and POPC vesicles were coated on the active surface and the control surface, respectively. Flow rate was 5 μl/min and 20–30 μl/min for equilibrium and kinetic measurements, respectively. Relative response (in the scale of 0–1) was calculated by dividing RU values by the maximal RU obtained for a particular protein under a given condition and used for most figures to minimize data variation caused by experimental factors, most notably different instrumental parameters of three separate SPR instruments used for measurements.
Stable Cell Line Preparation
A Jurkat cell line derivative, P116 (CRL-2676), was from ATCC and cultured in RPMI 1640 containing 5% FBS. GFP-ZAP70 WT and mutants were stably expressed in P116 by retroviral gene transduction. A retroviral construct, pLEGFP-N1-hZap70, was cloned by PCR and point mutations were introduced by site-directed mutagenesis. Retrovirus preparation and transduction were performed as previously described (Kim et al., 2013).
Rapamycin-inducible PI45P2 depletion and PIP3 enrichment
The PI45P2 depletion was performed in HeLa cells according to a reported procedure (Inoue et al., 2005; Varnai et al., 2006) using Lyn-based PM-anchored FKBP12-rapamycin binding (FRB) domain of mTOR (Lyn-FRB) and the FK506-binding protein-12-yeast inositol polyphosphate 5-phosphatase domain fusion protein (FKBP-Inp). PM translocation of mCherry-ZAP70-cSH2 WT and mutants was monitored in the presence of 2.5 μM rapamycin analog, A/C Heterodimerizer (Clontech). The PIP3 enhancement system consists of Lyn-FRB and YFP-tagged FKBP with an inter SH2 domain of p85β (Inoue and Meyer, 2008). Robustness of these systems was tested using microinjected PI45P2 (Yoon et al., 2011) and PIP3 (Liu et al., 2014) sensors.
Single cell calcium imaging
Cells were loaded with 2 μg/ml Fura-2 AM (Life Technologies) in phenol red-free DMEM containing 0.5% FBS for 30 min at 37°C, washed, and plated to a poly-L-lysine-coated culture plate for imaging. The changes in the fluorescence emission of calcium-bound and unbound form of Fura-2 were captured and the ratio of individual cells was analyzed using MetaFluor (Molecular devices) and Prism (GraphPad) software.
Tyrosine Phosphorylation Assay
Cells expressing WT or mutant GFP-ZAP70 were stimulated with OKT3 (3 μg/ml) at 37°C for indicated time. Cells were then lysed with the ice-cold radioimmunoprecipitation assay buffer supplemented with a mixture of protease and protein phosphatase inhibitors. Proteins were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and probed with antibodies against pLAT (Y191), pPLCγ1 (Y783), pERK1/2 (T202/Y204), pZAP70 (Y493) and β-actin. The chemiluminescence was detected with ImageQuant LAS 4000 (GE Healthcare).
IL-2 ELISA
Cells were added to 96 well plate (2 × 106 cells per well) coated with OKT3 and anti-CD28 antibodies and incubated for 24 h. The levels of IL-2 in culture supernatants were measured by sandwich ELISA (eBioscience).
Single Molecule Tracking by Total Internal Reflection Fluorescence Microscopy
Single molecule imaging was performed in P116 cells using a custom-built total internal reflection fluorescence microscope as described previously (Sheng et al., 2012). P116 cells stably expressing EGFP-ZAP70 WT (or mutants) were transiently transfected with SNAP®-TCR-ζ, which was subsequently labeled with SNAP-Cell® tetramethylrhodamine (TMR)-Star® (New England Biolabs). Labeled cells were washed, attached to a poly-L-lysine-coated glass plate, stimulated with 10 μg/ml OKT3, and ZAP70 and TCR-ζ molecules were simultaneously tracked. PIP3 depletion was achieved by pre-treating the cells with 50 μM LY294002 for 1 h before OKT3 stimulation. All particle tracking, data analysis and image processing were carried out with in-house programs written in MATLAB. Co-localization analysis of two molecules was performed with a fixed threshold criterion (i.e., <400 nm) for co-localization (Koyama-Honda et al., 2005). The same size of PM surface was analyzed for each data. The percentage of TCR-ζ molecules spending a given colocalzation time (>0.2 sec) with ZAP70 on the PM was calculated from the total number of TCR-ζ molecules and displayed as a histogram. Data were fit into a single exponential decay equation (i.e., P = Po e−kt) to determine the dissociation rate constant (k) values by non-linear least-squares analysis and the half-life values of co-localization were calculated as ln2/k. 50–100 images were analyzed for each data point.
Structural Modeling, Electrostatic Analysis, and Docking of SH2 Domains
The models for SH2 domains were created with Modeller 9.9 (Sali and Blundell, 1993). DelPhi (Rocchia et al., 2001) was used for assigning electrostatic potentials to the atoms of the structures using CHARMM force field (MacKerell et al., 1998). The calculated potentials were loaded into GRASP2 (Petrey and Honig, 2003) for subsequent visualization. Docking of the lipids to the structure of SH2 domains was performed with the DOCK program of the package DOCK 6, using Amber score program, which allows both the ligand and the active site of the receptor to be flexible (Graves et al., 2008).
NMR Data Acquisition and Processing
The 1H-, and 13C-HSQC experiments were recorded at 600- and 800-MHz on Bruker Avance spectrometers at 20°C. The samples contained 0.1 mM of 13C-labeled Fyn-SH2 in 25 mM potassium phosphate, 2 mM dithiothreitol and 5% deuterium oxide (pH 6.0). The chemical shift perturbations, upon addition of 1 mM IP4 were calculated from 1H and 13C chemical shifts using a weighted averaging scheme.
Supplementary Material
HIGHLIGHTS.
SH2 domains bind lipids with high affinity and specificity
Lipids coordinate SH2 domain-mediated protein-protein interactions.
Lipids spatiotemporally control ZAP70 signaling activities in T cells.
Lipid binding sites of SH2 domains represent a pharmacological target.
Acknowledgments
We thank Dr. Diana Murray for helpful discussion on non-specific electrostatic interactions, Dr. Takanari Inoue for a generous gift of PI45P2 depletion and PIP3 enrichment systems and Kayoung Lee for help with calcium imaging experiments. This work was in part supported by the grants from the National Institutes of Health (GM68849 and GM110128 for W.C. and GM030518, GM094597, and CA121852 for B.H.), National Research Foundation of Korea (NRF-2013R1A1A2074573) and the World Class University program R31-2008-000-10105-0 by the Korean government.
Footnotes
Supplemental Information includes Extended Experimental Procedures, seven figures, three tables, and four movies, and can be found with this article online.
AUTHOR CONTRIBUTION
MJP, RS, HK, SS, WN, EK, DGL, IS, LW and YC designed and performed biochemical studies and ZGW and YX performed imaging work. IK, CSH, and SR cloned the SH2 domains. AS and BH performed computational work and PTR and JL carried out NMR analysis. DJJ, MHJ, and YMK handled all cell studies. WC and YMK conceived the work and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YS, Cantley LG, Chen CS, Kim SR, Kwon KS, Rhee SG. Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:4465–4469. doi: 10.1074/jbc.273.8.4465. [DOI] [PubMed] [Google Scholar]
- Brauer PM, Tyner AL. RAKing in AKT: a tumor suppressor function for the intracellular tyrosine kinase FRK. Cell Cycle. 2009;8:2728–2732. doi: 10.4161/cc.8.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D. Signaling complexes: biophysical constraints on intracellular communication. Annu Rev Biophys Biomol Struct. 1998;27:59–75. doi: 10.1146/annurev.biophys.27.1.59. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sheng R, Kallberg M, Silkov A, Tun MP, Bhardwaj N, Kurilova S, Hall RA, Honig B, Lu H, et al. Genome-wide Functional Annotation of Dual-Specificity Protein- and Lipid-Binding Modules that Regulate Protein Interactions. Mol Cell. 2012;46:226–237. doi: 10.1016/j.molcel.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. Building signaling complexes at the membrane. Sci STKE. 2006;2006:pe7. doi: 10.1126/stke.3212006pe7. [DOI] [PubMed] [Google Scholar]
- Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Feng W, Zhang M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat Rev Neurosci. 2009;10:87–99. doi: 10.1038/nrn2540. [DOI] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves AP, Shivakumar DM, Boyce SE, Jacobson MP, Case DA, Shoichet BK. Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J Mol Biol. 2008;377:914–934. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groesch TD, Zhou F, Mattila S, Geahlen RL, Post CB. Structural basis for the requirement of two phosphotyrosine residues in signaling mediated by Syk tyrosine kinase. J Mol Biol. 2006;356:1222–1236. doi: 10.1016/j.jmb.2005.11.095. [DOI] [PubMed] [Google Scholar]
- Hatada MH, Lu X, Laird ER, Green J, Morgenstern JP, Lou M, Marr CS, Phillips TB, Ram MK, Theriault K, et al. Molecular basis for interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature. 1995;377:32–38. doi: 10.1038/377032a0. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Chalkia D, Ko KD, Bhardwaj G, Chang GS, van Rossum DB, Patterson RL. Phylogenetic Profiles Reveal Structural and Functional Determinants of Lipid-binding. J Proteomics Bioinform. 2009;2:139–149. doi: 10.4172/jpb.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huby RD, Iwashima M, Weiss A, Ley SC. ZAP-70 protein tyrosine kinase is constitutively targeted to the T cell cortex independently of its SH2 domains. J Cell Biol. 1997;137:1639–1649. doi: 10.1083/jcb.137.7.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods. 2005;2:415–418. doi: 10.1038/nmeth763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Meyer T. Synthetic activation of endogenous PI3K and Rac identifies an AND-gate switch for cell polarization and migration. PLoS One. 2008;3:e3068. doi: 10.1371/journal.pone.0003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Huh J, Hwang M, Kwon EH, Jung DJ, Brinkmann MM, Jang MH, Ploegh HL, Kim YM. Acidic amino acid residues in the juxtamembrane region of the nucleotide-sensing TLRs are important for UNC93B1 binding and signaling. J Immunol. 2013;190:5287–5295. doi: 10.4049/jimmunol.1202767. [DOI] [PubMed] [Google Scholar]
- Koyama-Honda I, Ritchie K, Fujiwara T, Iino R, Murakoshi H, Kasai RS, Kusumi A. Fluorescence imaging for monitoring the colocalization of two single molecules in living cells. Biophys J. 2005;88:2126–2136. doi: 10.1529/biophysj.104.048967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraskouskaya D, Duodu E, Arpin CC, Gunning PT. Progress towards the development of SH2 domain inhibitors. Chem Soc Rev. 2013;42:3337–3370. doi: 10.1039/c3cs35449k. [DOI] [PubMed] [Google Scholar]
- Ladbury JE, Arold S. Searching for specificity in SH domains. Chem Biol. 2000;7:R3–8. doi: 10.1016/s1074-5521(00)00067-3. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Lim WA, Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010;142:661–667. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BA, Nash PD. Evolution of SH2 domains and phosphotyrosine signalling networks. Philos Trans R Soc Lond B Biol Sci. 2012;367:2556–2573. doi: 10.1098/rstb.2012.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Sheng R, O’Connor MJ, Cui Y, Yoon Y, Kurilova S, Lee D, Cho W. Simultaneous in situ quantification of two cellular lipid pools using orthogonal fluorescent sensors. Angew Chem Int Ed Engl. 2014;53:14387–14391. doi: 10.1002/anie.201408153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, Mayer BJ. The SH2 domain: versatile signaling module and pharmaceutical target. Biochim Biophys Acta. 2005;1747:1–25. doi: 10.1016/j.bbapap.2004.10.005. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Manna D, Albanese A, Park WS, Cho W. Mechanistic basis of differential cellular responses of phosphatidylinositol 3,4-bisphosphate- and phosphatidylinositol 3,4,5-trisphosphate-binding pleckstrin homology domains. J Biol Chem. 2007;282:32093–32105. doi: 10.1074/jbc.M703517200. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- Petrey D, Honig B. GRASP2: visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]
- Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh LE, Chen CS, Cantley LC. Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell. 1995;83:821–830. doi: 10.1016/0092-8674(95)90195-7. [DOI] [PubMed] [Google Scholar]
- Rocchia W, Alexov E, Honig B. Extending the Applicability of the Nonlinear Poisson-Boltzmann Equation: Multiple Dielectric Constants and Multivalent Ions. J Phys Chem B. 2001;105:6507–6514. [Google Scholar]
- Sadowski I, Stone JC, Pawson T. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol Cell Biol. 1986;6:4396–4408. doi: 10.1128/mcb.6.12.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng R, Chen Y, Yung Gee H, Stec E, Melowic HR, Blatner NR, Tun MP, Kim Y, Kallberg M, Fujiwara TK, et al. Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain-containing scaffold proteins. Nat Commun. 2012;3:1249. doi: 10.1038/ncomms2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng R, Kim H, Lee H, Xin Y, Chen Y, Tian W, Cui Y, Choi JC, Doh J, Han JK, et al. Cholesterol selectively activates canonical Wnt signalling over non-canonical Wnt signalling. Nat Commun. 2014;5:4393. doi: 10.1038/ncomms5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin RV, Cho W. Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions: a surface plasmon resonance study on phospholipases A2. Biochemistry. 2001;40:4672–4678. doi: 10.1021/bi0020325. [DOI] [PubMed] [Google Scholar]
- Surdo PL, Bottomley MJ, Arcaro A, Siegal G, Panayotou G, Sankar A, Gaffney PR, Riley AM, Potter BV, Waterfield MD, et al. Structural and biochemical evaluation of the interaction of the phosphatidylinositol 3-kinase p85alpha Src homology 2 domains with phosphoinositides and inositol polyphosphates. J Biol Chem. 1999;274:15678–15685. doi: 10.1074/jbc.274.22.15678. [DOI] [PubMed] [Google Scholar]
- Tokonzaba E, Capelluto DG, Kutateladze TG, Overduin M. Phosphoinositide, phosphopeptide and pyridone interactions of the Abl SH2 domain. Chem Biol Drug Des. 2006;67:230–237. doi: 10.1111/j.1747-0285.2006.00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Thyagarajan B, Rohacs T, Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol. 2006;175:377–382. doi: 10.1083/jcb.200607116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, Weiss A. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol. 2010;2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters MJ, Lamson RE, Nakanishi H, Neiman AM, Pryciak PM. A membrane binding domain in the ste5 scaffold synergizes with gbetagamma binding to control localization and signaling in pheromone response. Mol Cell. 2005;20:21–32. doi: 10.1016/j.molcel.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Wu H, Feng W, Chen J, Chan LN, Huang S, Zhang M. PDZ domains of Par-3 as potential phosphoinositide signaling integrators. Mol Cell. 2007;28:886–898. doi: 10.1016/j.molcel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Yoon Y, Lee PJ, Kurilova S, Cho W. In situ quantitative imaging of cellular lipids using molecular sensors. Nat Chem. 2011;3:868–874. doi: 10.1038/nchem.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Asara JM, Tyner AL. Protein-tyrosine kinase 6 promotes peripheral adhesion complex formation and cell migration by phosphorylating p130 CRK-associated substrate. J Biol Chem. 2012;287:148–158. doi: 10.1074/jbc.M111.298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P, Meerschaert K, Reekmans G, Leenaerts I, Small JV, Vandekerckhove J, David G, Gettemans J. PIP(2)-PDZ domain binding controls the association of syntenin with the plasma membrane. Mol Cell. 2002;9:1215–1225. doi: 10.1016/s1097-2765(02)00549-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.