

Abstract
Psychiatric disorders are complex multifactorial illnesses involving chronic alterations in neural circuit structure and function as well as likely abnormalities in glial cells. While genetic factors are important in the etiology of most mental disorders, the relatively high rates of discordance among identical twins, particularly for depression and other stress-related syndromes, clearly indicate the importance of additional mechanisms. Environmental factors such as stress are known to play a role in the onset of these illnesses. Exposure to such environmental insults induces stable changes in gene expression, neural circuit function, and ultimately behavior, and these maladaptations appear distinct between developmental versus adult exposures. Increasing evidence indicates that these sustained abnormalities are maintained by epigenetic modifications in specific brain regions. Indeed, transcriptional dysregulation and the aberrant epigenetic regulation that underlies this dysregulation is a unifying theme in psychiatric disorders. Here, we provide a progress report of epigenetic studies of the three major psychiatric syndromes, depression, schizophrenia, and bipolar disorder. We review the literature derived from animal models of these disorders as well as from studies of postmortem brain tissue from human patients. While epigenetic studies of mental illness remain at early stages, understanding how environmental factors recruit the epigenetic machinery within specific brain regions to cause lasting changes in disease susceptibility and pathophysiology is revealing new insight into the etiology and treatment of these conditions.
Keywords: depression, schizophrenia, bipolar disorder, histone acetylation, histone methylation, DNA methylation, microRNA, 3D chromatin structure
Introduction
Psychiatric disorders, which are among the leading causes of disease burden worldwide, impose an ever-increasing burden on humanity. All major psychiatric syndromes are complex, heterogeneous conditions resulting from the interaction of several factors including genetic, neurobiological, cultural, and life experiences. Moreover, each of these syndromes is characterized by functional and transcriptional alterations in several limbic brain regions implicated in regulating stress responses, reward, and cognition (Box 1).
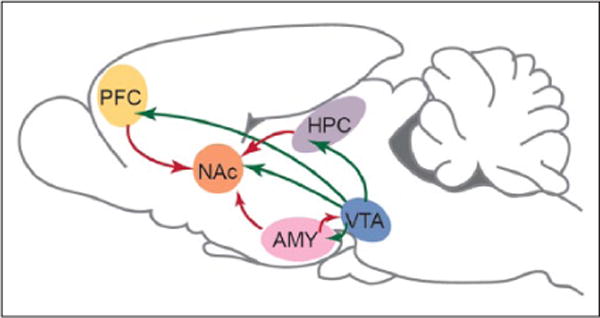
Box 1. Limbic Brain Circuitry Implicated in Mental Illness.
A majority of research on altered epigenetic regulation in psychiatric disorders has focused on changes within the brain’s limbic circuitry, depicted here in rodent brain. This circuitry involves highly interconnected brain structures important for interpreting and responding to rewarding and aversive stimuli and for several domains of cognitive function (e.g., attention, working memory, and declarative memory). Depicted are dopaminergic neurons (green), which project from the ventral tegmental area (VTA) of the midbrain to NAc, PFC, amygdala (AMY), and hippocampus (HPC), among other regions. The NAc receives excitatory glutamatergic innervation (red) from the HPC, PFC, and AMY, while the HPC, PFC, and AMY exhibit reciprocal glutamatergic projections. Not shown are regions of the hypothalamus, and serotoninergic and noradrenergic nuclei, which are also inter-connected with the limbic brain and influence psychiatric disorders. From Peña and others (2014) with permission.
Increasing evidence over the last decade has identified epigenetic mechanisms as important effectors in psychiatric conditions (Adachi and Monteggia 2014; Akbarian 2014; Guidotti and Grayson 2014; Nestler 2014; Peña and others 2014). Indeed, being at the foundation of gene regulation, epigenetic mechanisms are ideal candidates for the study of psychiatric syndromes that are caused by the interactions between genetic factors and environmental exposures. Epigenetic mechanisms refer to the highly complex organization of DNA in a cell nucleus and include many types of histone and DNA modifications as well as alterations in many types of non-histone proteins and noncoding RNAs (Jaenisch and Bird 2003; Jenuwein and Allis 2001). Work to date implicating epigenetic regulation in the context of psychiatric syndromes has come from both animal models and postmortem human brains, although to a different degree for various psychiatric disorders as will be seen.
Early development marks a time of dramatic changes in the brain as well as enhanced susceptibility to many environmental insults. Epigenetic mechanisms of gene regulation are a particularly attractive explanation for how early life exposures to stress, toxins, and other stimuli exert life-long effects on neuropsychiatric phenomena (Kundakovic and Champagne 2015; Meaney 2001; Peña and others 2014). Indeed, developmental exposures may have broader impact on epigenetic states and brain circuits than similar exposures later in life. It is therefore important, in characterizing the epigenetic contributions to mental illness, to perform studies across the life cycle.
The present review brings together findings relating to epigenetic mechanisms in the three major psychiatric syndromes: depression, schizophrenia (SCZ), and bipolar disorder. We begin with a brief summary of epigenetic regulation in general (Nestler, 2014) and then present a progress report of how epigenetic studies of mental illnesses, from both adult and developmental perspectives, are providing new insight into the biological basis of these complex disorders and their treatment (Akbarian 2014; Guidotti and others 2005; Peña and others 2014). Due to space limitations, we focus on histone and DNA modifications and do not include discussion of noncoding RNAs, which are also proving to be important in epigenetic regulation in psychiatric disorders (Issler and Chen 2015). We also focus on epigenetic regulation in brain, and make only passing mention of investigations of blood and peripheral tissues. It is possible that peripheral measures, even if the specific changes are different from those in brain, could reflect brain regulation, an important empirical question for future research.
Overview of Epigenetic Mechanisms
The three billion nucleotides of DNA in a mammalian genome would be ~2 meters long if stretched out linearly, yet fit within a microscopic cell nucleus due to an extraordinary degree of organization and compaction in chromatin—nuclear material composed of DNA, histones, and non-histone proteins (Jaenisch and Bird 2003). The fundamental unit of chromatin is the nucleosome, which consists of ~147 base pairs of DNA wrapped around a core histone octamer (~1.65 turns). Each octamer contains two copies each of the histones H2A, H2B, H3, and H4 (Fig. 1A). Epigenetic mechanisms control the spacing of nucleosomes and the degree to which they are condensed, which thereby determines gene activity. In simplified terms, chromatin exists across a continuum between an inactivated, condensed state (heterochromatin), which does not allow transcription of genes, and an activated, open state (euchromatin), which allows individual genes to be transcribed (Fig. 2). Regulation of the state of chromatin around specific genes, as well as in non-genic regions, is controlled by complex biochemical processes, which are described briefly here.
Figure 1.
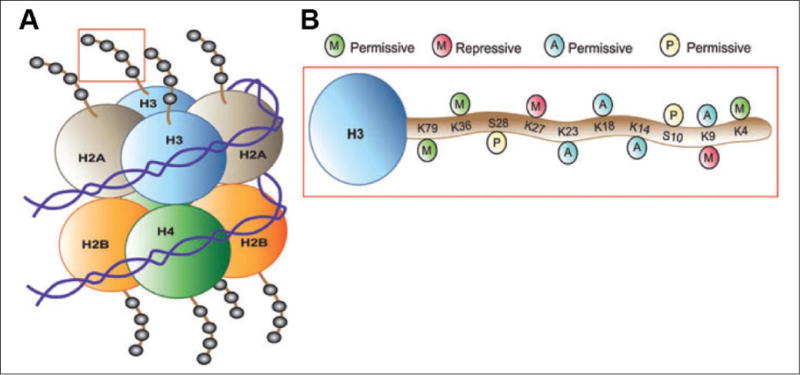
Scheme of posttranslational modifications of histones. (A) The nucleosome is the functional unit of chromatin, composed of 147 bp of DNA wrapped around a core octamer of histone proteins (two copies each of H2A, H2B, H3, and H4). The N-terminal tails of these histones face outward from the nucleosome. (B) Combinations of acetylation, phosphorylation, methylation, and so on, on histone tails (here, H3 is depicted) alter chromatin compaction and regulate gene expression. Histone modifications that weaken the interaction between histones and DNA or that promote the recruitment of transcriptional activating complexes (e.g., H3 acetylation at K23, K18, K14, and K9, as well as methylation at K79, K36, and K4 or phosphorylation at S28 and S10) correlate with permissive gene expression. Histone deacetylation, which strengthens histone–DNA contacts, or histone methylation on K27 or K9, which recruits repressive complexes to chromatin, promote a state of transcriptional repression. Adapted from Tsankova and others (2007) (permission not required).
Figure 2.
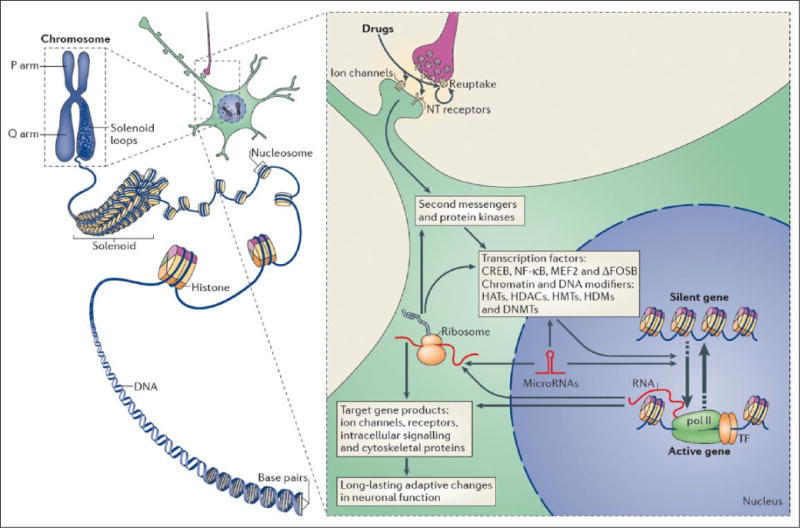
Epigenetic regulation in brain. Left: In eukaryotic cells, DNA wraps around histone octomers to form nucleosomes, which are then further organized and condensed to form chromosomes. Unraveling compacted chromatin makes the DNA of a specific gene accessible to the transcriptional machinery. Right: Stress and other environmental stimuli act in large part by altering synaptic function to alter intracellular signaling cascades, which leads to the activation or inhibition of transcription factors and of many other nuclear proteins; the detailed mechanisms involved in the latter remain poorly understood. This leads to the induction or repression of particular genes, including those for noncoding RNAs; altered expression of some of these genes can in turn further regulate gene transcription. It is hypothesized that some of these changes at the chromatin level are extremely stable and thereby underlie lifelong susceptibility to mental illness. CREB = cAMP response element binding protein; DNMTs = DNA methyltransferases; HATs = histone acetyltransferases; HDACs = histone deacetylases; HDMs = histone demethylases; HMTs = histone methyltransferases; MEF2 = myocyte enhancing factor-2; NFκB = nuclear factor κB; pol II = RNA polymerase II. From Robison and Nestler (2011) (permission not required).
Histones
The best characterized chromatin regulatory mechanism in brain is the posttranslational, covalent modification of histones at distinct amino acid residues on their N-terminal tails (Jenuwein and Allis 2001). Such modifications include acetylation, ubiquitination, or SUMOylation at lysine (K) residues, methylation at lysine or arginine residues, and phosphorylation at serine or threonine residues (e.g., Fig. 1B), among many others. Acetylation generally promotes decondensation of chromatin and increases gene activity by negating the positive charge of K residues in histone tails and increases spacing between nucleosomes. In contrast, histone methylation can either promote or repress gene activity, depending on the residue undergoing methylation. Phosphorylation of histones is also associated with chromatin inhibition or activation. The roles of other histone modifications are less well understood. The diversity of histone modifications supports the “histone code hypothesis,” which posits that the sum of modifications at a particular gene defines a specific epigenetic state of gene activation or silencing (Jenuwein and Allis 2001). However, as will be seen, such codes are likely to be highly complex and have yet to be identified.
The enzymes that add or remove these various covalent modifications of histones can be understood as “writers” and “erasers,” respectively. For example, histone acetyltransferases (HATs) catalyze acetylation and histone deacetylases (HDACs) catalyze deacetylation, while histone methyltransferases (HMTs) catalyze methylation and histone demethylases (HDMs) catalyze demethylation. The specificity of numerous HATs and HDACs for specific K residues remains incompletely understood. In contrast, distinct HMTs and HDMs control the methylation of specific K and arginine residues and even the valence of methylation, that is, mono-, di-, or tri-methylated states. The functional consequences of histone modifications are mediated in part through “readers”—proteins that bind to specific modified residues and effect transcriptional change (Jaenisch and Bird 2003; Jenuwein and Allis 2001). For example, different families of chromatin remodeling proteins, which use ATP-derived energy to alter nucleosome spacing and condensation, recognize specific forms of modified histones and enhance or repress the activity of nearby genes. The involvement of this diverse family of proteins is just now being studied in the nervous system (Sun and others 2015; Vogel-Ciernia and Wood 2014). Ultimately, hundreds of proteins are thought to be recruited to a gene in concert with its activation or repression, again emphasizing the extraordinary complexity of epigenetic mechanisms.
DNA Methylation
DNA methylation occurs with the addition of a methyl group to the C5 position of cytosine (5mC) predominantly at cytosine-guanine dyads (CpG sites) (Adachi and Monteggia 2014; Klose and Bird 2006). It plays a pivotal role in cell differentiation, imprinting, and X chromosome inactivation. DNA methylation within gene promoters generally exerts a repressive effect on gene transcription. It can either prevent the association of DNA-binding factors with their target sequence or bind to methyl-CpG-binding proteins to recruit transcriptional co-repressors to modify the surrounding chromatin into a silenced state. More recent findings have indicated that a significant portion of DNA methylation occurs at non-CpG sites and that DNA methylation can either induce or suppress gene expression depending on other factors (Lister and others 2013). Finally, as discussed in the next paragraph, additional forms of cytosine modifications have been described.
Compared with histone tail modifications, most of which are considered readily reversible, DNA methylation is viewed as a more stable epigenetic change. DNA methylation is catalyzed by DNA methyltransferases (DNMTs). Despite evidence for the dynamic control of DNA methylation in adult brain, including its reversibility, the enzymatic basis of demethylation remains incompletely understood. Putative demethylases are enzymes best studied for their role in DNA repair, such as the growth arrest and DNA damage (GADD45) family of proteins. Similarly, members of ten-eleven translocation (TET) proteins oxidize 5mC into 5-hydroxymethylcytosine (5hmC), and subsequently into 5-formylcytosine and 5-carboxylcytosine (Guo and others 2011; Kriaucionis and Heintz 2009; Moore and others 2013). Through deamination, glycosylation, or base excision repair, these newly discovered forms of cytosine modification can then be converted back into an unmethylated state. Interestingly, 5mC oxidation derivatives are expressed at highest levels in neurons, and in contrast to the generally repressive effect of 5mC on gene expression, 5hmC is more correlated with transactivation (Szulwach and others 2011). Most studies of DNA methylation in psychiatric disorders to date have not distinguished between 5mC and 5hmC, which is clearly a major need in the field.
Epigenetic Mechanisms of Depression
Depression is a complex and heterogeneous disorder, although stressful life events are a major factor in depression vulnerability. Indeed, depression is only ~40% heritable, which emphasizes the involvement of nongenetic factors. Most of what we know about the epigenetic basis of depression comes primarily from studies of animals exposed to stress, with several chronic stress paradigms having the most construct and face validity with respect to the human syndrome (Fig. 3). These studies can be divided into two major approaches: those that expose rodents to chronic stress during adulthood and those that expose rodents to chronic stress during development. Both likely recapitulate key features of the human syndrome, and it will be important in future studies to bridge the two approaches as well as validate them with analyses of postmortem human brain tissue.
Figure 3.
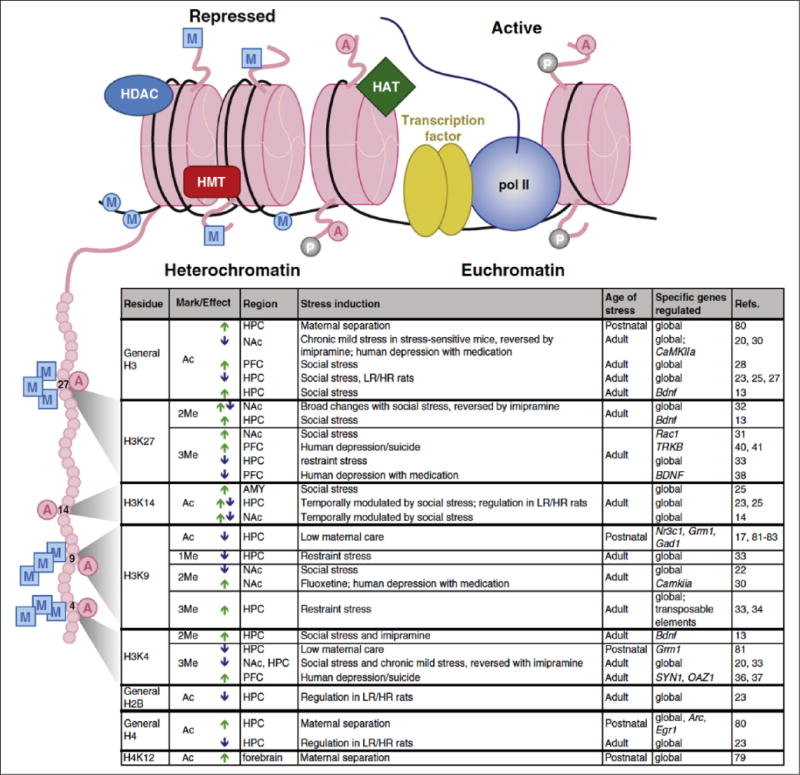
Examples of chromatin modifications regulated by stress or antidepressant treatment. Illustration (top) indicates histone octamers (pink) in heterochromatin (left) and euchromatin (right), along with associated proteins and histone tail/DNA modifications. Table (bottom) lists histone tail modifications of specific residues—depicted on the expanded histone tail illustration (left)—that are regulated by various stress paradigms or antidepressant treatments within the indicated brain regions. Arrows indicate an increase (green) or decrease (blue) in specific modifications. A = acetylation; P = phosphorylation; M (in a square) = histone methylation; M (in a circle) = DNA methylation; AMY = amygdala; HAT = histone acetyltransferase; HDAC = histone deacetylase; HPC = hippocampus; HMT = histone methyltransferase; HR and LR = high responding and low responding, respectively (with respect to baseline locomotor activity); pol II =RNA polymerase II. Modified from Peña and others (2014) with permission.
Histone Modifications in Adult Depression Models
The potential importance of epigenetic mechanisms in depression was suggested initially by observations that HDAC inhibition exerts antidepressant-like effects in adult rodent stress models (Covington and others 2009; Covington and others 2011b; Covington and others 2015; Schroeder and others 2007; Tsankova and others 2006). Systemic administration of highly nonspecific HDAC inhibitors (e.g., sodium butyrate), or direct injection of more selective inhibitors (e.g., MS275) into any of several brain regions, including nucleus accumbens (NAc), hippocampus, amygdala, or prefrontal cortex (PFC), alleviates depression-like symptoms in the chronic social defeat stress model in mice. Genome-wide studies of NAc gene expression in defeated mice treated systemically with fluoxetine or intra-NAc with MS275 demonstrated that both treatments reverse a large proportion of defeat-induced differential gene expression. Although each treatment also regulated subsets of unique genes, there was also significant overlap, suggesting that antidepressant effects of fluoxetine may in part be mediated by affecting histone acetylation (Covington and others 2009).
A major need of current research is to define the precise mechanisms by which histone acetylation controls depression- and antidepressant-related behavioral responses. There are numerous reports of stress or antidepressant regulation of specific HDAC isoforms; however, the mechanisms underlying this regulation and the specific target genes affected by each enzyme subtype remain largely unknown. Another need is to obtain genome-wide maps of any of several histone acetylation sites in several brain regions in depression models to define the genes and molecular pathways that mediate these responses.
Histone methylation is also implicated in depression. Chronic social defeat stress downregulates the histone methyltransferases G9a and G9a-like protein, which catalyze H3K9me2 (a major repressive mark) in NAc (Covington and others 2011a). Overexpression of G9a in this region is antidepressant and increased H3K9me2 at specific gene promoters is implicated in the antidepressant effect of fluoxetine (Robison and others 2013). Indeed, chronic exposure to fluoxetine reduces Camkii expression in NAc by reducing histone acetylation and increasing H3K3me2 levels at the Camkiia promoter in NAc. Interestingly, these effects are found in NAc of depressed humans exposed to antidepressants, suggesting that the stress-induced loss of repressive methylation is maladaptive and that the therapeutic effects of antidepressant drugs may act in part via the reinstatement of these marks at specific gene loci. Another gene that illustrates this mode of regulation is Ras. Reduced H3K9me2 at this gene in NAc of susceptible mice results in increased Ras expression, induction of ERK signaling, and, ultimately, CREB activation, which induces depression-like behavior (Covington and others 2011a).
Another repressive histone mark, H3K27me3, is increased upstream to the promoter of the Rac1 gene in susceptible mice and this is associated with a sustained reduction in Rac1 expression that influences characteristic dendritic spine changes in defeated mice (Golden and others 2013). These findings have been corroborated in depressed humans. H3K27me3 is implicated as well in the ability of chronic stress to suppress Bdnf expression in hippocampus (Tsankova and others 2006).
ChIP-chip analysis (chromatin immunoprecipitation followed by genome-wide promoter microarrays) examined stress-induced redistribution of H3K9me2 and H3K27me2 in NAc of mice subjected to chronic social defeat or protracted social isolation. Significant and dynamic changes in repressive histone methylation were observed in upstream regulatory regions in both models, with ~20% overlap (Wilkinson and others 2009). ChIP-seq was used to map H3K9me3, still another repressive histone mark, in hippocampus and found dramatic induction of the mark by restraint stress at repetitive elements (Hunter and others 2009; Hunter and others 2012), non-transcribed regions of the genome. Such an effect may influence genomic instability. Finally, whole forebrain overexpression of Setdb1, a histone methyltransferase that catalyzes H3K9me3, reduced depression-like behavior (Jiang and others 2008), suggesting that the increase in H3K9me3 after acute stress may represent an adaptive response.
Aside from the few examples cited above, human postmortem studies examining histone modifications in depression are sparse. Elevated levels of H3K4me3—a mark of gene activation—were reported at the synapsin gene family in PFC of depressed humans (Cruceanu and others 2013). There are also reports of altered H3K4me3 or H3K27me3 in promoter regions of several candidate genes (e.g., OAZ, SYN2, BDNF, TRKB) in postmortem PFC (Chen and others 2011; Fiori and others 2012), but thus far no genome-wide analysis of histone modifications in depressed human brain. This is a high priority for future research.
Chromatin Remodeling in Adult Depression Models
Very little is known concerning the role of chromatin remodeling complexes in depression or any other psychiatric syndrome. However, a recent study demonstrated that chronic social defeat stress induces a repressive chromatin remodeling complex in NAc, which by ChIP-seq was shown to mediate stress-repression of a set of genes important for mediating stress susceptibility (Sun and others 2015). Induction of the same complex was found in NAc of depressed humans, providing translational validation. Induction of this repressive complex at suppressed genes correlates with lower levels of activating histone marks (e.g., H3M4me3 and H4K16ac) and increased levels of certain repressive histone marks (e.g., H3K9me2), thus emphasizing the coordinated nature of epigenetic regulation (Fig. 4). These findings underscore the importance of mapping numerous epigenetic mechanisms genome-wide in order to define the combinatorial code of epigenetic changes associated with depression or antidepressant responses.
Figure 4.
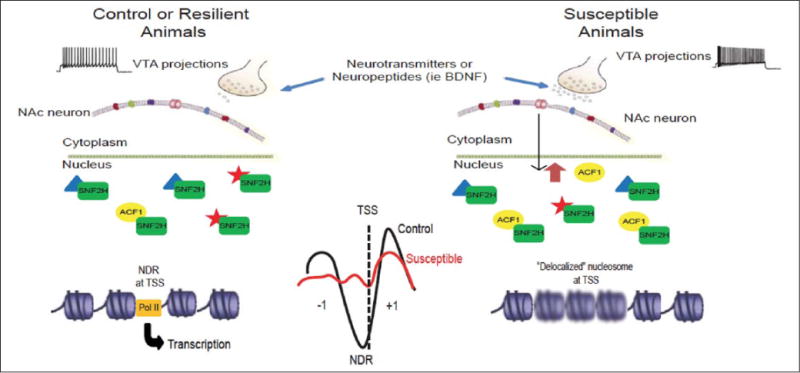
Hypothesized role of chromatin remodeling ACF complex in NAc in stress susceptibility. Chronic social defeat stress CSDS, via increased burst firing of VTA neurons and BDNF release, induces ACF1 expression in NAc. The resulting upregulation of ACF complex activity, possibly through changes in TSS (transcription start site) nucleosome positioning, represses a set of genes in NAc, the reduced expression of which contributes to susceptibility. Blurry nucleosomes in the right figure represent weakly positioned or delocalized nucleosomes at TSSs. From Sun and others (2015) (permission not required).
DNA Methylation in Adult Depression Models
In addition to the chromatin modifications described above, a growing body of evidence supports a role for DNA methylation in mediating the impact of stress on the brain. Chronic social defeat stress increases expression of Dnmt3a in NAc (LaPlant and others 2010). Overexpressing Dnmt3a in this region increases depression-like behavior while intra-NAc infusion of a DNMT inhibitor, RG108, exerts antidepressant-like effects. DNMT3a activity is generally associated with transcriptional repression suggesting that susceptibility may associate with down-regulation of transcriptional expression in NAc. Expression of DNMTs is altered in limbic and brain stem regions in depressed suicide completers (Poulter and others 2008). Genome-wide analysis of DNA methylation will be important in establishing the precise mechanisms of this epigenetic modification in defeat-induced susceptibility.
DNA methylation of several candidate genes, within NAc and several other brain regions, has been studied in stress models. Examples include glial cell-derived neurotrophic factor (Gdnf) (Uchida and others 2011) in NAc and corticotropin releasing factor (CRF) in paraventricular nucleus of hypothalamus (PVN) (Elliott and others 2010; Sterrenburg and others 2011). CRF, a critical regulator of hypothalamic-pituitary-adrenal (HPA)-axis activation and other stress actions in brain, is increased in PVN of mice that are susceptible to social defeat and this is accompanied by decreased DNA methylation at the Crf promoter. Both effects are reversed by chronic imipramine treatment (Elliott and others 2010). DNA methylation is also increased at the Crf promoter in PVN of female rats subjected to chronic unpredictable stress, suggesting that DNA methylation may play a role in determining sex-specific regulation of HPA-axis function (Sterrenburg and others 2011). An imperative for the field is to generate genome-wide maps, not only of 5mC, but also 5hmC, in several brain regions in chronic stress models in animals as well as in human brain.
Epigenetics and Developmental Vulnerability to Depression
Early life adversity, which can have lifelong effects on behavioral outcomes, has been modeled in rodents using prenatal stress (where pregnant dams are stressed) or separation of pups from their mothers (Turecki and Meaney 2014). Natural variations in maternal care, with the extremes classified as either low or high grooming, likewise associate with differential stress responses among adult offspring (Meaney 2001). Research over the last decade has implicated epigenetic alterations, a subset of which are likely permanent, in the enduring effects of such early life stress. Such epigenetic alterations presumably affect depression vulnerability both by mediating sustained alterations in the steady state expression levels of certain genes and by altering other genes’ inducibility in response to subsequent challenges later in life.
Several studies have examined the epigenetic consequences of prenatal stress on brain. In one series of experiments, investigators showed that maternal stress suppresses placental expression of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which normally protects the developing fetus from maternal glucocorticoids. Such 11β-HSD2 down-regulation is associated with hypermethylation of its gene promoter and with increased stress vulnerability of offspring animals (Jensen-Peña and others 2012; O’Donnell and others 2012). In other studies, early prenatal stress elevated DNA methylation at the NGF1-A binding region of the glucocorticoid receptor (GR; Nr3c1) promoter exon 17 in offspring hypothalamus, and decreased methylation of the Crf promoter, with no changes at Bdnf (Mueller and Bale 2008). Hypermethylation at NR3C1 was found similarly in infant cord blood from depressed mothers or those physically abused during pregnancy (Hompes and others 2013; Oberlander and others 2008; Radtke and others 2011). Finally, mice exposed to prenatal stress had elevated levels of Dnmt3a and Dnmt1 mRNA in PFC and hippocampus at birth, changes that persisted into adulthood and were associated with hypermethylation of the Reelin and Gad1 promoters, both implicated in SCZ (see below) (Matrisciano and others 2013).
There is also a large literature on the influence of maternal behavior on epigenetic endpoints in offspring brain. Maternal separation has been shown to alter levels of expression of several HDACs in PFC and other brain regions, although the genes influenced by such modifications remain unknown (Blaze and Roth 2013; Levine and others 2012). As in adults, treatment with nonselective HDAC inhibitors reverses the effects of maternal separation, while treatment with theophylline—which can activate HDACs in addition to its better described action as a phosphodiesterase inhibitor—had the opposite effect (Levine and others 2012).
Early maternal separation reduces DNA methylation in a known enhancer region for Avp expression in PVN, which is associated with increased Avp expression (Murgatroyd and others 2009). Maternal separation also alters DNA methylation and expression of Nr3c1 and Bdnf in PFC and hippocampus, changes which could contribute to depression-like behavior observed later in life (Kundakovic and others 2013). As well, DNA methylation is altered by extreme adversity (e.g., abuse) during early life. Maternal maltreatment of rat pups (tramping, dragging, rough handling) leads to lasting hypomethylation at the Bdnf promoter in PFC (Roth and others 2009). These effects are partially rescued by ICV treatment with zebularine, a DNMT inhibitor. A recent human study assessed the impact of child abuse on genome-wide DNA methylation signatures in gene promoters in hippocampus (Labonté and others 2012a). DNA methylation patterns were compared between suicide completers with a severe history of child abuse and healthy controls, and hundreds of differentially methylated sites were identified. Importantly, DNA methylation levels in gene promoters were inversely correlated with gene expression at a genome-wide level, supporting the globally repressive role of DNA methylation. Similar observations have been made in suicide completers (Labonté and others 2012b).
Likewise, low versus high maternal care alters the epigenetic status of numerous genes, the best studied of which is Nr3c1. Low maternal care decreases histone acetylation at the Nr3c1 exon 17 promoter in hippocampus (Weaver and others 2004), the same locus affected by prenatal stress and maternal separation (see above). This modification occurs coincidently with increased DNA methylation (McGowan and others 2011). The changes are associated with reduced GR expression and increased stress vulnerability. Treatment with the nonselective HDAC inhibitor, trichostatin A, infused either ICV or intra-hippocampus, reversed the effects of low maternal care on histone acetylation at Nr3c1 and stress behavior (Weaver and others 2004; Weaver and others 2006). The impact of maternal care on the establishment of DNA methylation profiles spreads across large genomic regions: microarray analysis of a 6.5 mb region centered on Nr3c1 showed that low maternal care induces hundreds of parallel DNA methylation changes colocalized with other chromatin modifications (Suderman and others 2012). These adaptations cluster in particular at protocadherin genes.
Similar alterations have been reported in hippocampus of suicide completers with a history of child abuse. Abused suicide completers exhibit lower expression levels of Nr3c1 compared to nonabused suicides and controls (Labonté and others 2012c; McGowan and others 2009). This regulation is associated with altered DNA methylation within respective promoters. Importantly, these alterations appear to be specific to early-life adversity as Nr3c1 transcriptional modifications found in brains of depressed patients without a history of child abuse do not associate with changes in DNA methylation (Alt and others 2010).
Maternal care in rats is reported to affect several additional genes, such as Gad1 and Grm1 in hippocampus (Bagot and others 2012; Zhang and others 2010) and Esr1 in the hypothalamic medial preoptic area (Peña and others 2013). Pups raised with low maternal care show lower expression levels of these genes, which are associated with reciprocal changes in promoter methylation and in some cases with altered levels of histone acetylation or methylation. Importantly, these epigenetic events have been related to behavioral outcomes.
Stress beyond the early neonatal period also leaves an epigenetic mark. Three weeks of adolescent isolation stress in a Disc1 mutant mouse induced mood-related behavioral alterations accompanied by hypermethylation of the tyrosine hydroxylase (Th) gene promoter in mouse VTA (Niwa and others 2013). Such hypermethylation at Th was sustained into adulthood and rescued by treatment with the GR antagonist RU38486.
This discussion shows robust effects of early life experiences on epigenetic endpoints and directly implicates epigenetic regulation of several candidate genes in stress-related phenomena. The next crucial step is the generation of genome-wide maps of histone and DNA modifications to obtain a complete view of the range of genes affected under these conditions.
Epigenetic Mechanisms of Schizophrenia
SCZ is a severely disabling disorder defined by positive symptoms such as delusions, hallucinations and disorganized thought, negative symptoms such as social withdrawal and apathy, and cognitive impairments. All antipsychotic medications, which produce symptomatic improvement for positive symptoms but far less efficacy for the other behavioral domains, antagonize (or are weak partial agonists at) the D2 dopamine receptor, with varied activity at a range of serotonergic and other receptors. While they have remained the primary therapeutic intervention for over half a century, most patients show an incomplete response. Rational drug development in SCZ remains challenging, given the absence of a unifying pathophysiology and a highly complex genetic risk architecture (Andreassen and others 2014; Neale and Sklar 2015; Rodriguez-Murillo and others 2012). However, SCZ is characterized by gene expression alterations in cerebral cortex and other brain regions and, given that transcriptional mechanisms depend on dynamic chromatin remodeling, genes with dysregulated expression in SCZ brain would be expected to show epigenetic alterations of their regulatory regions.
This view is supported primarily by postmortem human brain work, which has focused to date mainly on changes in DNA methylation at candidate gene promoters. One prominent example is RELN, which encodes reelin, whose promoter shows increased methylation in PFC and certain other brain regions of humans with SCZ (Abdolmaleky and others 2005; Grayson and others 2005). This hypermethylation is associated with reduced Reln expression and could be mediated by increased DNMT1 levels observed under these conditions (Veldic and others 2004). Importantly, these changes in reelin are unrelated to a history of antipsychotic drug exposure (Guidotti and others 2000). Given that reelin, a secreted protein, controls neuronal migration during development, these data support a scheme whereby deficiencies in reelin drive some of the developmental abnormalities associated with SCZ.
Another gene that exhibits increased DNA methylation in PFC in SCZ is SOX10, which encodes a transcription factor important in development (Iwamoto and others 2005; Kato and Iwamoto 2014). This hypermethylation is associated with reduced Sox10 expression and with altered expression levels of several genes associated with oligodendrocyte function (Iwamoto and others 2005). These findings are interesting in light of evidence that polymorphisms in SOX10 are reported to influence the age of onset of SCZ (Yuan and others 2014), and with considerable data implicating myelin abnormalities in this syndrome (Roussos and Haroutunian 2014). Still another example is the finding that several human leukocyte antigen (HLA) genes show altered DNA methylation in PFC in SCZ, which is noteworthy given the purported role of inflammation in disease pathogenesis (Pal and others 2015). A smaller number of studies have examined histone modifications in postmortem human brain samples, such as increased levels of several HMTs in the brains of SCZ patients (Chase and others 2013). In addition, altered levels of H3K9K14 acetylation in PFC in SCZ correlate with altered expression levels of the affected genes, which include GAD1 (glutamic acid decarboxylase-1, a key enzyme for GABA synthesis), HTR2C (serotonin 2C receptor), and PPM1E (protein phosphatase 1E) (Tang and others 2011).
Recent studies, when taken together, indicate robust epigenetic dysregulation of GAD1 in PFC in SCZ, including excessive levels of repressive DNA and histone methylation (Akbarian and Huang 2006; Dong and others 2009; Guidotti and Grayson 2014; Huang and Akbarian 2007; Ruzicka and others 2015), at the expense of certain activating histone marks such as H3K4me3 (Huang and others 2007). Superimposed on these highly localized molecular alterations of nucleosomal histones at GAD1 is a still poorly understood defect in the 3D architecture of the chromatin fiber containing this locus (see Box 2). This conclusion is based on the weakening of long range enhancer loopings that normally bypass many kilobases of linear genome to activate the GAD1 promoter in PFC in SCZ (Bharadwaj and others 2013) (Fig. 5A). Such changes in 3D chromatin architecture are not limited to GAD1, since they have been found for other SCZ risk genes, such as CACNA1C (Fig. 5B). We expect the field to continue to gain further insight into these and other epigenetic mechanisms governing dysregulated GABAergic gene expression, given that a very recent in vivo neuroimaging study provided the first empirical evidence for impairments in GABAergic transmission in PFC of patients with SCZ—a hypothesis that until now was primarily driven by molecular and cellular studies in diseased brain tissue (Frankle and others 2015).
Box 2. Three-Dimensional Studies of Chromatin Structure.
Interphase chromosomes are thought to be arranged in a probabilistic, nonrandom fashion inside the nucleus (far left). To examine chromosome loop formations, chromosome conformation capture (termed 3C) and its derivatives are employed. 3C libraries are generated from cross-linked chromatin that first is digested with high doses of a restriction enzyme followed by religation, then removal of protein and quantification of ligation products generated from noncontiguous sequences. Conventional 3C measures the looping between two specific candidate sequences (“one vs. one”) (top). Circular chromosome conformation capture (4C) libraries are creating from the 3C libraries by subjecting the 3C libraries to a second round of restriction digestion with a different enzyme. 4C captures the total set (genome-scale) of chromosomal loop formations for one specific locus (“one vs. all”) (middle). Finally, HiC libraries are created by restriction digestion of cross-linked DNA, filling in using biotin-CTP and re-ligating the genome (bottom). Biotin labeled interactions are precipitated and sequenced. Global interactions are agnostically interrogated across the genome (“all vs. all”) (bottom). Examples of the regulation of the 3D structure of genes in mental illness are given in Figure 5.
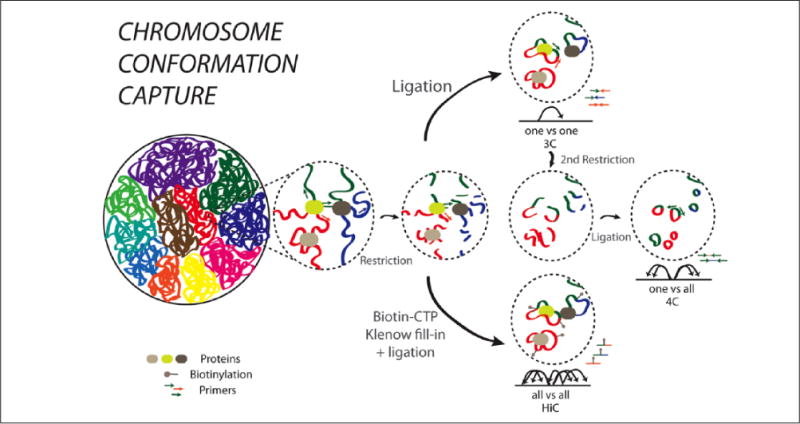
Figure 5.
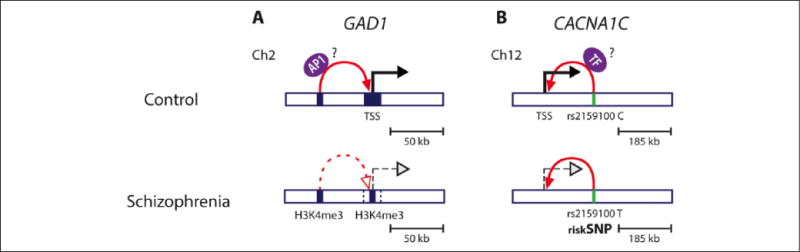
Higher order chromatin structure and schizophrenia. The role of higher order chromatin structure in transcriptional regulation of SCZ relevant genes has been shown for GAD1 (A) and CACNA1C (B). (A) GAD1, encoding GABA synthesis enzyme, is located on chromosome 2 (Ch2) and frequently down-regulated in cerebral cortex of SCZ patients (dashed black arrow) and this is associated with lower levels of active histone marks, including H3K4me3 (blue square) at the GAD1 transcription start site (TSS). The TSS region of GAD1 has been shown to physically interact with an AP1 motif-enriched enhancer region located 50 kb further upstream (also enriched in H3K4me3 mark, blue square). Evidence has been presented that a chromatin loop (red arrow), that may carry a cargo such as AP1 transcription factors (purple oval) into close proximity to the core promoter region facilitating GAD1 gene transcription, is weakened in brains of SCZ patients brain (dashed red arrow). This could contribute to lower GAD1 expression. (B) Several single nucleotide polymorphisms (SNPs) residing in noncoding regions of the CACNA1C gene on chromosome 12 (Ch12) have been associated with lower CACNA1C expression and SCZ risk. The rs215100 T SCZ risk allele (green bar) resides in an intronic enhancer region, 185 kb downstream from the CACNA1C TSS, which has been shown to physically interact with the CACNA1C TSS (solid red arrow). The T allele confers lower transcriptional activity (dashed black arrow) as compared to C allele (solid black arrow), presumably by affecting the binding of transcription factors (TF, purple oval) and their interaction via chromosomal loops with the promoter CACNA1C region.
There is a much larger literature on altered DNA methylation, and in some cases histone modifications, of gene promoters in blood or other peripheral tissues of individuals with SCZ. These studies are too numerous to describe here in detail. However, it is interesting that several genes that show alterations in brain—for example, RELN and GAD1—are also reported to differ in peripheral tissues (Aberg and others 2014; Auta and others 2013; Gavin and others 2009). Abnormal methylation status has been reported in blood for several additional candidate genes, such as BDNF (brain-derived neurotrophic factor), 5HTR1A (serotonin 1A receptor), and COMT (catechol-O-methyltransferase) (Carrard and others 2011; Ikegame and others 2013; Kundakovic and others 2015; Walton and others 2014). Given that the epigenetic status of genes is highly cell type-dependent, with such patterns differing dramatically even across distinct neuronal cell types, it would be surprising if shared abnormalities in brain and blood were common. On the other hand, this is ultimately an empirical question and it is conceivable that aspects of SCZ might be reflected across several tissues.
To date only very few studies have pursued DNA methylation or histone modification changes in SCZ on a genome-wide scale in brain tissue or peripheral cells (Aberg and others 2014; Dempster and others 2011; Kano and others 2013; Melas and others 2012; Mill and others 2008; Wockner and others 2014), and none has harnessed the full power of modern next-generation sequencing technology, which offers the most unbiased view of the distribution of an epigenetic mark across the entire genome. Federally funded consortia, including PsychENCODE (http://psychencode.org), are currently underway, with the mission to characterize the epigenome of healthy and diseased brain cells in hundreds of SCZ and control specimens using next-generation sequencing technology.
Importantly, the aforementioned genome-wide (published) studies and ongoing consortia virtually all focus on areas of PFC, with only few studies exploring other brain regions such as the cerebellar cortex (Wockner and others 2014). Such near-exclusive focus on a single brain region is unfortunate, as the neural circuits of psychosis most certainly include many subcortical areas too. However, it is encouraging that some of the candidate genes explored thus far, such as SOX10 or GAD1, resurfaced on the list of epigenetically dysregulated genes in genome-wide studies of human brain tissue (Wockner and others 2014) or of animal models (Connor and others 2012).
Finally, it should be mentioned that mutations in perhaps up to 50 genes, each encoding a different chromatin regulator, have been linked to a wide range of neurodevelopmental syndromes, including rare monogenic forms of SCZ (Ronan and others 2013). Chromatin defects in brain were traditionally considered static lesions of early development, but it is now clear that mutations and maladaptations of the epigenetic machinery cover a much wider continuum, including adult-onset neurodegenerative disease (Jakovcevski and Akbarian 2012; Klein and others 2011; Winkelmann and others 2012). For example, gene duplication of the HMTs, KMT1D and KMT2F, or the MYTL1 and ZNF804A transcription factors, have been linked to some cases of SCZ (Hess and Glatt 2014; Kirov and others 2012; Lee and others 2012; Takata and others 2014).
It is striking that, in contrast to depression, where most epigenetic investigations have focused on rodent models, the situation is reversed for SCZ for which there have been relatively few animal studies. This could relate to the far more challenging prospect of generating rodent models of SCZ. There are reports that prenatal stress alters cortical expression levels of several enzymes involved in DNA methylation, mirroring findings in SCZ brain (Guidotti and others 2014). Genome-wide DNA methylation maps were obtained for rat hippocampus in response to chronic olanzapine treatment (Melka and others 2014). As well, HDAC2 (Kurita and others 2012) and the transcription factor ΔFosB (Dietz and others 2014) have been implicated in antipsychotic drug action in rodents. A challenge for the future is to better utilize animal models, and perhaps cell models such as patient-derived neurons, in epigenetic studies of SCZ.
Epigenetic Mechanisms of Bipolar disorder
Bipolar illness shows considerable overlap with SCZ in terms of genetic risk architecture, neurobiology, and, to a certain degree, treatment (such as the use of antipsychotic drugs) (Neale and Sklar 2015). In this context, it is therefore not surprising that both genome-wide and candidate gene DNA methylation mapping in postmortem brains of individuals with SCZ or bipolar disorder with psychosis revealed, for both diagnostic categories, a similar degree of subtle (but significant) changes at many gene promoters (Dong and others 2014; Mill and others 2008; Tang and others 2011; Xin and others 2012).
One particularly interesting locus is HLA9, which showed aberrant DNA methylation patterns in multiple postmortem brain cohorts and in peripheral blood and, surprisingly, also in sperm of subjects diagnosed with bipolar disorder (Kaminsky and others 2012). The molecular mechanisms driving this multi-tissue epigenetic dysregulation remain unclear. Furthermore, the aforementioned GAD1 gene, which shows dysregulated expression and epigenetic regulation in SCZ, shows similar altered DNA methylation in hippocampus in bipolar disorder (Ruzicka and others 2015). While these findings collectively point to an emerging epigenetic risk architecture of bipolar disorder, a more definite assessment of the role of epigenetic dysregulation in bipolar disorder will have to await more comprehensive genome-wide maps not only of DNA methylation but also numerous histone modifications in larger cohorts of brains from subjects diagnosed with this syndrome. Careful transcriptome analyses of bipolar disorder (e.g., Cruceanu and others 2015) will also assist this effort as will more biologically driven diagnostic stratification of SCZ and bipolar patients.
Unexpectedly, regulators of H3K4 methylation recently emerged as a functional gene category showing one of the strongest links to the genetic risk architecture of bipolar disorder and related conditions such as SCZ, based on genome-wide association studies (Psychiatric Genetics Consortium 2015). This finding further underscores the critical role of epigenetic regulation in the biological basis of psychosis spectrum disorders.
Future Outlook
Although still in relatively early stages, work to date has demonstrated that many forms of epigenetic regulation are altered in limbic brain regions both in animal models of psychiatric disease and in postmortem tissue of humans with these disorders. These initial studies have identified several key challenges that will need to be addressed moving forward. A high priority for current research is to complete genome-wide assays for numerous chromatin mechanisms. Another high priority is to validate rodent findings in human tissue and, conversely, to recapitulate human findings in animal models so that causal data can be obtained. Such efforts are needed for a large number of histone modifications and chromatin remodeling complexes, only a handful of which have been examined to date by genome-wide methods: DNA methylation—both 5mC and 5hmC, which have not yet been separately mapped genome-wide in most disease models, as well as a host of noncoding RNAs. We do not review the latter here, but numerous reports of alterations in microRNAs have appeared for psychiatric syndromes in animal models and human tissue, and these too require further investigation (Issler and Chen 2015).
Another important question is how epigenetic regulation is translated into transcriptional change, not only steady state alterations in expression but also altered inducibility in response to a subsequent challenge as well as changes in alternative splicing, which are thought to be under the control of epigenetic regulation. As noted earlier, no single modification examined to date is deterministic for a change in gene activity. In fact, modifications that are most clearly associated with a functional change, for example, H3K4me3 in promoting transcription, are associated with no change or even opposite changes in transcription at many genes. Such findings are consistent with the required involvement of numerous modifications that work in concert. Deciphering such a code, or chromatin signatures, will be a very difficult, yet also highly important, goal for future research.
A technical challenge in this effort is the heterogeneous cell population of even brain micronuclei, which makes it impossible to derive data as clear-cut as for cell culture systems. Methodologies are underway to isolate specific cell types from brain (Jiang and others 2008) and to perform genome-wide ChIP-seq, RNA-seq, and DNA methylation assays on much less starting material (e.g., Adli and Bernstein 2011). In the meantime, nearly all bioinformatics tools for genome-wide analysis have been developed based on simpler cell culture data, which are not optimal to detect the more subtle signals from terminally differentiated neurons, particularly with the high background noise unavoidable with in vivo studies. Improved analytical tools will require creative collaborations between biologists and bioinformaticians (Maze and others 2014). A further technical challenge, but scientific imperative, is to relate chromatin modifications to a host of transcription factors (e.g., GR, CREB, ΔFosB, NFκB, β-catenin, and others) with which they act in concert to control disease-related behavioral abnormalities.
Beyond modifications of histones, chromatin remodeling, and DNA modifications, an important new effort in neuroepigenetics concerns the 3D organization of the genome in neurons and glia, mentioned briefly above. For example, chromosomal loop formations—which often require CCCTC-binding factor (CTCF)-binding factor, cohesins, and other proteins assembled into scaffolds and anchors—potentially bypass many kilobases, even mega-bases, of linear genome, thereby repositioning promoter-distal regulatory elements next to their target promoters. Exploration of regulatory DNA elements in the context of chromosomal loopings and higher order chromatin is beginning to assign regulation of genes and complex behavior for some of the noncoding sequences in the human genome (Bharadwaj and others 2013). Given that the majority of risk-associated polymorphisms in psychiatric disease, such as SCZ and bipolar disorder, are positioned in intergenic and intronic DNA, chromosome conformation capture assays, and other techniques that measure interaction and spatial proximity of noncontiguous DNA elements in brain cells (see Box 2) are likely to gain increasing traction in studies of human and animal brain tissue (Mitchell and others 2014).
A major limitation in the field is the difficulty in relating chromatin modifications at a given gene to a functional outcome. Studies to date have relied by necessity on overexpressing or knocking out, or pharmacologically inhibiting, a chromatin modifying enzyme (e.g., an HDAC, HMT, or DNMT) within a given brain region like the NAc and studying the behavioral consequences. However, such manipulations regulate the targeted chromatin modification at hundreds or thousands of genes. One approach to overcome this limitation is to use engineered zinc finger proteins (ZFPs), sequence-specific transcription activator-like effectors (TALEs), or CAS9/CRISPR, coupled to an enzymatic moiety, to target a particular chromatin modification to a given gene of interest within a region of adult brain. While still early in development, such approaches would represent an enormous advance in the field, to test, for example, whether a single histone modification at a particular gene truly regulates that gene and resulting behaviors. We recently demonstrated that this is possible using an engineered zinc finger protein: we targeted increased H3K9me2 to the FosB gene in mouse NAc in vivo, thus mimicking a defect seen in this region of depressed humans, and this action reduced ΔFosB expression and increased depression-like behavior (Heller and others 2014). Importantly, we provided evidence that the synthetic zinc finger increased H3K9me2 at the FosB locus selectively, with no effect on homologous loci. Further studies utilizing this and other locus-specific tools will dramatically advance studies in neuroepigenomics.
Note that there is virtually no mention of sex differences in epigenetic regulation in this review, because few if any animal studies have compared males and females, and few if any human studies have had sufficient power to compare the sexes. This is an urgent deficiency in the field given large sex differences in some psychiatric syndromes (e.g., depression is twofold more common in females). A focus on shared and different mechanisms of epigenetic regulation in males versus females is thus an extremely high priority for current research.
Conclusions
The ultimate goal of epigenetic studies of mental illness is to understand how genetic vulnerabilities interact with an individual’s life experiences to establish stable changes at precise genomic loci, which then control the levels of gene expression or inducibility. Together, this linking of genes and environment through epigenetic mechanisms determines that individual’s vulnerability to psychiatric syndromes over a lifetime. It is our expectation that these studies will reveal a host of genes whose products could serve as templates in future drug discovery efforts. It would also be interesting to determine whether drug effects on epigenetic endpoints in peripheral tissues (e.g., blood) might serve as useful biomarkers for clinical features of a given disorder, even if those changes in blood are different from those in brain. In these ways, epigenetic approaches promise unprecedented advances in our understanding, diagnosis, and treatment of psychiatric illness.
Acknowledgments
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institute of Mental Health.
Footnotes
Authors’ Note
Parts of this review are adapted from Nestler (2014) and Peña and others (2014) with permission.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Aberg KA, McClay JL, Nerella S, Clark S, Kumar G, Chen W, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014;71:255–64. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:60–6. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Adachi M, Monteggia LM. Decoding transcriptional repressor complexes in the adult central nervous system. Neuropharmacology. 2014;80:45–52. doi: 10.1016/j.neuropharm.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc. 2011;6:1656–68. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S. Epigenetic mechanisms in schizophrenia. Dialogues Clin Neurosci. 2014;16:405–17. doi: 10.31887/DCNS.2014.16.3/sakbarian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Alt SR, Turner JD, Klok MD, Meijer OC, Lakke EA, Derijk RH, et al. Differential expression of glucocor-ticoid receptor transcripts in major depressive disorder is not epigenetically programmed. Psychoneuroendocrinology. 2010;35:544–56. doi: 10.1016/j.psyneuen.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Thompson WK, Dale AM. Boosting the power of schizophrenia genetics by leveraging new statistical tools. Schizophrenia. 2014;40:13–7. doi: 10.1093/schbul/sbt168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auta J, Smith RC, Dong E, Tueting P, Sershen H, Boules S, et al. DNA-methylation gene network dysregulation in peripheral blood lymphocytes of schizophrenia patients. Schizophr Res. 2013;150:312–8. doi: 10.1016/j.schres.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot RC, Zhang TY, Wen X, Nguyen TT, Nguyen HB, Diorio J, et al. Variations in postnatal maternal care and the epigenetic regulation of metabotropic glutamate receptor 1 expression and hippocampal function in the rat. Proc Natl Acad Sci U S A. 2012;109(suppl 2):17200–7. doi: 10.1073/pnas.1204599109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj R, Jiang Y, Mao W, Jakovcevski M, Dincer A, Krueger W, et al. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci. 2013;33:11839–51. doi: 10.1523/JNEUROSCI.1252-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaze J, Roth TL. Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int J Dev Neurosci. 2013;31:804–10. doi: 10.1016/j.ijdevneu.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrard A, Salzmann A, Malafosse A, Karege F. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J Affect Disord. 2011;132:450–3. doi: 10.1016/j.jad.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Chase KA, Gavin DP, Guidotti A, Sharma RP. Histone methylation at H3K9: evidence for a restrictive epigenome in schizophrenia. Schizophr Res. 2013;149:15–20. doi: 10.1016/j.schres.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Ernst C, Turecki G. The epigenetic effects of antidepressant treatment on human prefrontal cortex BDNF expression. Int J Neuropsychopharmacol. 2011;14:427–9. doi: 10.1017/S1461145710001422. [DOI] [PubMed] [Google Scholar]
- Connor CM, Dincer A, Straubhaar J, Galler JR, Houston IB, Akbarian S. Maternal immune activation alters behavior in adult offspring, with subtle changes in the cortical transcriptome and epigenome. Schizophr Res. 2012;140:175–84. doi: 10.1016/j.schres.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, et al. Antidepressant actions of his-tone deacetylase inhibitors. J Neurosci. 2009;29:11451–60. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011a;71:656–70. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, Vialou VF, Laplant Q, Ohnishi YN, Nestler EJ. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci Lett. 2011b;493:122–6. doi: 10.1016/j.neulet.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, III, Maze I, Vialou V, Nestler EJ. Antidepressant action of HDAC inhibition in the prefrontal cortex. Neuroscience. 2015;298:329–35. doi: 10.1016/j.neuroscience.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruceanu C, Alda M, Nagy C, Freemantle E, Rouleau GA, Turecki G. H3K4 tri-methylation in synapsin genes leads to different expression patterns in bipolar disorder and major depression. Int J Neuropsychopharmacol. 2013;16:289–99. doi: 10.1017/S1461145712000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruceanu C, Tan PP, Rogic S, Lopez JP, Torres-Platas SG, Gigek CO, et al. Transcriptome sequencing of the anterior cingulate in bipolar disorder: dysregulation of G protein-coupled receptors. Am J Psychiatry. 2015 doi: 10.1176/appi.ajp.2015.14101279. Epub Aug 4. [DOI] [PubMed] [Google Scholar]
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–96. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz DM, Kennedy PJ, Sun H, Maze I, Gancarz AM, Vialou V, et al. ΔFosB induction in prefrontal cortex by antipsychotic drugs is associated with negative behavioral outcomes. Neuropsychopharmacology. 2014;39:538–44. doi: 10.1038/npp.2013.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, Grayson DR, Guidotti A, Costa E. Antipsychotic subtypes can be characterized by differences in their ability to modify GABAergic promoter methylation. Epigenomics. 2009;1:201–11. doi: 10.2217/epi.09.2. [DOI] [PubMed] [Google Scholar]
- Dong E, Ruzicka WB, Grayson DR, Guidotti A. DNA-methyltransferase 1 (DNMT1) binding to CpG rich GABAergic and BDNF promoters is increased in the brain of schizophrenia and bipolar disorder patients. Schizophr Res. 2014 doi: 10.1016/j.schres.2014.10.030. Epub Dec 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott EE, Ezra-Nevo GG, Regev LL, Neufeld-Cohen AA, Chen AA. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat Neurosci. 2010;13:1351–3. doi: 10.1038/nn.2642. [DOI] [PubMed] [Google Scholar]
- Fiori LM, Gross JA, Turecki G. Effects of histone modifications on increased expression of polyamine biosynthetic genes in suicide. Int J Neuropsychopharmacol. 2012;15:1161–6. doi: 10.1017/S1461145711001520. [DOI] [PubMed] [Google Scholar]
- Frankle WG, Cho RY, Prasad KM, Mason NS, Paris J, Himes ML, et al. In vivo measurement of GABA transmission in healthy subjects and schizophrenia patients. Am J Psychiatry. 2015 doi: 10.1176/appi.ajp.2015.14081031. Epub Jul 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin DP, Kartan S, Chase K, Jayaraman S, Sharma RP. Histone deacetylase inhibitors and candidate gene expression: an in vivo and in vitro approach to studying chromatin remodeling in a clinical population. J Psychiatr Res. 2009;43:870–6. doi: 10.1016/j.jpsychires.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K, et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med. 2013;19:337–44. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–6. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–9. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Dong E, Grayson DR, Veldic M, et al. GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology (Berl) 2005;180:191–205. doi: 10.1007/s00213-005-2212-8. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Dong E, Tueting P, Grayson DR. Modeling the molecular epigenetic profile of psychosis in prenatally stressed mice. Prog Mol Biol Transl Sci. 2014;128:89–101. doi: 10.1016/B978-0-12-800977-2.00004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Grayson DR. DNA methylation and demethylation as targets for antipsychotic therapy. Dialogues Clin Neurosci. 2014;16:419–29. doi: 10.31887/DCNS.2014.16.3/aguidotti. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–34. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller EA, Cates HM, Peña CJ, Sun H, Shao N, Feng J, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17:1720–1727. doi: 10.1038/nn.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JL, Glatt SJ. How might ZNF804A variants influence risk for schizophrenia and bipolar disorder? A literature review, synthesis, and bioinformatic analysis. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:28–40. doi: 10.1002/ajmg.b.32207. [DOI] [PubMed] [Google Scholar]
- Hompes T, Izzi B, Gellens E, Morreels M, Fieuws S, Pexsters A, et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiatric Res. 2013;47:880–91. doi: 10.1016/j.jpsychires.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Huang HS, Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS One. 2007;2:e809. doi: 10.1371/journal.pone.0000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–62. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A. 2009;106:20912–7. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RG, Murakami G, Dewell S, Seligsohn MA, Baker MER, Datson NA, et al. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc Natl Acad Sci U S A. 2012;109:17657–62. doi: 10.1073/pnas.1215810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegame T, Bundo M, Murata Y, Kasai K, Kato T, Iwamoto K. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. J Hum Genet. 2013;58:434–8. doi: 10.1038/jhg.2013.65. [DOI] [PubMed] [Google Scholar]
- Issler O, Chen A. Determining the role of microRNAs in psychiatric disorders. Nat Rev Neurosci. 2015;16:201–12. doi: 10.1038/nrn3879. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Yamada K, Takao H, Iwayama-Shigeno Y, Yoshikawa T, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J Neurosci. 2005;25:5376–81. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Peña C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One. 2012;7:e39791. doi: 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky Z, Tochigi M, Jia P, Pal M, Mill J, Kwan A, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2012;17:728–40. doi: 10.1038/mp.2011.64. [DOI] [PubMed] [Google Scholar]
- Kano S, Colantuoni C, Han F, Zhou Z, Yuan Q, Wilson A, et al. Genome-wide profiling of multiple histone methylations in olfactory cells: further implications for cellular susceptibility to oxidative stress in schizophrenia. Mol Psychiatry. 2013;18:740–2. doi: 10.1038/mp.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Iwamoto K. Comprehensive DNA methylation and hydroxymethylation analysis in the human brain and its implication in mental disorders. Neuropharmacology. 2014;80:133–9. doi: 10.1016/j.neuropharm.2013.12.019. [DOI] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–53. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundakovic M, Champagne FA. Early-life experience, epigenetics, and the developing brain. Neuropsychopharmacology. 2015;40:141–53. doi: 10.1038/npp.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundakovic M, Gudsnuk K, Herbstman JB, Tang D, Champagne FA. DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci U S A. 2015;112:6807–13. doi: 10.1073/pnas.1408355111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundakovic M, Lim S, Gudsnuk K, Champagne FA. Sex-specific and strain-dependent effects of early life adversity on behavioral and epigenetic outcomes. Front Psychiatry. 2013;4:78. doi: 10.3389/fpsyt.2013.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, García-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–54. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonté B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, et al. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry. 2012a;69:722–31. doi: 10.1001/archgenpsychiatry.2011.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonté B, Suderman M, Maussion G, Lopez JP, Navaro L, Yerko V, et al. Genome-wide methylation changes in the suicide brain. Am J Psychiatry. 2012b;170:511–20. doi: 10.1176/appi.ajp.2012.12050627. [DOI] [PubMed] [Google Scholar]
- Labonté B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, et al. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry. 2012c;72:41–8. doi: 10.1016/j.biopsych.2012.01.034. [DOI] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren B, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–43. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Mattai A, Long R, Rapoport JL, Gogtay N, Addington AM. Microduplications disrupting the MYT1L gene (2p25.3) are associated with schizophrenia. Psychiatr Genet. 2012;22:206–9. doi: 10.1097/YPG.0b013e328353ae3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A, Worrell TR, Zimnisky R, Schmauss C. Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment. Neurobiol Dis. 2012;45:488–98. doi: 10.1016/j.nbd.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–94. doi: 10.1016/j.neuropharm.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Shen L, Zhang B, Garcia BA, Shao NY, Mitchell A, et al. Analytical tools and current challenges in the modern era of neuroepigenomics. Nat Neurosci. 2014;17:1476–90. doi: 10.1038/nn.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–8. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Suderman M, Sasaki A, Huang TCT, Hallett M, Meaney MJ, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One. 2011;6:e14739. doi: 10.1371/journal.pone.0014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–92. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- Melas PA, Rogdaki M, Ösby U, Schalling M, Lavebratt C, Ekström TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012;26:2712–8. doi: 10.1096/fj.11-202069. [DOI] [PubMed] [Google Scholar]
- Melka MG, Laufer BI, McDonald P, Castellani CA, Rajakumar N, O’Reilly R, et al. The effects of olanzapine on genome-wide DNA methylation in the hippocampus and cerebellum. Clin Epigenetics. 2014;6:1. doi: 10.1186/1868-7083-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AC, Bharadwaj R, Whittle C, Rueger W, Mirnics K, Hurd Y, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry. 2014;75:961–9. doi: 10.1016/j.biopsych.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–65. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12:1559–66. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- Neale BM, Sklar P. Genetic analysis of schizophrenia and bipolar disorder reveals polygenicity but also suggests new directions for molecular interrogation. Curr Opin Neurobiol. 2015;30:131–8. doi: 10.1016/j.conb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology. 2014;76:259–68. doi: 10.1016/j.neuropharm.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Jaaro-Peled H, Tankou S, Seshadri S, Hikida T, Matsumoto Y, et al. Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science. 2013;339:335–9. doi: 10.1126/science.1226931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- O’Donnell KJ, Bugge Jensen A, Freeman L, Khalife N, O’Connor TG, Glover V. Maternal prenatal anxiety and downregulation of placental 11β-HSD2. Psychoneuroendocrinology. 2012;37:818–26. doi: 10.1016/j.psyneuen.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Pal M, Ebrahimi S, Oh G, Khare T, Zhang A, Kaminsky ZA, et al. High precision DNA modification analysis of HCG9 in major psychosis. Schizophr Bull. 2015 doi: 10.1093/schbul/sbv079. Epub Jun 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña CJ, Bagot RC, Labonté B, Nestler EJ. Epigenetic signaling in psychiatric disorders. J Mol Biol. 2014;426:3389–412. doi: 10.1016/j.jmb.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña CJ, Neugut YD, Champagne FA. Developmental timing of the effects of maternal care on gene expression and epigenetic regulation of hormone receptor levels in female rats. Endocrinology. 2013;154:4340–51. doi: 10.1210/en.2013-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter MO, Du L, Weaver IC, Palkovits M, Faludi G, Merali Z, et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol Psychiatry. 2008;64:645–52. doi: 10.1016/j.biopsych.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Psychiatric Genetics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, et al. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry. 2011;1:1–6. doi: 10.1038/tp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Vialou V, Sun HS, Labonté B, Golden S, Dias C, et al. Fluoxetine epigenetically alters the CaMKIIα promoter in nucleus accumbens to regulate FosB binding and antidepressant effects. Neuropsychopharmacology. 2013;39:1178–86. doi: 10.1038/npp.2013.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Murillo L, Gogos JA, Karayiorgou M. The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annu Rev Med. 2012;63:63–80. doi: 10.1146/annurev-med-072010-091100. [DOI] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–59. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Haroutunian V. Schizophrenia: susceptibility genes and oligodendroglial and myelin related abnormalities. Front Cell Neurosci. 2014;8:5. doi: 10.3389/fncel.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65:760–9. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka WB, Subburaju S, Benes FM. Circuit- and diagnosis-specific DNA methylation changes at γ-aminobutyric acid-related genes in postmortem human hippocampus in schizophrenia and bipolar disorder. JAMA Psychiatry. 2015;72:541–51. doi: 10.1001/jamapsychiatry.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Sterrenburg L, Gaszner B, Boerrigter J, Santbergen L, Bramini M, Elliott E, et al. Chronic stress induces sex-specific alterations in methylation and expression of corticotropin-releasing factor gene in the rat. PLoS One. 2011;6:e28128. doi: 10.1371/journal.pone.0028128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suderman M, McGowan PO, Sasaki A, Huang TC, Hallett MT, Meaney MJ, et al. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc Natl Acad Sci U S A. 2012;109(suppl 2):17266–72. doi: 10.1073/pnas.1121260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HS, Damez-Werno D, Scobie KN, Shao NY, Dias C, Rabkin J, et al. ACF chromatin remodeling complex mediates stress-induced depressive-like behavior. Nat Med. 2015 doi: 10.1038/nm.3939. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–16. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Xu B, Ionita-Laza I, Roos JL, Gogos JA, Karayiorgou M. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron. 2014;82:773–80. doi: 10.1016/j.neuron.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Dean B, Thomas EA. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry. 2011;1:e64. doi: 10.1038/tp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–25. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Turecki G, Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.11.022. Epub Dec 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, et al. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron. 2011;69:359–72. doi: 10.1016/j.neuron.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Veldic M, Caruncho HJ, Liu WS, Davis J, Satta R, Grayson DR, et al. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc Natl Acad Sci U S A. 2004;101:348–53. doi: 10.1073/pnas.2637013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA. Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology. 2014;80:18–27. doi: 10.1016/j.neuropharm.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton E, Liu J, Hass J, White T, Scholz M, Roessner V, et al. MB-COMT promoter DNA methylation is associated with working-memory processing in schizophrenia patients and healthy controls. Epigenetics. 2014;9:1101–7. doi: 10.4161/epi.29223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–5. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MB, Xiao G, Kumar A, Laplant Q, Renthal W, Sikder D, et al. Impairment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29:7820–32. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. 2012;21:2205–10. doi: 10.1093/hmg/dds035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wockner LF, Noble EP, Lawford BR, Young RM, Morris CP, Whitehall VL, et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry. 2014;4:e339. doi: 10.1038/tp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Chanrion B, O’Donnell AH, Milekic M, Costa R, Ge Y, et al. MethylomeDB: a database of DNA methylation profiles of the brain. Nucleic Acids Res. 2012;40:D1245–9. doi: 10.1093/nar/gkr1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Li W, Yu T, Zhang C, Wang D, Liu D, et al. SOX10 rs139883 polymorphism is associated with the age of onset in schizophrenia. J Mol Neurosci. 50:333–8. doi: 10.1007/s12031-013-9982-y. [DOI] [PubMed] [Google Scholar]
- Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30:13130–7. doi: 10.1523/JNEUROSCI.1039-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]