

Abstract
In humans, cytochrome P450 1A2 is the major enzyme metabolizing environmental arylamines or heterocyclic amines into carcinogens. Since evidence shows that planar triangle-shaped molecules are capable of selectively inhibiting P450 1A2, 16 triangular flavone, and coumarin derivatives were designed and synthesized for these studies. Among these compounds, 7,8-furanoflavone time-dependently inhibits P450 1A2 with a KI value of 0.44 μM. With a 5 min preincubation in the presence of NADPH, 0.01 μM 7,8-furanoflavone completely inactivates P450 1A2 but does not influence the activities of P450s 1A1 and 1B1. Another target compound, 7,8-pyrano-4-trifluoromethylcoumarin, is found to be a competitive inhibitor, showing high selectivity for the inhibition of P450 1A2 with a Ki of 0.39 μM, 155- and 52-fold lower than its Ki values against P450s 1A1 and 1B1, respectively. In yeast AhR activation assays, 7,8-pyrano-4-trifluoromethylcoumarin does not activate aryl hydrocarbon receptor when the concentration is lower than 1 μM, suggesting that this compound would not up-regulate AhR-caused P450 enzyme expression. In-cell P450 1A2 inhibition assays show that 7,8-pyrano-4-trifluoromethylcoumarin decreases the MROD activity in HepG2 cells at concentrations higher than 1 μM. Thus, using 7,8-pyrano-4-trifluoromethylcoumarin, a selective and specific P450 1A2 action suppression could be achieved, indicating the potential for the development of P450 1A2-targeting cancer preventive agents.
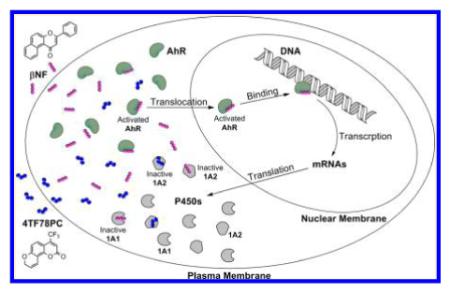
INTRODUCTION
P450 family 1 enzymes (1A1, 1A2, and 1B1) are important environmental xenobiotic-metabolizing enzymes. Different from P450s 1A1 and 1B1, P450 1A2 is responsible for the metabolism of aryl and heterocyclic amines. Some bioactive amines, such as 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), are metabolized by this enzyme into potent mutagenic or carcinogenic agents.1,2 Therefore, selective inhibition of P450 1A2 is a potential molecular target for chemoprevention of environmental arylamine-caused DNA mutations and carcino-genesis.3 Certain compounds with varying molecular structures have been shown to effectively inhibit P450 1A2, including aryl hydrocarbons, stilbenoids, flavonoids, coumarins, and anthra-quinone, etc.4–10 However, because of the high degree of similarity between P450 1A2 and P450s 1A1 and 1B1 (sharing 80% and 40% amino acid sequence identity, respectively), most P450 1A2 inhibitors also show some extent of inhibitory activity toward P450s 1A1 and 1B1.11,12 Although some efforts have been made to determine the differences between the active site cavities of these enzymes, and a few selective P450 1A2 inhibitors (selectivity indices around 5- to 20-fold) have been discovered, highly selective P450 1A2 inhibitors are still desired. In this work, employing the concept of ligand-based drug design, we performed a two-step design, synthesis, and evaluation and obtained new and selective P450 1A2 inhibitors.
A major concern preventing a P450 inhibitor from being used as a clinical drug is its potential ability to activate the aryl hydrocarbon receptor (AhR), which triggers the overexpression of P450 enzymes. Thus, the comprehensive action of a P450 inhibitor in human body may lead to up-regulation of P450 enzymes. As a typical example, β-naphthoflavone (βNF) inhibits P450 family I enzymes in enzymatic assays, but it promotes the activities of P450 family I enzymes in cell level assays.13,14 Therefore, an ideal inhibitor of P450 enzymes should not interrupt the AhR-signaling pathway, which is called “phase II” selectivity of P450 inhibitors. To identify the influence of tested inhibitors on P450 expression, a yeast AhR-signaling assay was performed.
RESULTS AND DISCUSSION
Common Pharmacophore Features of Selective P450 1A2 Inhibitors
Because of the great similarity between P450s 1A1 and 1A2, finding inhibitors that are selective for P450 1A2 over P450 1A1 is difficult. Recently, we completed a review of compounds that exhibit least 5 times stronger inhibition activity toward P450 1A2 compared to P450 1A1.15 In order to identify the common features of selective P450 1A2 inhibitors, 3D structures of 18 selected compounds were sketched using the ChemBio3D Ultra 12.0 (PerkinElmer, Inc., Waltham, MA). After energy minimization, these structures were aligned automatically by 100% steric field in the Alignment Module of Discovery Studio 3.5 (Accelrys, San Diego, CA). The superimposed image shown in Figure 1 indicates the shape and size of selective P450 1A2 inhibitors, denoting a planar triangular molecule with a size of 90 Å2. According to this structural characteristic and our lead compound 7,8-pyrano-flavone (2), we designed a group of flavone derivatives (3, 8, 11, and 12, Schemes 1 and 2) as potentially selective P450 1A2 inhibitors.
Figure 1.
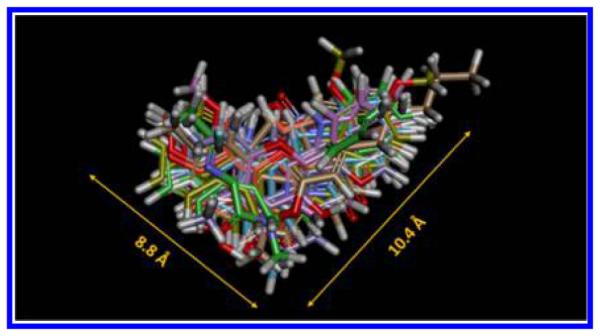
Superimposed image of 18 selective inhibitors of P450 1A2 over P450 1A1. The individual structures of these compounds are documented in the Supporting Information I.
Scheme 1. Synthetic Routes for Pyrano- and Furanoflavone Derivativesa.

a(a) Propargyl bromide, K2CO3 in acetone; (b) N,N-diethylaniline, 200 °C; (c) 1.5 equiv of CsF in N,N-diethylaniline, 200 °C; (d) benzyl bromide, NaH in DMSO; (e) 5% Pd/C in THF.
Scheme 2. Synthetic Routes for Pyridino- and Dioxoloflavone Derivativesa.

a(a) Propargyl bromide, K2CO3 in DMF; (b) AgSbF6 in N,N-diethylaniline, 110°C; (c) CH2Br2, Cs2CO3 in DMSO.
Synthesis of Pyrano- and Furanoflavone Derivatives
Starting with flavone propargyl ethers (1 and 4), the pyrano and furano products were successfully synthesized through a Claisen rearrangement at 200 °C. In the absence of any salt, this rearrangement reaction led to the pyrano product, like 7,8-pyranoflavone (2); while in the presence of 5% cesium fluoride it gave the furano product, like 7,8-furanoflavone (3).8,16 The compound 5-hydroxy-7-propargyloxyflavone (4) seemed to decompose at 200 °C in the presence of cesium fluoride. Thus, in order to obtain 5-hydroxy-7,8-furanoflavone (8), an indirect synthetic route was applied with a benzyl as the protecting group.
Synthesis of Pyridine- and Dioxoloflavone Derivatives
The alkylation of 6-aminoflavone by propargyl bromide in the presence of cesium carbonate produced the secondary amine (9) and the tertiary amine (10). Using AgSbF6 as catalyst, compound 9 was converted into 7,8-pyridinoflavone (11) completely through a similar Claisen rearrangement and annulation (Scheme 2). This reaction occurred in a relatively mild condition (heating at 110 °C), as recently reported.17 Starting from 7,8-hydroxyflavone, 7,8-dioxoloflavone (12) was facilely synthesized through an alkylation with dibromomethane.
Design and Synthesis of Pyrano-, Furano-, Pyridino-, and Dioxolocoumarin Derivatives
Coumarin, especially 4-phenylcoumarin, shares great structural similarity with flavone. Its derivative 4-phenyl-7,8-pyranocoumarin has a planar triangular shape and the same size as 7,8-pyranoflavone (Figure 2). On the basis of this observation, we designed and synthesized a group of coumarin derivatives as selective P450 1A2 inhibitor candidates (Scheme 3).
Figure 2.

Structural similarity of 7,8-pyranoflavone and 4-substituted 7,8-pyranocoumarin. (A) Structures of 7,8-pyranoflaovne and 4-phenyl-7,8-pyranocoumarin. (B) Superimposed image of 78PF (green) and 4P78PC (purple). Superimposed image was obtained through the automatic alignment by 100% steric field in the Alignment Module of Discovery Studio 3.5. 78PF and 4P78PC also have identical elemental composition and molecular weight.
Scheme 3. Synthetic Routes for Pyrano-, Furano-, Pyridine-, and Dioxolocoumarin Derivativesa.

a(a) Propargyl bromide, K2CO3 in acetone; (b) N,N-diethylaniline, 200 °C; (c) 1.5 equiv of CsF in N,N-diethylaniline, 200 °C; (d) propargyl bromide, K2CO3 in DMF; (e) AgSbF6 in DMSO, 110°C; (f) CH2Br2, Cs2CO3 in DMSO.
Pyrano- and furanocoumarin derivatives (17–24) were synthesized from coumarin propargyl ethers through the same methods used for 78PF (2) and 78FF (3), which were described above, in the absence or presence of cesium fluoride. 4-Methyl-7,8-pyridinocoumarin (27) was obtained through the Claisen rearrangement of monopropargyl substituted 7-amino-4-methylcoumarin (25) which is one of the products from propargylation of 7-amino-4-methylcoumarin. Employing the method to synthesize 7,8-dioxoloflavone, 4-phenyl-7,8-dioxolo-coumarin (28) was easily obtained from 7,8-dihydroxy-4-phenylcoumarin.
Inhibitory Activities of Triangular Flavone Derivatives toward P450s 1A1, 1A2, and 1B1
The inhibitory activities of the triangular flavone derivatives toward P450s 1A1, 1A2, and 1B1 were determined through P450 1A1-dependent ethoxyresorufin-O-deethylase (EROD), P450 1A2-dependent methoxyresorufin-O-demethylase (MROD), and P450 1B1-dependent EROD assays, respectively. The well-known P450 1A2 inhibitors, furafylline and α-naphthoflavone (αNF), served as positive controls.18,19 As shown in Table 1, all of the flavone derivatives, except 78PyF (compound 11), show somewhat higher degrees of inhibitory activity toward P450 1A2 than toward P450 1A1. This evidence proves our hypothesis that triangular αNF-like molecules are more selective in inhibiting P450 1A2 in comparison to P450 1A1. However, incorporation of an electron donor group as in 78PyF seems to subvert this selectivity. As to the inhibitory efficacy, αNF and 5H78PF are the most potent inhibitors toward P450 1A2, with Ki values of 20 and 14 nM, respectively. Comparing the Ki values between enzymes, 78FF (3) and 78DOC (12) exhibit 14-fold more selectivity toward P450 1A2 over 1A1 and exhibit 6- and 5.3-fold more selectivity toward P450 1A2 over P450 1B1, respectively. More interestingly, 78FF and 5H78FF (compounds 3 and 8) show time- and NADPH-dependent inhibition of P450 1A2 (Figure 3 and 4), which means these two molecules are potential mechanism-based inhibitors (MBIs) of this enzyme. Oxidation of an MBI by a certain P450 enzyme yields an active intermediate that covalently bonds with the same enzyme, leading to irreversible inactivation of the enzyme.20,21 Thus, an MBI exhibits stronger inhibitory activity than a competitive inhibitor with the same Ki. Since the inhibition mechanism for MBIs requires NADPH, DADPH dependency is one of the critical characteristics of these types of inhibitors. NADPH-dependency assays show that preincubation of 10 nM 78FF or 5H78FF with NADPH and P450 1A2 for 5 min induces 90% inhibition of the enzyme, while this does not occur in the case of P450s 1A1 and 1B1, suggesting that 78FF and 5H78FF are probably competitive inhibitors of P450s 1A1 and 1B1 (Figure 3).
Table 1.
Inhibitory Activity and Selectivity of Flavone Derivatives toward P450 1A2a
| Compound | Short Name |
Structure | Ki (μM) | Selective Index (SI) |
|||
|---|---|---|---|---|---|---|---|
| 1A1 | 1A2 | 1B1 | 1A1/1A2 | 1B1/1A2 | |||
| Furafylline |
|
>200 | 68.0 ±13.2 |
>200 | >2.9 | >2.9 | |
| α-Naphtho flavone |
αNF |
|
0.045 ±0.010 |
0.020 ±0.005 |
0.016 ±0.002 |
2.3 | 0.8 |
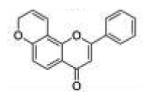
| 2 | 78PF |
|
0.27 ±0.02 |
0.058 ±0.018 |
0.053 ±0.032 |
4.7 | 0.9 |
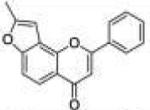
| 3 | 78FF |
|
0.43 ±0.19 |
0.030 ±0.002 |
0.18 ±0.05 |
14 | 6.0 |
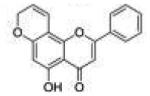
| 5 | 5H78PF |
|
0.11 ±0.02 |
0.014 ±0.004 |
0.0056 ±0.0020 |
7.9 | 0.4 |
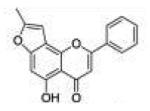
| 8 | 5H78FF |
|
0.47 ±0.12 |
0.044 ±0.023 |
0.10 ±0.06 |
11 | 2.3 |
| 11 | 78PyF |
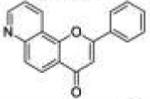
|
0.040 ±0.008 |
0.076 ±0.011 |
0.12 ±0.02 |
0.5 | 1.8 |
| 12 | 78DOF |
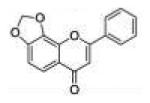
|
1.23 ±0.22 |
0.090 ±0.034 |
0.48 ±0.21 |
14 | 5.3 |
The Ki values are represented as the mean ± SD μM of three independent experiments.
Figure 3.

NADPH-dependency assays of 78FF and 5H78FF inhibition of P450s 1A1, 1A2, and 1B1: (black curves) blank control, the time-course curve of enzymatic reaction without an inhibitor; (light-gray curves) preincubation without NADPH, the time-course curve of enzymatic reaction with 10 nM 78FF or 5H78FF in the absence of 5 min NADPH preincubation; (medium-gray curves) preincubation with NADPH, the time-course curve of enzymatic reaction with 10 nM 78FF or 5H78FF in the presence of 5 min NADPH preincubation. For the assays on P450s 1A1 and 1B1, 10 nM 78FF or 5H78FF does not influence the enzymatic activities with or without NADPH preincubation. For the assays on P450 1A2, 10 nM 78FF or 5H78FF exhibits a significant inhibition toward the enzyme without NADPH preincubation, while 10 nM 78FF or 5H78FF exhibits >90% inhibition toward the enzyme with NADPH preincubation. This suggests that 78FF and 5H78FF inactivate P450 1A2 NADPH-dependently.
Figure 4.

(A) Production of resorufin by P450 1A2 in the absence (control) and presence of 0.0125, 0.025, 0.05, and 0.1 μM 78FF (3). The fluorescent product formation (resorufin anion) was monitored continuously over 6 min as described in the Experimental Section. 78FF inhibited the production of resorufin in a concentration-dependent manner, and the second-order product formation curves (y = ax2 + bx + c) were obtained using the Trendline tool of the Microsoft Excel program. The differential curve (y = 2ax + b) of each product formation curve represents the instantaneous enzymatic activity (v). (B) Dixon plots (1/v0 vs [I]). The first-order coefficient b in the above second-order equations in graph A represents enzymatic activity (v0) at the beginning of the enzymatic reaction. Dixon plots were used (by plotting the reciprocals of the enzymatic activity (1/v0) vs inhibitor concentrations [I]) to determine Ki values (x-intercepts) for the inhibitors. In this case, the Ki value of 78FF in inhibition of P450 1A2 was 0.45 μM. (C) Time- and concentration-dependent inhibition of P450 1A2-dependent MROD activity by 78FF. The enzymatic activity values were calculated through the first-order derivatives (y = 2ax + b) of the product formation curves at different times, and the percentages of activity remaining was obtained according to the formula (Ainhibitor/Acontrol)%. The activity loss with time indicates the time-dependent inhibition of P450 1A2 by 78FF. (D) Kitz–Wilson plots (1/kinact vs 1/[I]). kinact values were obtained using the curves in graph C as described in the Experimental Section, and [I] values were the final concentrations of 78FF. In this case, the final concentrations were 0.0125, 0.025, 0.05, and 0.1 μM. According to the linear equation shown in this graph, KI and limiting kinact values were calculated, respectively.
Inhibition Mechanism of 78FF and 5H78FF toward P450 1A2
The KI values of 78FF and 5H78FF in the inhibition of P450 1A2 are 0.44 and 0.12 μM, with the limiting kinact values of 0.42 and 0.26 min−1, respectively. Generally, 78FF and 5H78FF are strong time-dependent inhibitors with fast reaction rates with P450 1A2, since the limiting half inhibition times (limiting t1/2 values) are 1.65 and 2.60 min, respectively. By use of 78FF as an example, Figure 4 shows the method used to identify a time-dependent inhibitor (TDI) and to determine its KI, kinact, as well as Ki values. 78FF and 5H78FF contain a furan functional group which has been well investigated as a fragment of MBIs toward P450 enzymes, including P450 1A2.20,21 The grapefruit extract bergamottin is a typical furan-containing MBI, which inactivates several P450 enzymes in a mechanism-based manner.22 4-Ipomeanol, first isolated from sweet potatoes, is another furan-containing MBI.23 The inhibitory mechanism for this type of compound involves the oxidation of the furan fragment by the P450s producing a furan oxide which either directly or through a butenedial intermediate bonds with the enzyme.21,24
Inhibitory Activities of Triangular Coumarin Derivatives toward P450s 1A1, 1A2, and 1B1
Ten triangular coumarin derivatives were tested in P450 1A1-dependent EROD, P450 1A2-dependent MROD, and P450 1B1-dependent EROD assays in order to determine their inhibitory activities and selectivity toward P450 1A2. The inhibition potencies of these coumarin derivatives toward P450 1A2 were found to be weaker than those of flavone derivative. The Ki values of the flavone derivatives studied toward P450 1A2 were at the 10 nM level, while the Ki values of the coumarin derivatives were around 1 μM level. The most potent compound (20, 4TF78PC) toward P450 1A2 possesses a Ki value of 0.39 μM. However, the coumarin derivatives are much more selective toward P450 1A2 than P450s 1A1 and 1B1 compared to the flavone derivatives. The Ki value of 4TF78PC toward P450 1A2 is 155-fold and 52-fold lower than those toward P450s 1A1 and 1B1, respectively. Thus, the inhibitory activity and selectivity of 4TF78PC toward P450 1A2 are both better than those of furafylline, suggesting that 4TF78PC could be a promising new P450 1A2 inhibitor. In addition, the data show that pyranocoumarin derivatives are more potent than the other coumarin derivatives in inhibiting P450 1A2 and that the 4-position small hydrophobic group modification increases the inhibition of P450 1A2. No time-dependent inhibitors were identified in this group of tested compounds, even in the furano derivatives.
Effects of Compounds 3 and 20 on Activation of AhR
A yeast AhR-signaling assay was used to identify the impacts of desired compounds on the expression of P450 enzymes. AhR is a DNA transcription factor which could be activated by ligands like βNF, subsequently turning on the up-expression of P450s, including family I enzymes. Activation of AhR in yeast strain MYA-3637 was determined by a lacZ reporter assay according to the formation of o-nitrophenyl-β-galactoside (Abs405/Abs600), reflecting the level of P450 expression induced by AhR. Compounds 78FF (3) and 4TF78PC (20), the most selective inhibitors in their groups, were selected as the representatives of flavone derivatives and coumarin derivatives, respectively. The data obtained are shown in Figure 5. The positive control, 10−8 M βNF, exhibits a great activation of AhR-signaling pathway compared with the blank control (1% DMSO, v/v). Compound 78FF shows a considerable activation of AhR at concentrations higher than 10−7 M. Compound 4TF78PC does not activate AhR-signaling pathway at concentrations lower than 10−6 M (1 μM), suggesting that 4TF78PC does not lead to an increase in the expression of P450s caused by AhR pathway in cells at the dose of 1 μM. Thus, the effect of 1 μM 4TF78PC on the cells would be due to pure inhibition of P450 1A2.
Figure 5.
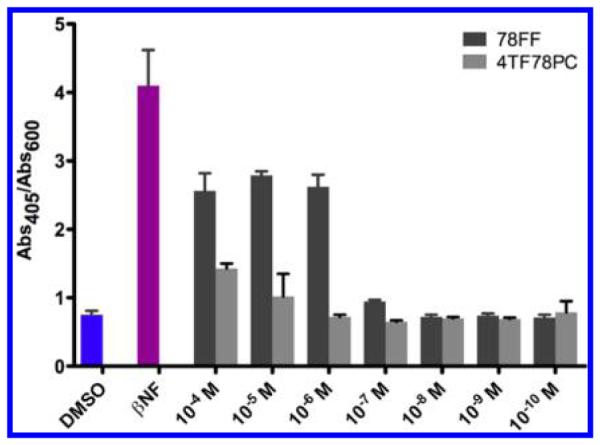
Activation of AhR-signaling pathway by various concentrations of 78FF and 4TF78PC in the yeast strain MYA-3637. DMSO serves as the blank control, and 10−8 M of βNF serves as the positive control. Abs405/Abs600 values are represented as the mean ± SD. Each experiment is repeated at least three times.
To determine the impact of 4TF78PC on the activation of AhR by βNF, a combination administration study in the yeast AhR-signaling assay was performed. As shown in Figure 6, various concentrations of 4TF78PC ranging from 10−4 to 10−10 M do not influence the activation of AhR by βNF, indicating that 4TF78PC could not be a high-affinity ligand of AhR, which leads to the “phase II” selectivity.
Figure 6.
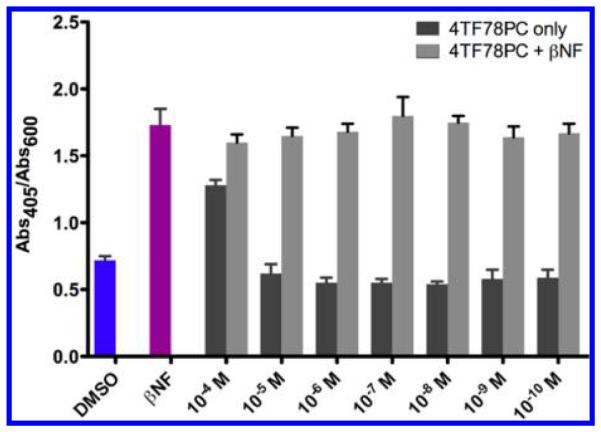
Influence of 4TF78PC on βNF-induced AhR activation −8 (10 M βNF) in the yeast strain MYA-3637. DMSO serves as the −8 are more efficient in the inhibition of P450 1A2 in comparison blank control, and 10−8 M βNF serves as the positive control. Abs405/ to the positive control furafylline. The most selective P450 1A2 Abs600 values are represented as the mean ± SD. Each experiment is repeated at least three times.
Inhibiting P450 1A2 Activity in HepG2 Cells
Since P450 1A2 is expressed mainly in the liver, the human hepatocellular carcinoma HepG2 cell line was used as an in-cell evaluation system.25 The ability of compound 20 (4TF78PC) in affecting the P450 1A2 enzyme activity was evaluated in HepG2 cells by measuring the MROD activity. In 24-well plates, HepG2 cells were incubated with a blank control (DMSO), a positive control (βNF, 10−5 M), and a series of concentrations of 4TF78PC for 24 h. Medium was decanted, and cells were washed once with PBS (phosphate buffered saline). A medium containing 5 μM methoxyresorufin was added to each well. The increase in fluorescence resulting from the conversion of methoxyresorufin into resorufin by P450 1A2 was measured at 10, 30, and 60 min. As shown in Figure 7, 10−5 M βNF increased the MROD activity in HepG2 cells, while 4TF78PC decreased the MROD activity at concentrations higher than 10−6 M. This result was consistently observed at the three time points, suggesting the effectiveness of 4TF78PC in inhibiting P450 1A2 at the cellular level.
Figure 7.

Effects of 4TF78PC on the MROD reaction in HepG2 cells. DMSO serves as the blank control, and 10−5 M βNF serves as the positive control. The MROD activity in cells is identified by the formation of fluorescent product resorufin, which was determined using the fluorescence intensity at 585 nm (excitation wavelength, 535 nm). The fluorescence intensity was measured at the time points of 10 min (A), 30 min (B), and 60 min (C) after adding the substrate methoxyresorufin. The fluorescence intensity values are presented as the mean ± SD (triplicate). Each experiment was repeated three times independently. The Student’s unpaired t test was employed for statistical analysis, and p < 0.05 was considered as statistically significant: *, compared with the blank control, p < 0.05.
To exclude the possibility that the inhibition of MROD in cells resulted from the toxicity of 4TF78PC to the cells, an Alamar blue viability assay of 4TF78PC in HepG2 cells was performed. The result is presented in Supporting Information S58. Up to the concentration of 10−5 M, 4TF78PC was not toxic to HepG2 cells. Thus, the inhibition of MROD activity in cells completely resulted from the inhibition of P450 1A2 by 4TF78PC.
CONCLUSION
In order to develop specific P450 1A2 inhibitors, we designed 14 planar triangular flavone and coumarin derivatives using a computer-assisted alignment assay. The desired compounds were synthesized with atom-economical and energy-saving methods with acceptable yields. All the target compounds except compound 11 (78PyF) show selective inhibition of P450 1A2 over P450s 1A1 and 1B1. All the tested compounds are more efficient in the inhibition of P450 1A2 in comparison to the positive control furafylline. The most selective P450 1A2 inhibitor in the flavone group is 7,8-furanoflavone (3). This compound is a time-dependent inhibitor of P450 1A2 compared to P450s 1A1 and 1B1. 4-Trifluomethyl-7,8-pyranocoumarin (20) is the most potent and selective P450 1A2 inhibitor in the coumarin group, with a Ki value of 0.39 μM. The selectivity indices of 4TF78PC toward P450 1A2 over P450s 1A1 and P450 1B1 are 155 and 52, respectively. The AhR-signaling pathway activation assays show that 4TF78PC does not activate AhR when the concentration is lower than 1 μM. In-cell P450 1A2 inhibition assays show that 4TF78PC decreases the MROD activity in HepG2 cells at concentrations higher than 1 μM. Thus, in this study, a specific selective P450 1A2 inhibitor, 4TF78PC, has been identified.
EXPERIMENTAL SECTION
Chemistry
The 7-hydroxyflavone and 5,7-dihydroxyflavone were purchased from INDOFINE Chemical Company, Inc. (Hillsborough, NJ), and other chemicals were purchased from Sigma-Aldrich Corporation (St. Louis, MO) and Fisher Scientific International, Inc. (Hampton, NH). All the thermal cyclization reactions were performed with a CEM Discover microwave synthesis system (Matthews, NC). Mass spectral data were determined using an Agilent 6890 GC instrument with a 5973 MS instrument. 1H NMR and 13C NMR spectra were recorded on a Bruker Fourier 300 MHz FT-NMR spectrometer. Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA). On the basis of elemental analyses, the purity of all tested compounds is higher than 95%.
General Procedure A: Synthesis of Propargyloxyflavones and Propargyloxycoumarins
To a solution of 2.00 mmol of hydroxyflavones or hydroxycoumarins in 40 mL of anhydrous acetone, 500 mg (3.62 mmol) of anhydrous potassium carbonate was added. After the reaction solution turned yellow, 1.0 mL (9.00 mmol) of 80% propargyl bromide solution in toluene was added. The reaction solution was heated to 50 °C in a heating mantle. Once the starting material’s spot disappeared on thin layer chromatography (TLC), the mixture was cooled to room temperature, filtered by gravity, and concentrated under vacuum to give the crude product. The pure product was obtained as crystals by recrystallization from 20 mL of anhydrous ethanol (yield, 66–78%).
2-Phenyl-7-(prop-2-yn-1-yloxy)-4H-chromen-4-one (7-Prop-argyloxyflavone, 1)
Starting with 7-hydroxyflavone, this compound was prepared using general procedure A (yield, 78%). Analytical data were reported previously.26
General Procedure B: Synthesis of Pyranoflavones (PFs) and Pyranocoumarins (PCs)
In a 10 mL reaction vessel, 200 mg of propargyloxyflavone or propargyloxycoumarin was suspended in 5 mL of N,N-diethylaniline. After heating in a CEM Discover microwave at 195 °C for an hour, the reaction solution was cooled to room temperature and transferred into a separatory funnel, to which 50 mL of dichloromethane (DCM) was added. The organic phase was washed successively with 5% potassium bisulfate (50 mL × 5) and brine (50 mL × 2). The DCM layer was then dried over anhydrous magnesium sulfate and concentrated under vacuum. The residue was purified by column chromatography using silica gel (solvent systems varied based on the products) to provide the pure pyranoflavone or pyranocoumarin compound (yield, 38–60%).
2-Phenylpyrano[2,3-f]chromen-4(8H)-one (7,8-Pyranoflavone, 78PF, 2)
Starting with 7-propargyloxyflavone, 120 mg (yield, 60%) of this compound was prepared using general procedure B. Column chromatography solvent system: petroleum ether/ethyl acetate 3:1. Analytical data were reported previously.8
General Procedure C: Synthesis of Furanoflavones (FFs) and Furanocoumarins (FCs)
In a 10 mL reaction vessel, an amount of 200 mg of the propargyloxyflavone or propargyloxycoumarin with 1.5 equiv of cesium fluoride was suspended in 5 mL of N,N-diethylaniline. After heating in a CEM Discover microwave at 200 °C for an hour, the reaction solution was cooled to room temperature and transferred into a separatory funnel, to which 50 mL of DCM was added. The organic phase was washed successively with 5% potassium bisulfate (50 mL × 5) and brine (50 mL × 2). The DCM layer was then dried over anhydrous magnesium sulfate and concentrated under vacuum. The residue was purified by column chromatography using silica gel (solvent systems varied based on the products) to provide the pure furanoflavone or furanocoumarin compound (yield, 36–61%).
8-Methyl-2-phenyl-4H-furo[2,3-h]chromen-4-one (7,8-Fura-noflavone, 78FF, 3)
Starting with 7-propargyloxyflavone, 96 mg (yield, 48%) of this compound was prepared using general procedure C. Column chromatography solvent system: petroleum ether/ethyl acetate 3:1. Mp 194–196 °C. GC/MS: 276 (M+, 100%), 248 (15), 174 (100), 145 (18). 1H NMR (CDCl3, 300 MHz) δ 8.06 (d, J = 8.7 Hz, 1H), 7.94 (m, 2H), 7.53 (m, 3H), 7.44 (dd, J = 8.7 Hz, J = 0.6 Hz, 1H), 6.85 (s, 1H), 6.80 (s, 1H), 2.54 (d, J = 0.6 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 178.45, 162.53, 158.03, 156.75, 149.98, 131.86, 131.48, 129.08, 126.15, 120.43, 119.14, 118.57, 109.60, 107.75, 100.13, 14.15. Anal. Calcd for C18H12O3: C, 78.25; H, 4.38. Found: C, 78.05; H, 4.48.
5-Hydroxy-2-phenyl-7-(propargyloxy)-4H-chromen-4-one (5-Hydroxy-7-propargyloxyflavone, 4)
Starting with 5,7-dihydroxyflavone, this compound was prepared using general procedure C (yield, 73%). Analytical data were reported previously.26
5-Hydroxy-2-phenylpyrano[2,3-f]chromen-4(8H)-one (5-Hydroxy-7,8-pyranoflavone, 5H78PF, 5)
Starting with 5-hydroxy-7-propargyloxyflavone, 80 mg (yield, 40%) of this compound was prepared using general procedure B. Column chromatography solvent system: petroleum ether/DCM 1:1. Analytical data were reported previously.8
5-(Benzyloxy)-2-phenyl-7-(prop-2-yn-1-yloxy)-4H-chromen-4-one (5-Benzyloxy-7-propargyloxyflavone, 6)
To a solution of 250 mg (0.86 mmol) of 5-hydroxy-7-propargyloxyflavone (4) in 10 mL of anhydrous DMSO, 41 mg (1.03 mmol) of sodium hydride (60% dispersion in mineral oil) and 112 μL (0.95 mmol) of benzyl bromide were added. The reaction solution was allowed to stir at room temperature overnight. Once the starting material’s spot disappeared on TLC, the mixture was quenched with 20 mL of water and washed with 50 mL of ethyl acetate twice (50 mL × 2). The ethyl acetate phase was combined, washed with brine (50 mL × 5), dried over anhydrous magnesium sulfate, and concentrated under vacuum. The residue was purified by column chromatography using silica gel (petroleum ether/ethyl acetate 4:1) to provide 260 mg (yield, 79%) of title compound 6 as colorless crystals. Mp 189–191 °C. GC/MS: 382 (M+, 100%). 1H NMR (CDCl3, 300 MHz) δ 7.88 (m, 2H), 7.62 (d, J = 7.2, 2H), 7.50 (m, 3H), 7.40 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.5 Hz, 1H), 6.68 (s, 1H), 6.67 (d, J = 2.4 Hz, 1H), 6.48 (d, J = 2.1 Hz, 1H), 5.25 (s, 2H), 4.75 (d, J = 2.4 Hz, 2H), 2.60 (t, J = 2.4, 1H). 13C NMR (CDCl3, 75 MHz) δ 177.33, 161.66, 160.81, 159.76, 159.62, 136.32, 131.51, 131.25, 128.96, 128.63, 127.70, 126.60, 126.02, 110.24, 109.15, 98.30, 94.39, 77.38, 70.79, 56.17.
5-(Benzyloxy)-8-methyl-2-phenyl-4H-furo[2,3-h]chromen-4-one (5-Benzyloxy-7,8-furanoflavone, 7)
Starting with 5-benzyloxy-7-propargyloxyflavone (6), 90 mg (yield, 45%) of this compound was prepared using general procedure C. Column chromatography solvent system: petroleum ether/ethyl acetate 4:1. Mp 162–164 °C. GC/MS: 382 (M+, 100%), 305 (28). 1H NMR (CDCl3, 300 MHz) δ 7.92 (m, 2H), 7.66 (d, J = 7.2, 2H), 7.54 (m, 3H), 7.41 (m, 2H), 7.30 (m, 1H), 6.96 (d, J = 0.6 Hz, 1H), 6.77 (s, 1H), 6.69 (t, J = 1.2 Hz, 1H), 5.29 (s, 2H), 2.48 (d, J = 1.2 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 177.98, 160.26, 157.48, 155.91, 155.16, 151.09, 136.67, 131.59, 131.26, 129.02, 128.59, 127.65, 126.79, 125.97, 112.11, 111.50, 109.51, 99.81, 94.20, 71.53, 13.97.
5-Hydroxy-8-methyl-2-phenyl-4H-furo[2,3-h]chromen-4-one (5-Hydroxy-7,8-furanoflavone, 5H78FF, 8)
To a solution of 80 mg (0.21 mmol) of 5-benzyloxy-7,8-furanoflavone (7) in 10 mL of THF, 40 mg of 5% Pd/C was added. The reaction solution was stirred at room temperature under H2. Once the starting material’s spot disappeared on TLC, the reaction mixture was filtrated by gravity and concentrated under vacuum. The crude product was purified by recrystallization from 5 mL of anhydrous ethanol to provide 50 mg (yield, 82%) of 5-hydroxy-7,8-furanoflavone (8) as colorless crystals. Mp 206–208 °C. GC/MS: 292 (M+, 100%), 190 (40), 102 (40). 1H NMR (CDCl3, 300 MHz) δ 12.64 (s, 1H), 7.92 (m, 2H), 7.56 (m, 3H), 6.86 (d, J = 0.9 Hz, 1H), 6.76 (s, 1H), 6.63 (t, J = 0.9 Hz, 1H), 2.47 (d, J = 1.2 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 183.32, 163.49, 159.19, 157.74, 155.00, 148.95, 131.96, 131.20, 129.18, 126.27, 110.02, 107.30, 106.42, 99.25, 95.23, 13.96. Anal. Calcd for C18H12O4: C, 73.97; H, 4.14. Found: C, 73.42; H, 4.14.
2-Phenyl-7-(prop-2-yn-1-ylamino)-4H-chromen-4-one (N-Propargyl-7-aminoflavone, 9) and 7-(Di(prop-2-yn-1-yl)-amino)-2-phenyl-4H-chromen-4-one (N,N-Dipropargyl-7-ami-noflavone, 10)
To a solution of 500 mg (2.11 mmol) of 7-aminoflavone in 10 mL of DMF, 500 mg (3.62 mmol) of potassium carbonate and 1.0 mL (9.00 mmol) of 80% propargyl bromide solution in toluene were added. The reaction was stirred at room temperature for at least 1 week. When the starting material completely disappeared, the reaction mixture was quenched with 150 mL of DCM. After filtration by gravity, the organic phase was washed with saturated sodium bicarbonate (50 mL × 5), dried over anhydrous magnesium sulfate, and concentrated under vacuum. The residue was purified by column chromatography using silica gel (DCM/methanol 50:1) to give compounds 9 and 10 as yellowish and colorless crystals, respectively (yield, 55% and 14%).
N-Propargyl-7-aminoflavone (9)
Mp 189–191 °C. GC/MS: 275 (M+, 100%), 246 (15), 208 (35), 172 (16). 1H NMR (DMSO-d6, 300 MHz) δ 8.02 (m, 2H), 7.77 (d, J = 8.7, 1H), 7.55 (m, 3H), 7.25 (t, J = 5.7 Hz, 1H), 6.82–6.74 (m, 3H), 4.05 (dd, J = 5.4 Hz, J = 2.1 Hz, 2H), 3.21 (t, J = 2.1, 1H). 13C NMR (DMSO-d6, 75 MHz) δ 176.52, 161.64, 158.49, 153.32, 131.97, 131.74, 129.50, 126.43, 126.08, 114.12, 113.72, 107.12, 97.12, 81.45, 74.18, 32.21.
N,N-Dipropargyl-7-aminoflavone (10)
Mp 178–180 °C. GC/MS: 312 ([M – 1]+, 100%), 274 (100), 246 (15), 172 (35). 1H NMR (CDCl3, 300 MHz) δ 8.23 (d, J = 8.7, 1H), 7.93 (m, 2H), 7.66 (t, J = 1.5 Hz, 1H), 7.54 (m, 3H), 7.45 (dd, J = 8.4 Hz, J = 1.8 Hz, 1H), 6.83 (s, 1H), 4.81 (d, J = 2.1 Hz, 2H), 4.55 (d, J = 2.1 Hz, 2H), 2.51 (t, J = 2.4, 1H), 2.37 (t, J = 2.4, 1H). 13C NMR (CDCl3, 75 MHz) δ 177.73, 163.73, 156.36, 153.52, 145.78, 131.76, 131.59, 129.11, 126.47, 126.33, 122.20, 114.97, 107.78, 78.48, 77.24, 75.37, 73.39, 54.02, 39.99.
2-Phenyl-4H-pyrano[2,3-f]quinolin-4-one (7,8-Pyridinoflavone, 78PyF, 11)
In a 10 mL reaction vessel, 100 mg (0.36 mmol) of N-propargyl-7-aminoflavone (9) and 200 mg (0.58 mmol) of AgSbF6 were dissolved in 5 mL of DMSO. After heating in a CEM Discover microwave at 110 °C for an hour, the reaction solution was cooled to room temperature and transferred into a separatory funnel, to which 50 mL of DCM was added. The organic phase was washed successively with saturated sodium bicarbonate (25 mL × 5) and brine (25 mL × 2). The DCM layer was then dried over anhydrous magnesium sulfate and concentrated under vacuum. The residue was purified by column chromatography using silica gel (DCM/methanol 50:1) to provide 7,8-pyridinoflavone as colorless crystals (yield, 52%). Mp 185–187 °C. GC/MS: 273 (M+, 100%), 245 (20), 171 (85), 143 (15), 115 (25). 1H NMR (DMSO-d6, 300 MHz) δ 9.11 (dd, J = 4.2 Hz, J = 1.5 Hz, 1H), 9.06 (d, J = 8.4 Hz, 1H), 8.25–8.18 (m, 3H), 7.97 (d, J = 9.0 Hz, 1H), 7.78 (dd, J = 8.4 Hz, J = 4.2 Hz, 1H), 7.62 (m, 3H), 7.22 (s, 1H). 13C NMR (DMSO-d6, 75 MHz) δ 177.02, 162.76, 153.74, 152.80, 150.20, 132.36, 131.62, 131.41, 129.69, 126.95, 126.90, 124.11, 123.13, 120.16, 119.61, 108.77. Anal. Calcd for C18H11NO2: C, 79.11; H, 4.06; N, 5.13. Found: C, 77.73; H, 4.37; N, 4.98.
8-Phenyl-6H-[1,3]dioxolo[4,5-h]chromen-6-one (7,8-Dioxoloflavone, 78DOF, 12)
To a solution of 500 mg (1.97 mmol) of 7,8-dihydroxyflavone in 25 mL of dibromomethane and 5 mL of DMSO, 1.0 g (3.08 mmol) of cesium carbonate was added. The reaction solution was heated to 70 °C in a heating mantle. Once the starting material’s spot disappeared on TLC, the mixture was cooled to room temperature, filtered by gravity, and then concentrated under vacuum. The residue was dissolved with 150 mL of ethyl acetate, and the organic solution was washed successively with 5% potassium bisulfate (50 mL × 3), saturated sodium bicarbonate (50 mL × 3), and brine (50 mL × 2). After drying and filtration, the organic phase was concentrated under vacuum. The pure 7,8-dioxoloflavone was obtained as colorless crystals by recrystallization from 30 mL of acetone (yield, 72%). Mp >200 °C. GC/MS: 266 (M+, 100%), 238 (15), 164 (90), 106 (18). 1H NMR (CDCl3, 300 MHz) δ 7.91–7.88 (m, 2H), 7.79 (d, J = 8.7 Hz, 1H), 7.53–7.47 (m, 3H), 6.94 (d, J = 8.4 Hz, 1H), 6.72 (s, 1H), 6.21 (s, 2H). 13C NMR (CDCl3, 75 MHz) δ 177.41, 162.55, 152.44, 141.14, 134.78, 131.65, 131.42, 129.05, 126.19, 120.13, 119.94, 107.14, 106.95, 103.27. Anal. Calcd for C16H10O4: C, 72.18; H, 3.79. Found: C, 71.92; H, 3.87.
4-Phenyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (4-Phenyl-7-propargyloxycoumarin, 13)
Starting with 7-hydroxy-4-phenylcoumarin, this compound was prepared using general procedure A (yield, 71%). Mp 108–110 °C. GC/MS: 276 (M+, 100%), 247 (80), 189 (30), 165 (20), 139 (25). 1H NMR (CDCl3, 300 MHz) δ 7.53– 7.49 (m, 3H), 7.46–7.40 (m, 3H), 7.01 (d, J = 2.4, 1H), 6.86 (dd, J = 9.0 Hz, J = 2.4 Hz, 1H), 6.23 (s, 1H), 4.77 (d, J = 2.4 Hz, 2H), 2.58 (t, J = 2.4, 1H). 13C NMR (CDCl3, 75 MHz) δ 161.08, 160.49, 155.74, 155.71, 135.44, 129.67, 128.87, 128.38, 128.10, 113.22, 112.76, 112.35, 102.38, 77.38, 76.60, 56.20.
3-Methyl-4-phenyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (3-Methyl-4-phenyl-7-propargyloxycoumarin, 14)
Starting with 7-hydroxy-3-methyl-4-phenylcoumarin, this compound was prepared using general procedure A (yield, 74%). Mp 135–137 °C. GC/MS: 290 (M+, 100%), 261 (45), 189 (10). 1H NMR (CDCl3, 300 MHz) δ 7.55–7.44 (m, 3H), 7.23–7.20 (m, 2H), 6.97 (d, J = 2.7, 1H), 6.93 (d, J = 8.7, 1H), 7.76 (dd, J = 8.7 Hz, J = 2.7 Hz, 1H), 4.73 (d, J = 2.4 Hz, 2H), 2.55 (t, J = 2.4, 1H), 1.96 (s, 3H). 13C NMR (CDCl3, 75 MHz) δ 162.64, 159.41, 153.76, 150.69, 135.08, 128.84, 128.62, 128.31, 128.03, 119.97, 115.03, 112.45, 101.80, 77.55, 76.40, 56.15, 14.47.
4-Methyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (4-Methyl-7-propargyloxycoumarin, 15)
Starting with 7-hydroxy-4-methylcoumarin, this compound was prepared using general procedure A (yield, 66%). Mp 136–138 °C. GC/MS: 214 (M+, 100%), 185 (54), 158 (50), 147 (70), 91 (45). 1H NMR (CDCl3, 300 MHz) δ 7.55–7.51 (m, 1H), 6.95–6.91 (m, 2H), 6.16 (d, J = 1.2, 1H), 4.77 (d, J = 2.1 Hz, 2H), 2.58 (t, J = 2.4, 1H), 2.41 (d, J = 0.9 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 161.14, 160.34, 155.02, 152.47, 125.65, 114.25, 112.71, 112.40, 102.14, 77.43, 76.52, 56.17, 18.69.
7-(Prop-2-yn-1-yloxy)-4-(trifluoromethyl)-2H-chromen-2-one (7-Propargyloxy-4-trifluoromethylcoumarin, 16)
Starting with 7-hydroxy-4-trifluoromethylcoumarin, this compound was prepared using general procedure A (yield, 76%). Mp 113–114 °C. GC/MS: 267 ([M – 1]+, 100%), 239 (92), 212 (60), 173 (40), 145 (50), 115 (45), 69 (40). 1H NMR (CDCl3, 300 MHz) δ 7.67–7.63 (m, 1H), 7.00–6.97 (m, 2H), 6.64 (d, J = 0.6, 1H), 4.79 (d, J = 2.4 Hz, 2H), 2.59 (t, J = 2.4, 1H). 13C NMR (CDCl3, 75 MHz) δ 161.23, 159.24, 156.07, 141.50 (q, J = 32 Hz), 126.45 (q, J = 2.3 Hz), 121.55 (q, J = 274 Hz), 113.82, 112.73 (q, J = 5.8 Hz), 107.70, 102.68, 76.98, 76.93, 56.30.
4-Phenylpyrano[2,3-f]chromen-2(8H)-one (4-Phenyl-7,8-pyranocoumarin, 4P78PC, 17)
Starting with 4-phenyl-7-propargyl-oxycoumarin, 108 mg (yield, 54%) of this compound was prepared using general procedure B. Column chromatography solvent system: petroleum ether/ethyl acetate 6:1. Mp 154–156 °C. GC/MS: 276 (M+, 100%), 247 (80), 189 (30), 165 (20), 139 (25). 1H NMR (CDCl3, 300 MHz) δ 7.51–7.48 (m, 3H), 7.44–7.39 (m, 2H), 7.20 (d, J = 8.7 Hz, 1H), 7.03 (dtd, J = 10.2 Hz, J = 1.8 Hz, J = 0.6, 1H), 6.50 (dd, J = 8.7 Hz, J = 0.6, 1H), 6.20 (s, 1H), 5.88 (dt, J = 10.2 Hz, J = 3.6, 1H), 4.94 (dd, J = 3.6 Hz, J = 1.8 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 160.91, 157.16, 156.17, 150.03, 135.57, 129.58, 128.81, 128.38, 127.18, 121.87, 117.69, 113.05, 112.58, 111.78, 110.46, 66.07. Anal. Calcd for C18H12O3: C, 78.25; H, 4.38. Found: C, 78.23; H, 4.48.
3-Methyl-4-phenylpyrano[2,3-f]chromen-2(8H)-one (3-Methyl-4-phenyl-7,8-pyranocoumarin, 3M4P78PC, 18)
Starting with 3-methyl-4-phenyl-7-propargyloxycoumarin, 76 mg (yield, 38%) of this compound was prepared using general procedure B. Column chromatography solvent system: petroleum ether/DCM 1:1. Mp 113– 115 °C. GC/MS: 290 (M+, 100%), 261 (45), 189 (10). 1H NMR (CDCl3, 300 MHz) δ 7.54–7.45 (m, 3H), 7.21–7.18 (m, 2H), 7.03 (dtd, J = 10.2 Hz, J = 1.8 Hz, J = 0.6, 1H), 6.72 (d, J = 8.7 Hz, 1H), 6.55 (dd, J = 8.7 Hz, J = 0.6, 1H), 5.86 (dt, J = 10.2 Hz, J = 3.6, 1H), 4.90 (dd, J = 3.6 Hz, J = 1.8 Hz, 2H), 1.94 (s, 3H). 13C NMR (CDCl3, 75 MHz) δ 162.42, 155.96, 151.12, 148.20, 135.19, 128.79, 128.54, 128.30, 127.06, 121.78, 119.33, 117.78, 114.83, 112.32, 109.95, 65.94, 14.47. Anal. Calcd for C19H14O3: C, 78.61; H, 4.86. Found: C, 77.07; H, 5.03.
4-Methylpyrano[2,3-f]chromen-2(8H)-one (4-Methyl-7,8-pyranocoumarin, 4M78PC, 19)
Starting with 4-methyl-7-propargyloxycoumarin, 80 mg (yield, 40%) of this compound was prepared using general procedure B. Column chromatography solvent system: petroleum ether/ethyl acetate 8:1. Mp 154–155 °C. GC/MS: 214 (M+, 100%), 185 (65). 1H NMR (CDCl3, 300 MHz) δ 7.33 (d, J = 8.7 Hz, 1H), 6.98 (d, J = 10.2 Hz, 1H), 6.72 (d, J = 8.7 Hz, 1H), 6.13 (s, 1H), 5.86 (dt, J = 10.2 Hz, J = 3.3, 1H), 4.92 (dd, J = 3.0 Hz, J = 1.8 Hz, 2H), 2.37 (s, 3H). 13C NMR (CDCl3, 75 MHz) δ 161.00, 156.97, 152.87, 149.37, 124.56, 121.84, 117.72, 114.07, 112.54, 111.90, 110.28, 65.99, 18.78. Anal. Calcd for C13H10O3: C, 72.89; H, 4.71. Found: C, 72.61; H, 4.74.
4-(Trifluoromethyl)pyrano[2,3-f]chromen-2(8H)-one (4-Trifluoromethyl-7,8-pyranocoumarin, 4TF78PC, 20)
Starting with 7-propargyloxy-4-trifluoromethylcoumarin, 76 mg (yield, 38%) of this compound was prepared using general procedure B. Column chromatography solvent system: petroleum ether/ethyl acetate 8:1. Mp 157–159 °C. GC/MS: 267 ([M – 1]+, 100%), 239 (65), 171 (25), 115 (20), 63 (28). 1H NMR (CDCl3, 300 MHz) δ 7.45 (dq, J = 9.0 Hz, J = 1.8, 1H), 6.94 (dt, J = 10.2 Hz, J = 2.1 Hz, 1H), 6.77 (d, J = 9.0 Hz, 1H), 6.60 (s, 1H), 5.89 (dt, J = 10.2 Hz, J = 3.6, 1H), 4.98 (dd, J = 3.3 Hz, J = 2.1 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 159.12, 157.99, 150.21, 141.86 (q, J = 32 Hz), 125.40 (J = 2.3 Hz), 122.46, 121.56 (q, J = 274 Hz), 117.01, 113.61, 112.08 (q, J = 5.8 Hz), 110.62, 107.52, 66.26. Anal. Calcd for C13H7F3O3: C, 58.22; H, 2.63; F, 21.25. Found: C, 57.99; H, 2.47; F, 21.12.
8-Methyl-4-phenyl-2H-furo[2,3-h]chromen-2-one (4-Phenyl-7,8-furanocoumarin, 4P78FC, 21)
Starting with 4-phenyl-7-propargyloxycoumarin, 86 mg (yield, 43%) of this compound was prepared using general procedure C. Column chromatography solvent system: petroleum ether/ethyl acetate 6:1. Mp 131–133 °C. GC/MS: 276 (M+, 75%), 248 (100), 189 (18). 1H NMR (CDCl3, 300 MHz) δ 7.53–7.46 (m, 5H), 7.30 (d, J = 9.0 Hz, 1H), 7.26 (d, J = 8.7 Hz, 1H), 6.78 (s, 1H), 6.33 (s, 1H), 2.52 (s, 3H). 13C NMR (CDCl3, 75 MHz) δ 161.08, 157.09, 157.01, 156.79, 147.67, 136.02, 129.55, 128.80, 128.48, 121.85, 118.60, 113.46, 112.64, 107.75, 100.21, 14.16. Anal. Calcd for C18H12O3: C, 78.25; H, 4.38. Found: C, 77.76; H, 4.49.
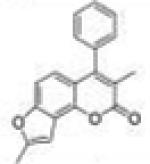
3,8-Dimethyl-4-phenyl-2H-furo[2,3-h]chromen-2-one (3-Methyl-4-phenyl-7,8-furanocoumarin, 3M4P78FC, 22)
Starting with 3-methyl-4-phenyl-7-propargyloxycoumarin, 122 mg (yield, 61%) of this compound was prepared using general procedure C. Column chromatography solvent system: petroleum ether/DCM 1:1. Mp 165– 166 °C. GC/MS: 290 (M+, 100%), 262 (65), 189 (18). 1H NMR (CDCl3, 300 MHz) δ 7.58–7.47 (m, 3H), 7.28–7.24 (m, 2H), 7.18 (dd, J = 8.7 Hz, J = 0.9 Hz, 1H), 6.82 (d, J = 8.7 Hz, 1H), 6.77 (t, J = 0.9, 1H), 2.51 (d, J = 0.9, 3H), 2.01 (s, 3H). 13C NMR (CDCl3, 75 MHz) δ 162.65, 156.51, 156.35, 151.91, 145.70, 135.70, 128.81, 128.52, 128.33, 121.95, 120.12, 118.04, 115.18, 107.49, 100.03, 14.69, 14.17. Anal. Calcd for C19H14O3: C, 78.61; H, 4.86. Found: C, 78.24; H, 5.14.
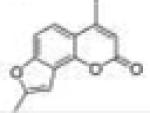
4,8-Dimethyl-2H-furo[2,3-h]chromen-2-one (4-Methyl-7,8-furanocoumarin, 4M78FC, 23)
Starting with 4-methyl-7-propargyl-oxycoumarin, 90 mg (yield, 45%) of this compound was prepared using general procedure C. Column chromatography solvent system: petroleum ether/ethyl acetate 8:1. Mp 172–173 °C. GC/MS: 214 (M+, 100%), 186 (85). 1H NMR (CDCl3, 300 MHz) δ 7.41 (d, J = 8.7 Hz, 1H), 7.33 (dd, J = 8.7 Hz, J = 0.6 Hz, 1H), 6.71 (s, 1H), 6.23 (d, J = 1.2 Hz, 1H), 2.50 (d, J = 0.6 Hz, 3H), 2.47 (d, J = 0.9 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 161.15, 156.99, 156.65, 153.72, 147.02, 119.24, 118.39, 114.42, 112.58, 107.76, 100.18, 19.40, 14.13. Anal. Calcd for C13H10O3: C, 72.89; H, 4.71. Found: C, 72.51; H, 4.96.
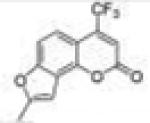
8-Methyl-4-(trifluoromethyl)-2H-furo[2,3-h]chromen-2-one (4-Trifluoromethyl-7,8-furanocoumarin, 4TF78FC, 24)
Starting with 7-propargyloxy-4-trifluoromethylcoumarin, 72 mg (yield, 36%) of this compound was prepared using general procedure C. Column chromatography solvent system: petroleum ether/ethyl acetate 10:1. Mp 180–181 °C. GC/MS: 268 (M+, 100%), 239 (90). 1H NMR (CDCl3, 300 MHz) δ 7.55 (dq, J = 8.7 Hz, J = 1.8 Hz, 1H), 7.41 (dd, J = 8.7 Hz, J = 0.9 Hz, 1H), 6.74 (m, 2H), 2.53 (d, J = 0.9 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 159.29, 157.44, 157.38, 148.04, 142.56 (q, J = 32 Hz), 121.68 (q, J = 274 Hz), 119.78 (J = 2.3 Hz), 118.743, 113.15 (q, J = 5.8 Hz), 108.84, 108.05, 100.05, 14.12. Anal. Calcd for C13H7F3O3: C, 58.22; H, 2.63; F, 21.25. Found: C, 58.17; H, 2.51; F, 21.48.
4-Methyl-7-(prop-2-yn-1-ylamino)-2H-chromen-2-one (N-Propargyl-7-amino-4-methylcoumarin, 25) and 7-(Di(prop-2-yn-1-yl)amino)-4-methyl-2H-chromen-2-one (N,N-Diproparg-yl-7-amino-4-methylcoumarin, 26)
To a solution of 500 mg (2.86 mmol) of 7-amino-4-methylcoumarin in 10 mL of DMF, 500 mg (3.62 mmol) of potassium carbonate and 1.0 mL (9.00 mmol) of 80% propargyl bromide solution in toluene were added. The reaction was stirred at room temperature for at least 1 week. When the starting material completely disappeared, the reaction mixture was quenched with 150 mL of DCM. After filtration by gravity, the organic phase was washed with saturated sodium bicarbonate (50 mL × 5), dried over anhydrous magnesium sulfate, and concentrated under vacuum. The residue was purified by column chromatography using silica gel (DCM/methanol 100:1) to give compounds 25 and 26 as yellowish and colorless crystals, respectively (yield, 58% and 16%).
N-Propargyl-7-amino-4-methylcoumarin (25)
Mp 127–129 °C. GC/MS: 213 (M+, 100%), 185 (33), 146 (55). 1H NMR (DMSO-d6, 300 MHz) δ 7.48 (d, J = 8.7, 1H), 6.99 (t, J = 5.7 Hz, 1H), 6.66 (dd, J = 8.7 Hz, J = 2.1 Hz, 1H), 6.50 (d, J = 2.1, 1H), 5.97 (s, 1H), 3.98 (dd, J = 5.7 Hz, J = 2.1 Hz, 2H), 3.16 (t, J = 2.1, 1H), 2.32 (s, 3H). 13C NMR (DMSO-d6, 75 MHz) δ 161.07, 155.78, 154.20, 151.89, 126.41, 111.15, 110.00, 108.68, 97.88, 81.60, 74.03, 32.19, 18.50.
7-(Di(prop-2-yn-1-yl)amino)-4-methyl-2H-chromen-2-one (26)
Mp 177–179 °C. GC/MS: 251 (M+, 100%), 222 (10), 184 (15), 154 (13), 128 (10). 1H NMR (DMSO-d6, 300 MHz) δ 7.60 (d, J = 9.0, 1H), 6.93 (dd, J = 9.0 Hz, J = 2.4 Hz, 1H), 6.80 (d, J = 2.4, 1H), 6.08 (s, 1H), 4.30 (d, J = 2.1 Hz, 4H), 3.23 (s, 2H), 2.36 (s, 3H). 13C NMR (DMSO-d6, 75 MHz) δ 160.90, 155.13, 153.94, 150.45, 126.41, 111.33, 111.14, 110.14, 100.69, 79.85, 75.72, 40.36, 18.46.
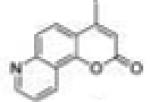
4-Methyl-2H-pyrano[2,3-f]quinolin-2-one (4-Methyl-7,8-pyridinocoumarin, 4M78PyC, 27)
In a 10 mL reaction vessel, 100 mg (0.47 mmol) of N-propargyl-7-amino-4-methylcoumarin (25) and 200 mg (0.58 mmol) of AgSbF6 were dissolved in 5 mL of DMSO. After heating in a CEM Discover microwave at 110 °C for an hour, the reaction solution was cooled to room temperature and transferred into a separatory funnel, to which 50 mL of DCM was added. The organic phase was washed successively with saturated sodium bicarbonate (25 mL × 5) and brine (25 mL × 2). The DCM layer was then dried over anhydrous magnesium sulfate and concentrated under vacuum. The residue was purified by column chromatography using silica gel (DCM/methanol 200:1) to provide 4-methyl-7,8-pyridinocoumarin as colorless crystals (yield, 48%). Mp >220 °C decomposed. GC/MS: 211 (M+, 100%), 183 (95), 154 (35), 127 (15).1 H NMR (DMSO-d6/CDCl3 1:1, 300 MHz) δ 8.98 (dd, J = 4.2 Hz, J = 1.5 Hz, 1H), 8.73 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.89 (d, J = 9.0 Hz, 1H), 7.61 (dd, J = 8.4 Hz, J = 4.2 Hz, 1H), 6.43 (d, J = 0.9 Hz, 1H), 2.53 (d, J = 0.9 Hz, 3H). 13C NMR (DMSO-d6/CDCl3 1:1, 75 MHz) δ 159.76, 153.76, 152.61, 149.79, 149.09, 130.67, 125.40, 124.70, 122.45, 118.33, 115.78, 114.83, 19.19. Anal. Calcd for C13H9NO2: C, 73.92; H, 4.29; N, 6.63. Found: C, 71.44; H, 4.44; N, 6.31.
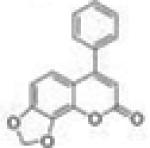
6-Phenyl-8H-[1,3]dioxolo[4,5-h]chromen-8-one (4-Phenyl-7,8-dioxolocoumarin, 4P78DOC, 28)
To a solution of 500 mg (1.97 mmol) of 7,8-dihydroxy-4-phenylcoumarin in 25 mL of dibromomethane and 5 mL of DMSO, 1.0 g (3.08 mmol) of cesium carbonate was added. The reaction mixture was heated to 70 °C in a heating mantle. Once the starting material’s spot disappeared on TLC, the mixture was cooled to room temperature, filtered by gravity, and then concentrated under vacuum. The residue was dissolved in 150 mL of ethyl acetate, and the organic solution was washed successively with 5% potassium bisulfate (50 mL × 3), saturated sodium bicarbonate (50 mL × 3), and brine (50 mL × 2). After drying and filtration, the organic phase was concentrated under vacuum to give pure 4-phenyl-7,8-dioxolocoumarin as colorless crystals (yield, 80%). Mp 152–154 °C. GC/MS: 266 (M+, 100%), 238 (80), 152 (25). 1H NMR (CDCl3, 300 MHz) δ 7.51–7.40 (m, 5H), 6.98 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 8.4 Hz, 1H), 6.19 (s, 1H), 6.16 (s, 2H). 13C NMR (CDCl3, 75 MHz) δ 159.71, 156.15, 151.41, 138.58, 135.63, 134.30, 129.66, 128.81, 128.38, 121.47, 115.24, 112.51, 105.18, 103.15. Anal. Calcd for C16H10O4: C, 72.18; H, 3.79. Found: C, 72.11; H, 4.01.
Materials and Equipment in Bioassays
Gentest human CYP1A1, CYP1A2, and CYP1B1 supersomes were purchased from BD Biosciences (Franklin Lakes, NJ). D-Glucose 6-phosphate sodium salt, β-nicotinamide adenine dinucleotide phosphate sodium salt (NADP+), and glucose 6-phosphate dehydrogenase were purchased from Sigma-Aldrich Corporation. Other reagents in bioassays were purchased from Fisher Scientific International, Inc. Figures were plotted with Prism 6 (GraphPad Software, Inc., La Jolla, CA) as well as Microsoft Excel 2013.
Fluorimetric Enzyme Inhibition Assays of P450s 1A1, 1A2, and 1B1
The inhibition activities of the target compounds toward P450s 1A1-, 1A2-, and 1B1-dependent enzymatic reactions were tested through standard methods as previously described.27,28 These studies included P450 1A1-dependent EROD, P450 1A2-dependent MROD, and P450 1B1-dependent EROD assays. In brief, potassium phosphate buffer (1760 μL of a 0.1 M solution, pH 7.6) was placed in a 1.0 cm quartz cuvette, and 10 μL of a 1.0 M MgCl2 solution, 10 μL of the 1.0 mM corresponding resorufin substrate solution (final concentration of 5 μM) in DMSO, 10 μL of the microsomal P450 protein (final concentration, 5 nM), and 10 μL of an inhibitor solution in DMSO were added. For the controls, 10 μL of pure DMSO was added in place of the inhibitor solution. The reaction was initiated by the addition of 200 μL of an NADPH regenerating solution.29 The final assay volume was 2.0 mL. The production of resorufin anion was monitored by a spectrofluorimeter (OLIS DM 45 spectrofluorimetry system) at 535 nm excitation and 585 nm emission, with a slit width of 2 nm. The reactions were performed at 37 °C. For each inhibitor, a number of assay runs were performed using gradually diluted inhibitor solutions. At least four concentrations of each inhibitor showing 20–80% inhibition were tested.
Data Analysis
Ki Values
The initial data obtained from the above assays were a series of time-course curves in the presence of various inhibitor concentrations and in the absence of the inhibitor as the control. In Microsoft Excel 2013, the Trendline tool was used to fit these data (fluorescence intensity vs time) in order to obtain the best-fit second-order curves (y = ax2 + bx + c). The first-order coefficient b in the above second-order equation represented enzymatic activity (v0) at time spot 0. Dixon plots were used (by plotting the reciprocals of the enzymatic activity (1/v0) vs inhibitor concentrations [I]) in order to determine Ki values (x-intercepts) for the inhibitors. The data are represented as the mean ± SD μM of three independent experiments. The Ki values of tested compounds for inhibition of P450s 1A1, 1A2, and 1B1 are tabulated in Tables 1 and 2. For a competitive inhibitor, the Ki value is equal to the IC50 value.
Table 2.
Inhibitory Activity and Selectivity of Coumarin Derivatives toward P450 1A2a
| Compoun d |
Short Name |
Structure | Ki (μM) | Selective Index (SI) |
|||
|---|---|---|---|---|---|---|---|
| 1A1 | 1A2 | 1B1 | 1A1/1A2 | 1B1/1A2 | |||
| 17 | 4P78PC |
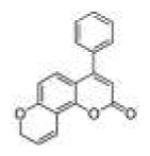
|
25.2±2.6 | 1.29±0.33 | 5.04±0.76 | 20 | 3.9 |
| 18 | 3M4P78 PC |
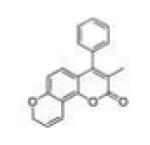
|
18.4±0.4 | 1.72±0.27 | 21.2±2.3 | 11 | 12 |
| 19 | 4M78PC |
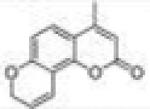
|
110±14 | 1.51±0.18 | 12.0±3.0 | 73 | 7.9 |
| 20 | 4TF78PC |
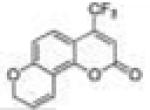
|
60.6±6.2 | 0.39±0.08 | 20.4±4.8 | 155 | 52 |
| 21 | 4P78FC |
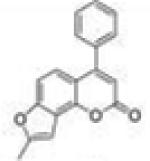
|
12.0±0.3 | 5.02±0.16 | 4.62±0.78 | 2.4 | 0.9 |
| 22 | 3M4P78 FC |
|
11.6±0.4 | 3.70±0.73 | 22.6±6.1 | 3.1 | 6.1 |
| 23 | 4M78FC |
|
37.9±5.3 | 1.15±0.17 | 7.46±0.52 | 33 | 6.5 |
| 24 | 4TF78FC |
|
60.7±9.5 | 1.35±0.13 | 46.8±9.0 | 45 | 35 |
| 27 | 4M78Py C |
|
101±17 | 14.2±4.0 | 46.6±9.1 | 7.1 | 3.3 |
| 28 | 4P78DO C |
|
>200 | 3.55±0.84 | 30.1±4.6 | 56 | 8.5 |
The Ki values are represented as the mean ± SD μM of three independent experiments.
KI and Limiting kinact Values
The first-order derivatives (y = 2ax + b) of the above second-order curves (y = ax2 + bx + c) represent the enzymatic activity over time. The semilog plots of the percent relative activity (Y = log[(y/y0) × 100]) versus time demonstrate the centesimal loss of enzymatic activity with time. The linear portions of the above semilog plots were used to determine t1/2 values (the time required for the enzyme to lose half of its original activity, which equals 0.693/kinact) at various concentrations for the observed time-dependent losses of activity. To obtain KI and limiting kinact values, 1/kinact values were plotted versus reciprocals of the inhibitor concentration (1/[I]) (Kitz–Wilson plots). The limiting kinact value was the abscissa intercept of this plot, and the KI value was calculated from the ordinate intercept (−1/KI). The KI and limiting kinact values of the time-dependent inhibitors (78FF and 5H78FF) for P450 1A2 are described in context.
NADPH-Dependency Assay
All assay solution components had the same concentrations as in the above fluorimetric enzyme inhibition assays. For preincubation assays in the presence of NADPH, potassium phosphate buffer (1560 μL, pH 7.6) was placed in a 1.0 cm quartz cuvette followed by 10 μL of a 1.0 M MgCl2 solution, 10 μL of the microsomal P450 protein, 10 μL of inhibitor solution in DMSO (the concentration leading to approximate 20% enzymatic activity inhibition), and 200 μL of an NADPH regenerating solution. The assay mixture was incubated for 5 min at 37 °C before reaction initiation by the addition of 200 μL of buffer and 10 μL of the corresponding substrate solution (final concentration, 5 μM). For the preincubation assays in the absence of NADPH, potassium phosphate buffer (1760 μL, pH 7.6) was placed in a 1.0 cm quartz cuvette followed by 10 μL of a 1.0 M MgCl2 solution, 10 μL of the microsomal P450 protein, and 10 μL of an inhibitor solution in DMSO (the concentration leading to approximate 20% enzymatic activity inhibition). The assay mixture was incubated for 5 min at 37 °C, before reaction initiation by the addition of 200 μL of the NADPH regenerating solution and 10 μL of the corresponding substrate solution (final concentration, 5 μM). The final assay volume for both assays was 2 mL. The production of P450-dependent reaction products was monitored as described above. The reactions were performed at 37 °C for 6 min.
Yeast AhR-Signaling Assay
The commercially available yeast strain MYA-3637 was used to test the target compounds. Since the integrated human AhR gene is expressed from a bidirectional GAL (galactose regulated) promoter in this strain, and the lacZ reporter plasmid carries the TRP1 marker, the yeast was grown on synthetic minimal medium lacking tryptophan and glucose substituted by galactose as the carbon source for this assay. The maintenance and the assay of the cells have been described previously.30 Briefly, an amount of 2 μL of different concentrations of tested compounds and/or β-naphtoflavone (positive control) dissolved in DMSO was added to a 200 μL of overnight yeast culture in a 96-well plate in quadruplicate. The next day, in order to take into account the uneven cell growth or seeding, the absorbance at 600 nm of each well was determined after careful mixing. Then an amount of 100 μL of LacZ lysis buffer containing 0.2% (wt/vol) sarcosyl (sodium lauroyl sarcosinate), 1 mM DTT (dithiothreitol), and 0.4 mg/mL of ONPG (the substrate for β-galactosidase, O-nitrophenol-β-D-galactopyranoside) was added into the 200 μL of cell suspension for each well as quickly as possible. After a yellow color developed, the absorbance reading at 405 nm was recorded and normalized to 600 nm reading for each well. The formation of O-nitrophenol resulting in yellow color development was detected as Abs405 by Synergy 4 hybrid plate reader (BioTek) and normalized to Abs600 for each well, taking into account the differences in cell densities. Thus, the ratio Abs405/Abs600 reffects the activation of AhR in a single yeast cell. Each experiment was repeated at least three times.
Cell Culture
Human hepatocellular carcinoma HepG2 cells (ATCC no. HB-8065) were maintained in DMEM (Dulbecco’s modified Eagle medium) supplemented with 10% fetal bovine serum and 20 μM L-glutamine. Cells were grown in 25 cm2 tissue culture flasks at 37 °C under a 5% CO2 humidified environment.
MROD Assay in HepG2 Cells
2 × 105 of HepG2 cells in 1 mL of medium were plated overnight onto 24-well plates and exposed to 10 μL of DMSO (blank control), 10 μL of β-naphthoflavone solution (final concentration 10−5 M, positive control) in DMSO, or 10 μL of 4TF78PC solution (compound 20, final concentration 10−4, 10−5, 10−6, and 10−7 M) in DMSO. After 24 h, the medium was decanted, and cells were washed once with PBS. To each well, 1 mL of medium containing 5 × 10−6 M methoxyresorufin was added, and fluorescence density of the metabolic product (resorufin) was measured using a CytoFluor multiwell plate reader (Applied Biosystems, Foster City, CA), with an excitation of 535 nm and emission at 585 nm, at the time spots of 10, 30, and 60 min.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by DoD Award W81XWH-11-1-0105. We thank NIH-MBRS SCORE (Grant S06 GM 08008) for support of the preliminary work done on this project by the Foroozesh research group. We also thank the Louisiana Cancer Research Consortium and the NIH-RCMI Grant 2G12MD007595 from the National Institute on Minority Health and Health Disparities for their support of the Major Instrumentation and the Cell and Molecular Biology Cores at Xavier University of Louisiana. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the Louisiana Cancer Research Consortium, the DoD, or the NIH.
ABBREVIATIONS USED
- IQ
2-amino-3-methylimidazo[4,5-f ]quinoline
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- AhR
aryl hydrocarbon receptor
- DMSO
dimethyl sulfoxide
- βNF
β-naphthoflavone
- EROD
ethoxyresorufin-O-deethylase
- MROD
methoxyresorufin-O-demethylase
- αNF
α-naphthoflavone
- TDI
time-dependent inhibitor
- MBI
mechanism-based inhibitor
- NaH
sodium hydride
- DMSO
dimethyl sulfoxide
- DCM
dichloromethane
- TLC
thin layer chromatography
- DTT
dithiothreitol
- ONPG
O-nitrophenol-β-D-galacto-pyranoside
- DMEM
Dulbecco’s modified Eagle medium
- PBS
phosphate buffered saline
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b00494.
1H and 13C NMR spectra, elemental analysis results, the structures of 18 selective P450 inhibitors used for the alignment study, and the data of HepG2 cell viability assay (PDF)
Molecular formula strings (CSV)
REFERENCES
- (1).Kim D, Guengerich FP. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu. Rev. Pharmacol. Toxicol. 2005;45:27–49. doi: 10.1146/annurev.pharmtox.45.120403.100010. [DOI] [PubMed] [Google Scholar]
- (2).Rendic S, Guengerich FP. Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 2012;25:1316–1383. doi: 10.1021/tx300132k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kleiner HE, Reed MJ, DiGiovanni J. Naturally occurring coumarins inhibit human cytochromes P450 and block benzo[a]-pyrene and 7,12-dimethylbenz[a]anthracene DNA adduct formation in MCF-7 cells. Chem. Res. Toxicol. 2003;16:415–422. doi: 10.1021/tx025636d. [DOI] [PubMed] [Google Scholar]
- (4).Shimada T, Murayama N, Okada K, Funae Y, Yamazaki H, Guengerich FP. Different mechanisms for inhibition of human cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic inhibitors. Chem. Res. Toxicol. 2007;20:489–496. doi: 10.1021/tx600299p. [DOI] [PubMed] [Google Scholar]
- (5).Kim S, Ko H, Park JE, Jung S, Lee SK, Chun YJ. Design, synthesis, and discovery of novel trans-stilbene analogues as potent and selective human cytochrome P450 1B1 inhibitors. J. Med. Chem. 2002;45:160–164. doi: 10.1021/jm010298j. [DOI] [PubMed] [Google Scholar]
- (6).Mikstacka R, Baer-Dubowska W, Wieczorek M, Sobiak S. Thiomethylstilbenes as inhibitors of CYP1A1, CYP1A2 and CYP1B1 activities. Mol. Nutr. Food Res. 2008;52(Suppl. 1):S77–83. doi: 10.1002/mnfr.200700202. [DOI] [PubMed] [Google Scholar]
- (7).Mikstacka R, Rimando AM, Dutkiewicz Z, Stefanski T, Sobiak S. Design, synthesis and evaluation of the inhibitory selectivity of novel trans-resveratrol analogues on human recombinant CYP1A1, CYP1A2 and CYP1B1. Bioorg. Med. Chem. 2012;20:5117–5126. doi: 10.1016/j.bmc.2012.07.012. [DOI] [PubMed] [Google Scholar]
- (8).Liu J, Taylor SF, Dupart PS, Arnold CL, Sridhar J, Jiang Q, Wang Y, Skripnikova EV, Zhao M, Foroozesh M. Pyranoflavones: a group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J. Med. Chem. 2013;56:4082–4092. doi: 10.1021/jm4003654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu J, Nguyen TT, Dupart PS, Sridhar J, Zhang X, Zhu N, Stevens CL, Foroozesh M. 7-Ethynylcoumarins: selective inhibitors of human cytochrome P450s 1A1 and 1A2. Chem. Res. Toxicol. 2012;25:1047–1057. doi: 10.1021/tx300023p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sridhar J, Liu J, Foroozesh M, Klein Stevens CL. Inhibition of cytochrome p450 enzymes by quinones and anthraquinones. Chem. Res. Toxicol. 2012;25:357–365. doi: 10.1021/tx2004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Walsh AA, Szklarz GD, Scott EE. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013;288:12932–12943. doi: 10.1074/jbc.M113.452953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhou S-F, Yang L-P, Wei MQ, Duan W, Chan E. Insights into the structure, function, and regulation of human cytochrome P450 1A2. Curr. Drug Metab. 2009;10:713–729. doi: 10.2174/138920009789895552. [DOI] [PubMed] [Google Scholar]
- (13).Sinal CJ, Webb CD, Bend JR. Differential in vivo effects of alpha-naphthoflavone and beta-naphthoflavone on CYP1A1 and CYP2E1 in rat liver, lung, heart, and kidney. J. Biochem. Mol. Toxicol. 1999;13:29–40. doi: 10.1002/(sici)1099-0461(1999)13:1<29::aid-jbt4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- (14).Nannelli A, Rossignolo F, Tolando R, Rossato P, Longo V, Gervasi PG. Effect of beta-naphthoflavone on AhR-regulated genes (CYP1A1, 1A2, 1B1, 2S1, Nrf2, and GST) and antioxidant enzymes in various brain regions of pig. Toxicology. 2009;265:69–79. doi: 10.1016/j.tox.2009.09.010. [DOI] [PubMed] [Google Scholar]
- (15).Liu J, Sridhar J, Foroozesh M. Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules. 2013;18:14470–14495. doi: 10.3390/molecules181214470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ishii H, Ishikawa T, Takeda S, Ueki S, Suzuki M. Cesium fluoride-mediated Claisen Rearrangement of aryl propargyl ether. Exclusive formation of 2-methylarylfuran and its availability as a masked salicylaldehyde. Chem. Pharm. Bull. 1992;40:1148–1153. [Google Scholar]
- (17).Majumdar KC, Nandi RK, Ganai S, Taher A. Regioselective synthesis of annulated quinoline and pyridine derivatives by silver-catalyzed 6-endo-dig cycloisomerization. Synlett. 2011;2011:116–120. [Google Scholar]
- (18).Sesardic D, Boobis AR, Murray BP, Murray S, Segura J, de la Torre R, Davies DS. Furafylline is a potent and selective inhibitor of cytochrome P450IA2 in man. Br. J. Clin. Pharmacol. 1990;29:651–663. doi: 10.1111/j.1365-2125.1990.tb03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shimada T, Tanaka K, Takenaka S, Murayama N, Martin MV, Foroozesh MK, Yamazaki H, Guengerich FP, Komori M. Structure-function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem. Res. Toxicol. 2010;23:1921–1935. doi: 10.1021/tx100286d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hollenberg PF, Kent UM, Bumpus NN. Mechanism-based inactivation of human cytochromes p450s: experimental characterization, reactive intermediates, and clinical implications. Chem. Res. Toxicol. 2008;21:189–205. doi: 10.1021/tx7002504. [DOI] [PubMed] [Google Scholar]
- (21).Peterson LA. Reactive metabolites in the biotransformation of molecules containing a furan ring. Chem. Res. Toxicol. 2013;26:6–25. doi: 10.1021/tx3003824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lin HL, Kenaan C, Hollenberg PF. Identification of the residue in human CYP3A4 that is covalently modified by bergamottin and the reactive intermediate that contributes to the grapefruit juice effect. Drug Metab. Dispos. 2012;40:998–1006. doi: 10.1124/dmd.112.044560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Alvarez-Diez TM, Zheng J. Mechanism-based inactivation of cytochrome P450 3A4 by 4-ipomeanol. Chem. Res. Toxicol. 2004;17:150–157. doi: 10.1021/tx034143l. [DOI] [PubMed] [Google Scholar]
- (24).Taxak N, Kalra S, Bharatam PV. Mechanism-based inactivation of cytochromes by furan epoxide: unraveling the molecular mechanism. Inorg. Chem. 2013;52:13496–13508. doi: 10.1021/ic401907k. [DOI] [PubMed] [Google Scholar]
- (25).Ciolino HP, MacDonald CJ, Memon OS, Bass SE, Yeh GC. Sulindac regulates the aryl hydrocarbon receptor-mediated expression of Phase 1 metabolic enzymes in vivo and in vitro. Carcinogenesis. 2006;27:1586–1592. doi: 10.1093/carcin/bgi359. [DOI] [PubMed] [Google Scholar]
- (26).Sridhar J, Ellis J, Dupart P, Liu J, Stevens CL, Foroozesh M. Development of flavone propargyl ethers as potent and selective inhibitors of cytochrome P450 enzymes 1A1 and 1A2. Drug Metab. Lett. 2012;6:275–284. doi: 10.2174/1872312811206040007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Buters JT, Schiller CD, Chou RC. A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem. Pharmacol. 1993;46:1577–1584. doi: 10.1016/0006-2952(93)90326-r. [DOI] [PubMed] [Google Scholar]
- (28).Burke MD, Thompson S, Weaver RJ, Wolf CR, Mayers RT. Cytochrome P450 specificities of alkoxyresorufin O-dealkylation in human and rat liver. Biochem. Pharmacol. 1994;48:923–936. doi: 10.1016/0006-2952(94)90363-8. [DOI] [PubMed] [Google Scholar]
- (29).Goyal N, Liu J, Lovings L, Dupart P, Taylor S, Bellow S, Mensah L, McClain E, Dotson B, Sridhar J, Zhang X, Zhao M, Foroozesh M. Ethynylflavones, highly potent, and selective inhibitors of cytochrome P450 1A1. Chem. Res. Toxicol. 2014;27:1431–1439. doi: 10.1021/tx5001865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fox JE, Burow ME, McLachlan JA, Miller CA., 3rd Detecting ligands and dissecting nuclear receptor-signaling pathways using recombinant strains of the yeast Saccharomyces cerevisiae. Nat. Protoc. 2008;3:637–645. doi: 10.1038/nprot.2008.33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.