

Abstract
Objective:
To delineate the developmental and progressive neurodegenerative features in 9 young adults with the atypical form of Chediak-Higashi disease (CHD) enrolled in a natural history study.
Methods:
Patients with atypical clinical features, but diagnostically confirmed CHD by standard evaluation of blood smears and molecular genotyping, underwent complete neurologic evaluation, MRI of the brain, electrophysiologic examination, and neuropsychological testing. Fibroblasts were collected to investigate the cellular phenotype and correlation with the clinical presentation.
Results:
In 9 mildly affected patients with CHD, we documented learning and behavioral difficulties along with developmental structural abnormalities of the cerebellum and posterior fossa, which are apparent early in childhood. A range of progressive neurologic problems emerge in early adulthood, including cerebellar deficits, polyneuropathies, spasticity, cognitive decline, and parkinsonism.
Conclusions:
Patients with undiagnosed atypical CHD manifesting some of these wide-ranging yet nonspecific neurologic complaints may reside in general and specialty neurology clinics. The absence of the typical bleeding or infectious diathesis in mildly affected patients with CHD renders them difficult to diagnose. Identification of these individuals is important not only for close surveillance of potential CHD-related systemic complications but also for a full understanding of the natural history of CHD and the potential role of the disease-causing protein, LYST, to the pathophysiology of other neurodevelopmental and neurodegenerative disorders.
Chediak-Higashi disease (CHD; OMIM #214500) is a rare, autosomal recessive disorder with hematologic, pigmentary, and neurologic manifestations caused by biallelic pathogenic variants in the lysosomal trafficking regulator gene LYST.1 Typical patients with CHD present with immunodeficiency and hemophagocytic lymphohistiocytosis (HLH), or the accelerated phase,2,3 along with variable degrees of bleeding, due to a delta storage pool deficiency of platelets, and oculocutaneous albinism.4 Without therapy, children with classical CHD die of infections or from complications of HLH in the first decade. Bone marrow transplantation (BMT) prevents both of these outcomes, but does not stop the progression of neurologic deficits.5 The neurologic involvement is characterized by subtle and nonspecific early neurodevelopmental issues including learning and behavioral deficits in childhood followed in early adulthood by a progressive neurodegeneration with varying degrees of cerebellar dysfunction, peripheral neuropathy, spasticity, dystonia, parkinsonism, impaired cognition, and premature death.6
Adults with mild forms of CHD, despite harboring biallelic LYST variants, may escape early detection and consideration for BMT, because early life-threatening infections, bleeding, and HLH complications are muted.7,8 Nevertheless, such patients develop striking neurologic declines that may not be recognized as part of the CHD phenotype. Here, we review the natural history and the spectrum of neurologic involvement in 9 mildly affected adult patients with CHD, including their history, clinical, brain MRI, electrophysiologic, molecular, and cell biological findings.
METHODS
Standard protocol approvals, registrations, and patient consents.
Patients with a presumed diagnosis of CHD were referred to and enrolled in clinical protocol 00-HG-0153, Investigations into Chediak-Higashi Syndrome and Related Disorders (clinicaltrials.gov identifier NCT00005917) approved by the National Human Genome Research Institute Institutional Review Board. Written informed consent was obtained. All evaluations took place at the NIH Clinical Center between 2005 and 2014. Under a separate consent, skin biopsies were performed to obtain fibroblasts.
Laboratory investigations.
Specialized laboratory investigations included monitoring for HLH, peripheral blood smears to confirm the presence of giant inclusions within neutrophils, microscopic hair examination, and whole mount electron microscopy of platelets to quantify dense bodies.
Neurophysiology.
Nerve conduction studies were performed mainly on the median and sural sensory nerves and the peroneal and median motor nerves. Measurements used standard methodology with a Nicolet Viking Select machine (Cardinal Health, Dublin, OH) and were compared to department-based normative values.9 Needle EMG was performed using a concentric needle and filter settings of 2K–10K with spontaneous and motor unit activity recorded according to standard methodology.
MRI.
The standard MRI examination of the brain consisted of sagittal T1-weighted, axial T2-weighted, axial T1-weighted, axial diffusion tensor, postcontrast axial T1-weighted, postcontrast axial fluid-attenuated inversion recovery, and postcontrast sagittal 3D T1-weighted images; 2 patients had similar examinations without administration of contrast material, and one patient was not scanned due to intense claustrophobia. The field strength was either 1.5T or 3T. We used elements of previously published metrics of posterior fossa traits to evaluate the sagittal configuration of the posterior fossa.10 A superior cerebellar vermis angle was measured as the intersection of 2 line segments, one segment originating at the opening of the aqueduct into the fourth ventricle to the vein of Galen and the other projecting from the vein of Galen through the straight sinus into the torcula herophili. This angle was measured after reformatting the 3D T1-weighted images into an exact midsagittal plane, as defined by the plane of the falx cerebri. Measurements in patients were compared against sex-matched normal controls (in a 2:1 ratio) enrolled in other research protocols who had undergone similar imaging studies that included axial or sagittal 3D T1-weighted images.
Cells.
Primary dermal fibroblasts were cultured from a forearm skin biopsy, as described.8,11 Cell culture conditions are described in the e-Methods on the Neurology® Web site at Neurology.org.
Light microscopy.
Bright field images of hair were obtained using a Zeiss (Oberkochen, Germany) Axiovert 200M with a Plan NeoFluar 40×/1.3 oil differential interference contrast objective. Images were obtained with an Axiocam HRc color camera with Axiovision software version 4.5.0 (Carl Zeiss, Microimaging Inc., Jena, Germany).
Molecular analysis, quantitative real-time PCR (qPCR), high-molecular-weight immunoblotting, and confocal laser-scanning microscopy.
See the e-Methods for a description of molecular analysis, qPCR, high-molecular-weight immunoblotting, and confocal laser-scanning microscopy.
RESULTS
Patient cohort.
Nine patients (8 male, 1 female; age 16–35 years) were diagnosed with CHD by identifying giant inclusions within leukocytes on standard peripheral blood smears; the diagnoses were confirmed by biallelic genetic LYST variants (table 1). No patient had experienced life-threatening infections or symptoms of the accelerated phase; none had undergone BMT. Hair (showing characteristic pigment granulation), skin (showing hypopigmentation), and blood smear (showing giant granules in leukocytes) findings were all consistent with CHD (figure 1). Other clinical characteristics are summarized in table 1. Patient CHD-20 lived for over 3 decades without a BMT and without HLH symptoms or signs. Despite these clinical indications of mild disease, this patient had classical CHD by molecular analysis (LYST gene and LYST protein analysis); he died of HLH at age 34 years.
Table 1.
Clinical and molecular characteristics of atypical Chediak-Higashi disease
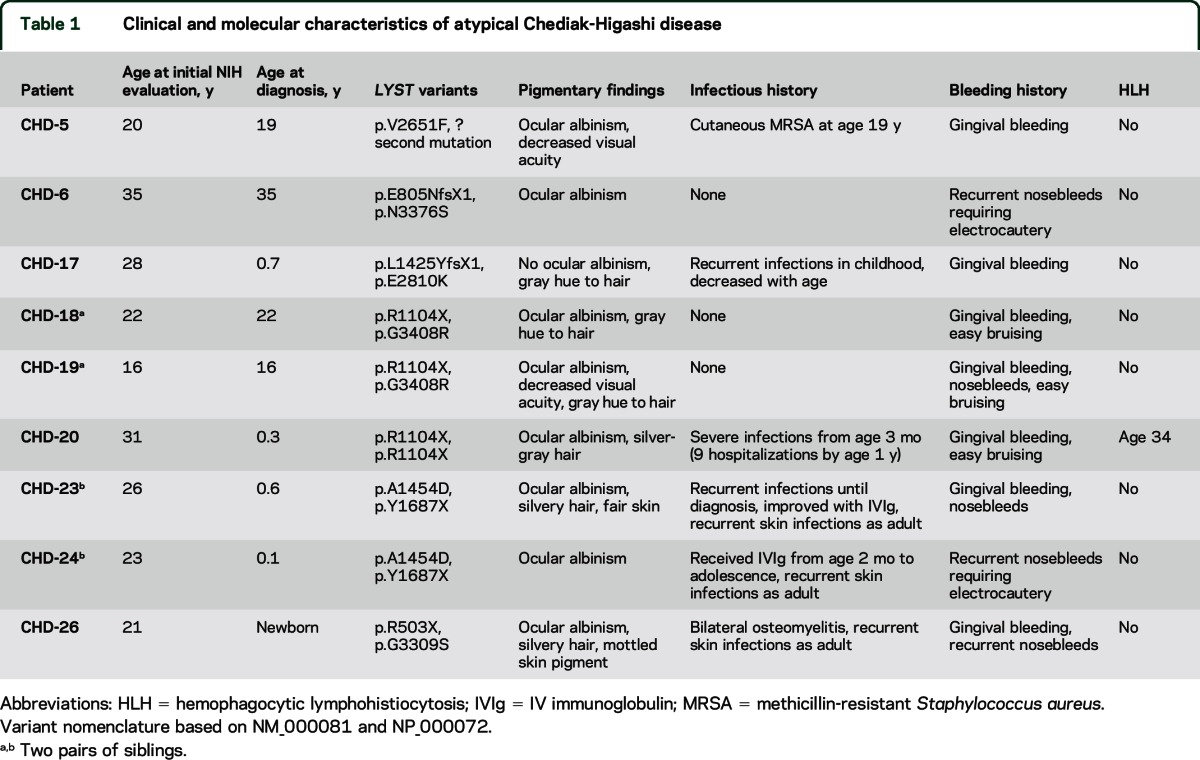
Figure 1. Hair and skin pigmentation, blood smears, and hair microscopy of representative patients with atypical Chediak-Higashi disease (CHD).

(A) Hair and skin pigmentation. (B) Characteristic giant inclusions (arrows) within neutrophils are numerous and variable in size. (C) A solitary giant inclusion is typically seen within lymphocytes. (D) Light microscopy reveals atypical pigment clumping within hair shafts. Note uniform distribution of pigment in normal hair (CHD-5 inset).
Molecular and cellular studies.
Biallelic variants in LYST were identified in every patient (figure e-1A) except CHD-5, who was heterozygous for a novel missense variant; a second variant could not be detected. However, gDNA (derived from fibroblasts) sequencing showed 2 variant peaks of equal intensity (figure e-1B, arrow), indicating the presence of equal allelic amounts of DNA. In contrast, cDNA (derived from fibroblast mRNA) sequencing showed a higher intensity band for the variant missense allele (T) compared to the normal allele (G) (figure e-1B), indicating that the normal allele may carry a cryptic variant (deep intronic, or 3′ or 5′ UTR) causing mRNA decay. In fact, qPCR showed a significant reduction (63%–69%) of LYST mRNA using 3 different probes (p < 0.0001) (figure e-1B).
The pathogenicity of the 6 missense variants in our patients was examined using a 5′ nuclease allelic discrimination (TaqMan) assay. None of these LYST variants was present in a DNA panel of 100 healthy individuals, indicating they are not common polymorphisms. Querying the ExAC databases of whole exome data revealed that most of the LYST variants were not present, and a few variants had very small allele frequencies (p < 0.0001), where none were homozygotes. In addition, the deleteriousness of the missense variants was assessed using 3 analysis programs, SIFT, PolyPhen-2, and Align-GVGD. All variants except p.E2810K were deemed damaging by all 3 programs. SIFT predicted p.E2810K to be tolerated, while PolyPhen-2 considered it possibly damaging and Align-GVGD predicted it to be deleterious.
The molecular alterations in our patients with mild CHD translated into reduced LYST protein expression in their cultured fibroblasts. We used CHD-4 (early-onset CHD, harboring deleterious variants p.R514X and p.F3298SfsX6)12 as a negative control, lacking LYST protein expression (figure 2A, lane 2). CHD-5 showed LYST expression comparable to that of CHD-6, i.e., 51% of control (lanes 3 and 4), while the very mildly affected CHD-17, CHD-18, CHD-23, and CHD-26 had the highest protein expression (figure 2A, lanes 5, 6, 8, and 9). CHD-20, with null LYST variants, expressed no protein (figure 2A, lane 7).
Figure 2. LYST protein expression and lysosome size and distribution in Chediak-Higashi disease (CHD) fibroblasts.

(A) High-molecular-weight immunoblots of protein extracts from normal control fibroblasts (lane 1), negative control fibroblasts CHD-4 (lane 2), and 7 different CHD patients' fibroblast cultures. β-Actin was used as a loading control. Values given are percentage expression of LYST normalized to β-actin and relative to control. (B) Fibroblasts are stained with LAMP-1 antibodies (green) to visualize lysosomal membranes and with an F-actin marker (phalloidin, red) to visualize the cell boundaries. Control fibroblasts show a typical distribution of lysosomes throughout the cell. Classic CHD-4 fibroblasts show enlarged lysosomes restricted to the perinuclear area. CHD-5 fibroblasts show enlarged lysosomes in the perinuclear area and normal-sized lysosomes in the periphery. CHD-6 fibroblasts show slightly enlarged lysosomes. CHD-17 and CHD-18 fibroblasts have slightly enlarged lysosomes with a typical distribution of lysosomes throughout the cell. Marked white dotted squares in left panels are zoomed out in right panels. Scale bar 20 μm.
Cultured fibroblasts from patients with CHD.
The cellular phenotype of the patients with CHD reflected the severity of their molecular defects. In general, CHD cells have enlarged lysosomes and lysosome-related organelles (LROs) in various cell types such as fibroblasts, melanocytes, and polymorphonuclear leukocytes.8,13 We previously established that the size of lysosomes and LROs correlated with the severity of the disease,8 and we found this to be true for the lysosomes in fibroblasts of our atypical CHD cases (figure 2B). We included previously described CHD-4 (classical CHD) and CHD-6 (mild late-onset CHD) for comparison.8 CHD-4 fibroblasts exhibited giant lysosomes clustered around the nucleus while CHD-6 fibroblasts had a mixture of normal and slightly enlarged lysosomes in the perinuclear and peripheral areas. CHD-5 fibroblasts exhibited a cellular phenotype between that of CHD-4 and CHD-6 cells, with enlarged lysosomes in the perinuclear area and normal-sized lysosomes in the periphery. CHD-17 and CHD-18 closely resembled the cellular phenotype of control fibroblasts, with normal-sized lysosomes distributed in the perinuclear and peripheral regions.
Clinical neurology.
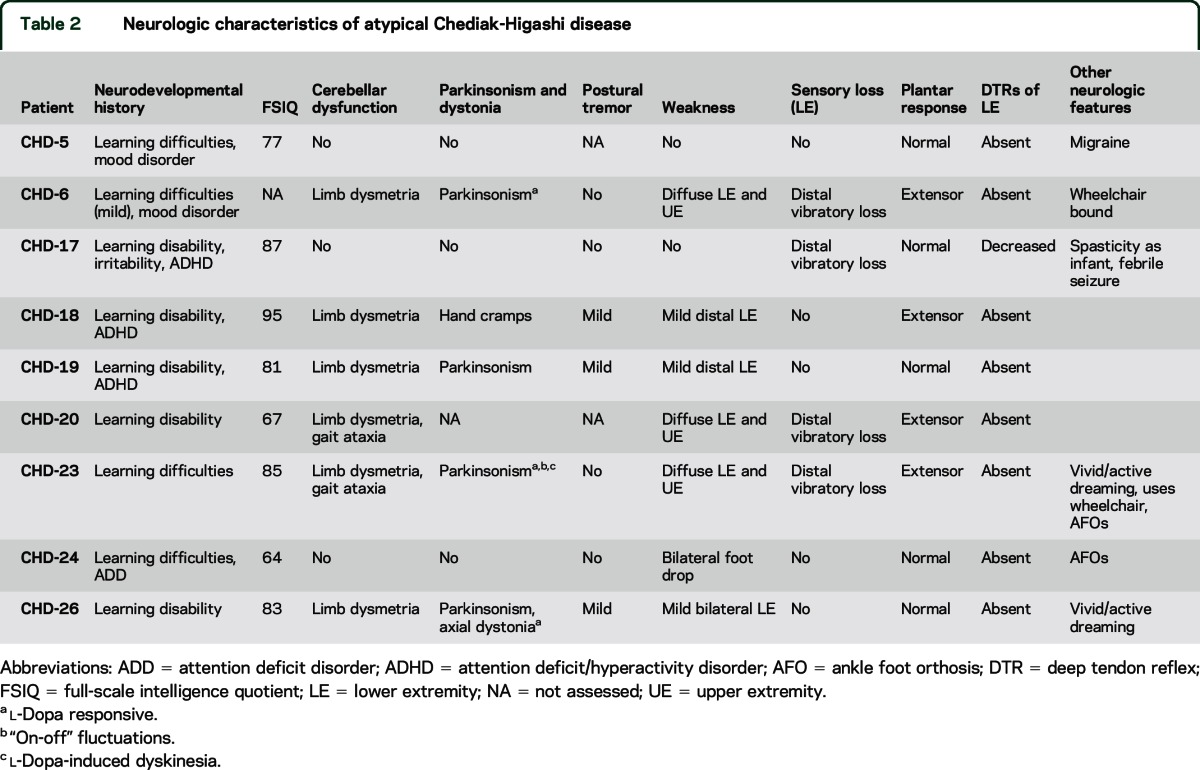
All 9 patients exhibited developmental impairment manifested by learning disabilities (table 2). Full-scale IQ (FSIQ) at baseline NIH visit evaluated using either the Wechsler Abbreviated Scale of Intelligence14 or Wechsler Adult Intelligence Scale, 3rd edition,15 varied from 64 to 95, with an average baseline FSIQ of 80. Six patients had cerebellar signs, largely limb dysmetria, and 4 had parkinsonism; 3 were l-dopa responsive, and 2 displayed “on-off” fluctuations and peak-dose dyskinesia. Three patients had a mild postural tremor, 7 displayed weakness, and 4 had subtle sensory deficits manifesting as loss of distal vibratory sensation. Plantar responses were extensor in 4 patients, and all had absent or decreased deep tendon reflexes in the lower extremities, reflecting concurrent central and peripheral nervous system involvement. Two patients, both with parkinsonism, experienced active vivid dreaming clinically suggestive of REM sleep behavior disorder (RBD).
Table 2.
Neurologic characteristics of atypical Chediak-Higashi disease
Brain imaging.
MRI examinations revealed 1 patient with mild cerebral atrophy, 1 with mild cerebellar atrophy, 2 with mild cerebral and cerebellar atrophy, and 1 with moderate cerebral and cerebellar atrophy (figure 3, E, F, and I). In general, atrophy correlated with longer disease duration, more severe cognitive disabilities, or motor dysfunction. There were no supratentorial focal lesions, heterotopia, structural defects, or white matter signal abnormalities.
Figure 3. Brain MRIs of patients with atypical Chediak-Higashi disease (CHD).

Midline MRI sagittal images for 6 patients with atypical CHD obtained at the time of their initial evaluation. (A–F) Patients CHD-6, 19, 24, 17, 23, and 20. The tentorial angle (in degrees) and lines converging on the tentorial angle are represented for each image. (G) Similarly processed image in one representative age-matched control. (H, I) Coronal MRI views at the level of the cerebellar hemispheres for mild (case CHD-23) and advanced (case CHD-20) neurologic impairment demonstrate cerebral and cerebellar atrophy in the more severely affected patient.
However, a group-specific morphologic variation in the configuration of the posterior fossa was observed in midsagittal images. The angle enclosing the superior cerebellar vermis was more acute in patients with CHD (mean 64°, range 52°–79°) (figure 3, A–F) than in age- and sex-matched controls (mean 86°, range 72°–97°), with p = 0.00002 by 2-tailed heteroscedastic t test.
Electrophysiology.
Seven patients underwent studies of nerve conduction and needle EMG; 5 had follow-up studies within 1–2 years. Table e-1 gives the initial study values, as well as 1- and 2-year follow-up values when available. At baseline, 2 patients had no detectable sural responses and 3 patients had low responses. Median nerve sensory response was low in one patient. For the motor nerves, one patient had a low peroneal response. All patients had normal median motor responses. The conduction velocities were all normal except for the median sensory and peroneal motor response in CHD-20, likely related to axonal loss and not in the demyelinating range.
Needle EMG was performed in proximal and distal leg muscles in 6 patients. Three patients had acute or chronic neurogenic changes in the distal leg muscles, although one individual had no evidence of a neuropathy by nerve conduction studies. Two patients had electrodiagnostic evidence of an axonal sensorimotor polyneuropathy. Three individuals had an axonal sensory polyneuropathy.
On longitudinal follow-up, after 2 years, 2 individuals had developed an axonal neuropathy by nerve conduction studies. One patient also had a very low median motor response, but a limited electrodiagnostic study could not further define the cause. Only 2 patients had repeat EMG studies of 2 leg muscles; both showed progressive neurogenic changes that could be associated with additional motor neuron pathology.
DISCUSSION
Among the ∼500 cases of CHD reported,16 genotype and phenotype correlate well.7,8,17 In general, patients with classical CHD have 2 severe/pathogenic LYST variants, while mildly affected individuals have one severe and one milder variant (e.g., missense). While LYST protein expression in cultured fibroblasts reflects the severity of the non-neurologic aspects of the disease (figure 2A),11 the neurologic complications of CHD affect all patients without regard to the severity of their LYST variants.
LYST contains 3,801 amino acids, has a molecular weight of 430 kDa, and possesses ARM/HEAT repeat motifs that mediate membrane interactions and WD-40 repeats that function in protein interactions.18 Adjacent to the WD-40 repeats is the amino acid sequence WIDL, together characterizing the Beige and Chediak (BEACH) group of proteins.16,18 LYST is widely expressed, including brain and bone marrow. LYST functions in trafficking lysosomes and regulating lysosomal morphology16; the absence of LYST results in reduced lysosome fission without altering fusion, and overexpression of LYST results in a faster rate of lysosome fission.19 CHD fibroblasts have giant vesicles that stain with LRO markers, clustering in the perinuclear cytoplasm, and fail to distribute normally into the periphery of cells.
The bloated lysosome-like organelles of CHD cells account for many of the manifestations of the disease.20 Enlarged melanosomes in melanocytes move poorly along dendritic processes, and skin melanosomes cannot transfer melanin to surrounding keratinocytes,4,8,20 resulting in hypopigmentation. The trafficking defect in CHD megakaryocytes results in absent or deficient platelet dense bodies, impairing the secondary aggregation response and resulting in bruising and mucous membrane bleeding.20 In dendritic cells, giant lysosomes cannot migrate to and ingest pathogens, accounting for susceptibility to infections. The cause of the accelerated phase in CHD could be related to defective trafficking of perforin and, consequently, impaired apoptosis induced by lymphocytic cells.
The basis for nervous system involvement in CHD remains enigmatic. We do not know if progressive neurodegeneration emerges from the accumulation of undigested substrate in giant lysosomes, from impaired LRO trafficking along neuritic processes of Purkinje cells, corticospinal neurons, anterior horn cells, and other neuronal populations, or from dysfunctional vesicle incorporation into membranes.
In an early phase of CHD, features of developmental delay, learning difficulties, or attention deficit/hyperactivity disorder are nearly universal and reflect a developmental disturbance, supported by our finding of structural variations in the configuration of the posterior fossa. This nonspecific abnormality likely reflects variable degrees of cerebellar hypoplasia, especially in the midline cerebellum. We found similar MRI findings in all patients with CHD (including infants) prior to any hematologic complications compared to patients with classical CHD who had undergone BMT (unpublished observation).
A second phase of the illness appears around late adolescence, and manifests as a gradual and progressive neurologic decline. Progressive atrophic MRI changes in the cerebrum and cerebellum correlate with clinical worsening and increased disability. Generally, mild and nonspecific incoordination appears at school age, while well-defined midline cerebellar dysfunction and limb dysmetria, common features in mild CHD, occur in early adulthood. LYST variants in humans and in a missense LystI3618R murine model of CHD21 appear to target Purkinje cells. Young adults with CHD have diminished or absent deep tendon reflexes resulting from a length-dependent, predominantly axonal polyneuropathy, commonly presenting as foot drop. Once present, progression occurs rapidly. Long tract signs, including spasticity and extensor plantar responses, are common but often obscured by weakness and atrophy associated with the coexisting peripheral neuropathy. In late stages, individuals lose the ability to ambulate and perform activities of daily living. We speculate that LYST dysfunction uncovers vulnerabilities in membrane repair, bioenergetics, vesicular trafficking, and cytoskeleton maintenance. In vitro studies demonstrate failure of cultured melanocytes from patients with CHD to properly traffic and release their pigment cargo to the extracellular space.8 Both human CHD cells and those of the beige murine CHD model have defective lysosomal exocytosis and plasma membrane repair22; these deficiencies likely contribute to neuronal degeneration and neurologic dysfunction.
CHD may present as parkinsonism. Other patients may experience limb dystonia. Four of our patients, and others in the literature,23–27 have levodopa-responsive parkinsonism not unlike Parkinson disease (PD).28 Two of our patients undergoing l-dopa replacement therapy for longer than 3 years experienced “on-off” fluctuation and l-dopa-induced dyskinesia consistent with a progressive presynaptic dopaminergic deficit. Two patients experienced RBD, a condition long regarded as an early feature of synucleinopathies, suggesting that LYST dysfunction may also influence the proteosomal degradation system and autophagy. LRO dysfunction occurs in several monogenic forms of PD,29 most notably in Gaucher disease. Reduced vesicle fission in CHD is likely to adversely affect other neuronal processes such as vesicular packaging of dopamine, which might contribute to neurodegeneration.30,31
While classical hematologic and pigmentary features of CHD are recapitulated in the beige mouse model due to deletions within Lyst,32 the neurologic characteristics of mild CHD are best exemplified in another CHD murine model with a homozygous LystI3618R missense variant. These animals, with a silvery gray coat and prolonged bleeding, have no evidence of ocular albinism, blood cell dysmorphology, susceptibility to infection, or development of HLH.21 Instead, they have an age-dependent neurologic phenotype consisting of late-onset cerebellar dysfunction and motor coordination deficits. Their brains revealed no abnormalities in young mice, but by 5 months, giant inclusions appeared, primarily in the cerebellum. The Purkinje cells are laden with giant vesicular inclusions at the base of their dendritic arborization. With advancing age, these changes evolved into substantial Purkinje cell degeneration.21
For decades, the neurologic findings of CHD were masked by the disease's early, fatal infections and HLH. Now, bone marrow transplantation and mildly affected individuals are making the neurologic aspects of CHD apparent. Ascertainment remains challenging for patients with mild disease, largely because of the vague, varied, and nonspecific nature of their neurologic phenotypes. Neurologists should suspect CHD when they encounter individuals with subtle neurologic signs and any combination of skin or ocular pigment dilution, bleeding tendency, or unusual infectious history. Given their early learning disabilities, cognitive issues, and soft neurologic findings, many young patients with mild CHD may carry static diagnoses such as developmental delay or cerebral palsy. However, the neurologic involvement of CHD progresses inexorably. For some patients with CHD with parkinsonism, standard dopaminergic therapy produces substantial, life-changing benefits, at least in the short term. For others, proper adaptive equipment, vocational counseling, and physical and occupation rehabilitation can improve quality of life. Finally, our patient CHD-20 is a powerful reminder that the accelerated phase can occur at any age, and patients should be closely monitored for any suggestion of HLH.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Roxanne Fischer and Isa Bernardini for clinical and laboratory work and the patients and families who participated in this research.
GLOSSARY
- BMT
bone marrow transplantation
- CHD
Chediak-Higashi disease
- FSIQ
full-scale IQ
- HLH
hemophagocytic lymphohistiocytosis
- LRO
lysosome-related organelle
- LYST
lysosomal trafficking regulator
- PD
Parkinson disease
- qPCR
quantitative PCR
- RBD
REM sleep behavior disorder
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
W.J.I., W.W., A.R.C., W.A.G., and C.T. designed the study and drafted the first draft of the manuscript. All authors collected and interpreted data. All authors participated in manuscript revision.
STUDY FUNDING
Supported by the Intramural Research Programs of the National Human Genome Research Institute, the National Institute of Neurological Disorders and Stroke, and the Hatfield Clinical Center, NIH, Bethesda, MD.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 1996;14:307–311. [DOI] [PubMed] [Google Scholar]
- 2.Blume RS, Wolff SM. The Chediak-Higashi syndrome: studies in four patients and a review of the literature. Medicine 1972;51:247–280. [PubMed] [Google Scholar]
- 3.Jessen B, Maul-Pavicic A, Ufheil H, et al. Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome. Blood 2011;118:4620–4629. [DOI] [PubMed] [Google Scholar]
- 4.Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab 1999;68:283–303. [DOI] [PubMed] [Google Scholar]
- 5.Tardieu M, Lacroix C, Neven B, et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood 2005;106:40–42. [DOI] [PubMed] [Google Scholar]
- 6.Introne WJ, Westbroek W, Golas GA, Adams D. Chediak-Higashi syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle: University of Washington; 2009:1993–2015. [PubMed] [Google Scholar]
- 7.Karim MA, Suzuki K, Fukai K, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet 2002;108:16–22. [PubMed] [Google Scholar]
- 8.Westbroek W, Adams D, Huizing M, et al. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol 2007;127:2674–2677. [DOI] [PubMed] [Google Scholar]
- 9.Liveson JA. Laboratory Reference for Clinical Neurophysiology. New York: Oxford University Press; 1992. [Google Scholar]
- 10.Markunas CA, Enterline DS, Dunlap K, et al. Genetic evaluation and application of posterior cranial fossa traits as endophenotypes for Chiari type I malformation. Ann Hum Genet 2014;78:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manoli I, Golas G, Westbroek W, et al. Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome 1. Am J Med Genet A 2010;152A:1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zarzour W, Kleta R, Frangoul H, et al. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Mol Genet Metab 2005;85:125–132. [DOI] [PubMed] [Google Scholar]
- 13.Burkhardt JK, Wiebel FA, Hester S, Argon Y. The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J Exp Med 1993;178:1845–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wechsler D. Wechsler Abbreviated Scale of Intelligence. San Antonio: Psychological Corporation; 1999. [Google Scholar]
- 15.Wechsler D. Wechsler Adult Intelligence Scale, 3rd ed San Antonio: Psychological Corporation; 1997. [Google Scholar]
- 16.Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol 2008;15:22–29. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Guiu I, Anton AI, Garcia-Barbera N, et al. Chediak-Higashi syndrome: description of two novel homozygous missense mutations causing divergent clinical phenotype. Eur J Heamatol 2014;92:49–58. [DOI] [PubMed] [Google Scholar]
- 18.Cullinane AR, Schaffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic 2013;14:749–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durchfort N, Verhoef S, Vaughn MB, et al. The enlarged lysosomes in beige j cells result from decreased lysosome fission and not increased lysosome fusion. Traffic 2012;13:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet 2008;9:359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudelius M, Osanger A, Kohlmann S, et al. A missense mutation in the WD40 domain of murine Lyst is linked to severe progressive Purkinje cell degeneration. Acta Neuropathol 2006;112:267–276. [DOI] [PubMed] [Google Scholar]
- 22.Huynh C, Roth D, Ward DM, Kaplan J, Andrews NW. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc Natl Acad Sci USA 2004;101:16795–16800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobi C, Koerner C, Fruehauf S, Rottenburger C, Storch-Hagenlocher B, Grau AJ. Presynaptic dopaminergic pathology in Chediak-Higashi syndrome with parkinsonian syndrome. Neurology 2005;64:1814–1815. [DOI] [PubMed] [Google Scholar]
- 24.Silveira-Moriyama L, Moriyama TS, Gabbi TV, Ranvaud R, Barbosa ER. Chediak-Higashi syndrome with parkinsonism. Mov Disord 2004;19:472–475. [DOI] [PubMed] [Google Scholar]
- 25.Hauser RA, Friedlander J, Baker MJ, Thomas J, Zuckerman KS. Adult Chediak-Higashi parkinsonian syndrome with dystonia. Mov Disord 2000;15:705–708. [DOI] [PubMed] [Google Scholar]
- 26.Uyama E, Hirano T, Ito K, et al. Adult Chediak-Higashi syndrome presenting as parkinsonism and dementia. Acta Neurol Scand 1994;89:175–183. [DOI] [PubMed] [Google Scholar]
- 27.Pettit RE, Berdal KG. Chediak-Higashi syndrome: neurologic appearance. Arch Neurol 1984;41:1001–1002. [DOI] [PubMed] [Google Scholar]
- 28.Bhambhani V, Introne WJ, Lungu C, Cullinane A, Toro C. Chediak-Higashi syndrome presenting as young-onset levodopa-responsive parkinsonism. Mov Disord 2013;28:127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron 2010;68:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guillot TS, Miller GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol 2009;39:149–170. [DOI] [PubMed] [Google Scholar]
- 31.Anderson DG, Mariappan SV, Buettner GR, Doorn JA. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J Biol Chem 2011;286:26978–26986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barbosa MD, Nguyen QA, Tchernev VT, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 1996;382:262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.