

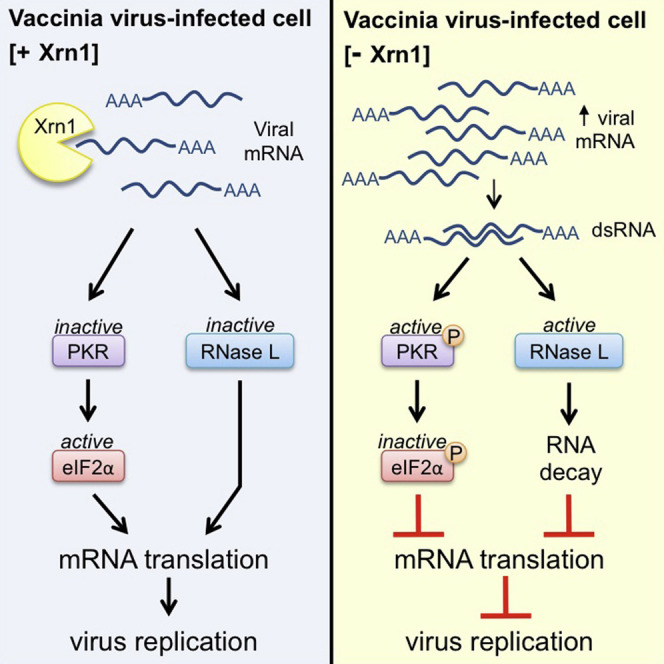
Summary
By accelerating global mRNA decay, many viruses impair host protein synthesis, limiting host defenses and stimulating virus mRNA translation. Vaccinia virus (VacV) encodes two decapping enzymes (D9, D10) that remove protective 5′ caps on mRNAs, presumably generating substrates for degradation by the host exonuclease Xrn1. Surprisingly, we find VacV infection of Xrn1-depleted cells inhibits protein synthesis, compromising virus growth. These effects are aggravated by D9 deficiency and dependent upon a virus transcription factor required for intermediate and late mRNA biogenesis. Considerable double-stranded RNA (dsRNA) accumulation in Xrn1-depleted cells is accompanied by activation of host dsRNA-responsive defenses controlled by PKR and 2′-5′ oligoadenylate synthetase (OAS), which respectively inactivate the translation initiation factor eIF2 and stimulate RNA cleavage by RNase L. This proceeds despite VacV-encoded PKR and RNase L antagonists being present. Moreover, Xrn1 depletion sensitizes uninfected cells to dsRNA treatment. Thus, Xrn1 is a cellular factor regulating dsRNA accumulation and dsRNA-responsive innate immune effectors.
Graphical Abstract
Highlights
-
•
Vaccinia virus (VacV) replication requires the host Xrn1 mRNA decay enzyme
-
•
The 5′-3′ mRNA exonuclease Xrn1 limits dsRNA accumulation
-
•
In the absence of Xrn1, host dsRNA-responsive innate immune defenses are activated
-
•
VacV antagonists of dsRNA-responsive host defenses are Xrn1 dependent
dsRNA produced during virus infection is a potent innate immune stimulus. Burgess and Mohr show that the host mRNA exonuclease Xrn1 stimulates vaccinia virus replication by restricting dsRNA accumulation. This reveals a role for the host mRNA decay machinery in regulating dsRNA accumulation and counteracting innate anti-viral responses.
Introduction
By modulating mature mRNA abundance, regulated mRNA decay provides a powerful means to control gene expression post-transcriptionally (Garneau et al., 2007, Parker and Song, 2004). While the fate of distinct eukaryotic mRNAs is typically controlled by critical structural features, including cis-acting genetic elements, the 3′ polyadenylated tail, and the m7-GTP cap protecting the mRNA 5′ terminus, the irreversible degradation required for mRNA turnover and surveillance pathways is catalyzed by the 5′-3′ exonuclease Xrn1 in a rapid, processive manner (Arribas-Layton et al., 2013, Jonas and Izaurralde, 2013, Nagarajan et al., 2013). In addition to deadenylation-dependent mRNA decay, where decapped messages bearing an exposed 5′ monophosphate recruit Xrn1 through interactions with the DCP2/DCP1a/Hedls complex (Braun et al., 2012), Xrn1 degrades 3′ fragments produced by endonucleolytic cleavage associated with mRNA quality control processes and regulates stability of specific messages such as those containing AREs (Stoecklin et al., 2006) or targeted by siRNAs (Orban and Izaurralde, 2005). Not only does Xrn1-dependent mRNA degradation effectively sculpt the proteome by influencing the mRNA population available for translation both spatially and temporally, but it also plays a significant role controlling how cells and organisms respond to stress, including virus infection (Beckham and Parker, 2008, Mohr and Sonenberg, 2012).
To thwart production of host defense molecules and stimulate viral mRNA translation, viruses often subvert cellular mRNA decay pathways and manipulate Xrn1 (Gaglia and Glaunsinger, 2010, Read, 2013). While some RNA viruses circumvent Xrn1 action to preserve their genomic integrity (Chapman et al., 2014, Dougherty et al., 2011, Silva et al., 2010), others that produce m7GTP-capped mRNAs harness the mRNA exonucleolytic powers of Xrn1 to accelerate host and viral mRNA decay (Gaglia et al., 2012). Besides restricting host protein synthetic capabilities by reducing mRNA abundance, accelerating viral mRNA turnover sharpens transitions between different kinetic classes of temporally transcribed mRNAs and shapes the viral developmental gene expression profile (Kwong and Frenkel, 1987, Read and Frenkel, 1983). This is exemplified by mRNA endonucleases encoded by certain herpesviruses, which produce exposed 5′-monophosphate-containing RNA fragments that are degraded by Xrn1 (Covarrubias et al., 2011, Elgadi et al., 1999, Everly et al., 2002, Gaglia et al., 2012). Other viruses including influenza and coronaviruses also encode mRNA endonucleases (Jagger et al., 2012, Kamitani et al., 2006, Plotch et al., 1981); however, a role for Xrn1 and the host decay machinery has only been shown for the SARS coronavirus nsp1 (Gaglia et al., 2012). In contrast, vaccinia virus (VacV) encodes two nudix domain-containing polypeptides related to the cellular Dcp2 decapping enzyme that accelerate mRNA turnover (Parrish and Moss, 2006, Parrish and Moss, 2007, Parrish et al., 2007).
As large DNA viruses that replicate exclusively within the cytoplasm, poxviruses like VacV encode the components required to produce capped, polyadenylated mRNAs (Moss, 2013). A virus-encoded heterodimeric cap methyltransferase (Morgan et al., 1984, Niles et al., 1989, Shuman et al., 1980, Venkatesan et al., 1980) and a poly(A) polymerase (Gershon et al., 1991, Moss et al., 1975, Nevins and Joklik, 1977) effectively mark nascent mRNAs with structural features vital for their stability and capacity to be translated. These mRNAs accumulate in discrete subcellular replication compartments together with select host proteins, including translation initiation factors (Katsafanas and Moss, 2007, Walsh et al., 2008). Remarkably, the D9 and D10 open reading frames (ORFs) encode proteins that stimulate mRNA turnover in infected and uninfected cells and function as decapping enzymes in vitro (Parrish and Moss, 2006, Parrish and Moss, 2007, Parrish et al., 2007). While D9 is expressed early in the viral lifecycle, D10 is expressed later, and its expression correlates with the virus-induced suppression of host protein synthesis (Parrish and Moss, 2006). Indeed, the kinetics of host protein synthesis suppression was delayed in cells infected with a D10-deficient virus, and a D10 mutant virus was attenuated for virulence in mice (Liu et al., 2014, Parrish and Moss, 2006). D10 may also regulate viral gene expression since it prefers m7GpppG over m7GpppA substrates in vitro, and the latter are only found on intermediate and late genes (Parrish et al., 2007). While decapped mRNAs like those produced by D9/10 are posited targets for Xrn1, precisely how Xrn1 might impact infected cell biology has not been investigated.
Here, we show that Xrn1 plays an unexpected role in VacV biology, as all ongoing protein synthesis ceased in Xrn1-depleted primary human fibroblasts infected with VacV, severely restricting virus growth. This occurred prior to completion of the viral lifecycle and was exacerbated by the absence of D9 decapping enzyme. Moreover, it coincided with dsRNA accumulation and activation of host dsRNA-responsive defenses controlled by PKR, which phosphorylates and inactivates the critical translation initiation factor eIF2, and 2′-5′ oligoadenylate synthetase (OAS), which stimulates rRNA cleavage by RNase L. Significantly, Xrn1 depletion even sensitized uninfected cells to dsRNA treatment. Thus, a key host mRNA decay enzyme, Xrn1, is required to regulate cytoplasmic dsRNA accumulation and signaling through critical host dsRNA-responsive innate immune sensing pathways in uninfected and VacV-infected cells. As VacV, like many viruses, encodes a dsRNA binding protein to limit dsRNA accumulation and signaling, our work showing the host Xrn1 functionally controls dsRNA homeostasis in infected cells, despite the presence of a viral dsRNA binding protein, challenges existing notions regarding the potency of viral dsRNA antagonists. Furthermore, it establishes that a 5′-3′ mRNA exonuclease plays a surprising role limiting dsRNA accumulation in infected and uninfected cell biology.
Results
Inhibition of Infected Cell Protein Synthesis and VacV Replication by Xrn1 Depletion
Although factors important for RNA metabolism and translation, including Xrn1, were identified in a high-throughput format, genome-wide, RNAi screen for host proteins affecting GFP-expressing VacV spread in an established, transformed cell line, the reliance of the virus on host mRNA decay pathways for infectious virus production was not further investigated (Sivan et al., 2013). To interrogate the role of Xrn1 in productive viral growth, normal primary human fibroblasts (NHDFs) were treated with control non-silencing (ns) siRNA or one of two independent Xrn1-specific siRNAs and infectious virus production quantified by plaque assay in permissive BSC40 cells. Compared to control ns siRNA-treated cultures, both individual Xrn1-specific siRNAs effectively depleted Xrn1 protein levels and reduced viral replication and spread 100- to 500-fold (Figures 1 A and 1B). To address the possibility that Xrn1 depletion might interfere with the VacV-induced suppression of host protein synthesis, mock- and VacV-infected NHDFs were metabolically labeled with 35S-containing amino acids and the proteins synthesized separated by SDS-PAGE and visualized by autoradiography. In control ns siRNA-treated cultures, the global protein synthesis profile in mock-infected cells was effectively suppressed by VacV infection, which resulted in high-level viral protein synthesis (Figure 1C, compare lane 1 versus 5). Surprisingly, while Xrn1 depletion had little detectable impact on protein synthesis in mock-infected NHDFs (Figure 1C, lane 1 versus 2), it dramatically reduced all protein synthesis in VacV-infected cells (lane 5 versus 6). This phenotype was dependent on viral gene expression, as it was not observed in cells infected with UV-inactivated virus (Figure 1C, lane 3 versus 4).
Figure 1.

Inhibition of Protein Synthesis and VacV Replication by Xrn1 Depletion
(A) NHDFs were transfected with non-silencing (ns) control or one of two Xrn1-specific siRNAs (−1 and −2). After 3 days, total protein was collected and Xrn1 levels analyzed by immunoblotting. Tubulin served as a loading control.
(B) NHDFs treated with siRNAs as in (A) were infected with VacV (MOI = 5 × 10−4). Infectious virus produced after 3 days was quantified by plaque assay. Means of three independent experiments are plotted ± SEM. ∗∗ indicates p ≤ 0.01 by paired Student’s t test compared to control siRNA-treated samples.
(C) NHDFs treated with siRNAs as in (A) were mock-infected or infected with VacV or UV-inactivated VacV (MOI = 5). At 18 hr post-infection (hpi), cells were metabolically pulse-labeled with [35S]Met-Cys for 30 min. Total protein was collected and separated by SDS-PAGE and 35S-labeled proteins visualized by exposing the fixed, dried gel to X-ray film. Molecular mass standards (in kDa) are shown to the right (upper panel). The same lysates were analyzed by immunoblotting (IB) with the indicated antibodies (lower panel). PABP1 served as a loading control.
(D) As in (C), except NHDFs were infected with WT VacV or D9 (ΔD9)- or D10 (ΔD10)-deficient VacV viruses (MOI = 3). HSC70 was used as a loading control. See also Figure S1.
As D9 and D10 decapping enzymes stimulate mRNA decay (Parrish et al., 2007), uncapped mRNAs could accumulate in Xrn1-depleted, VacV-infected cells and perhaps inhibit translation or result in cell stress. To determine if either D9 or D10 were required to globally inhibit translation in Xrn1-depleted NHDFs, the dependence of ongoing protein synthesis on Xrn1 was examined in cells infected with a D9- (ΔD9) or D10-deficient (ΔD10) virus. However, rather than suppressing the phenotype, the inhibition of protein synthesis in Xrn1-depleted cells was even more pronounced in cells infected with ΔD9 compared to ΔD10 or WT VacV (Figures 1D and S1). Thus, while the inhibition of protein synthesis in Xrn1-depleted, VacV-infected NHDFs was readily observed in the absence of D9 or D10, Xrn1 and the VacV-encoded D9 decapping enzyme showed a synthetic genetic interaction, consistent with their participation in a common pathway (Figures 1D and S1).
The global inhibition of ongoing protein synthesis in Xrn1-depleted cells infected with VacV is reminiscent of the cellular response to physiological stresses achieved by inactivation of eIF2, a critical translation initiation factor required to load 40S ribosome subunits with initiator tRNA, via phosphorylation of S51 on its α subunit (Mohr and Sonenberg, 2012, Walsh and Mohr, 2011). Significantly, the inhibition of protein synthesis in Xrn1-depleted, VacV-infected cells correlated with phosphorylated eIF2α accumulation (Figure 1C). While levels of phosphorylated eIF2α observed in control ns siRNA-treated NHDFs infected with ΔD9 or ΔD10 were modestly elevated compared to WT VacV (Figure 1D, compare lanes 7 and 10 versus 4), they were not sufficient to detectably suppress protein synthesis. Moreover, phosphorylated eIF2α abundance was further augmented in ΔD9- and ΔD10-infected cells by Xrn1 depletion (Figure 1D, compare lane 7 versus 8 and 9; lane 10 versus 11 and 12). In all cases, the substantial increase in eIF2α phosphorylation in Xrn1-depleted, VacV-infected cells was unexpected, as VacV, like numerous viruses, encodes multiple functions thought to prevent eIF2α phosphorylation (Walsh et al., 2013, Walsh and Mohr, 2011).
Accumulation of Phosphorylated eIF2α Is Dependent upon a Virus-Specific Transcription Factor in Xrn1-Depeted Cells
To more precisely define the point in the viral lifecycle where mRNA translation was inhibited in Xrn1-depleted cells, protein synthesis was analyzed by separating metabolically radiolabeled proteins by SDS-PAGE at different times post-infection. Little or no qualitative differences were detected between overall protein banding profiles of proteins produced in VacV-infected cells treated with control ns siRNA versus Xrn1 siRNA at all time points. From 9 hpi onward, however, radiolabeled VacV proteins were readily detected, and total 35S incorporation into proteins throughout the lane was reduced in Xrn1-depleted cultures versus those treated with control ns siRNA (Figure 2 A). The magnitude to which protein synthesis was suppressed in Xrn1-depleted cultures versus control siRNA-treated cultures increased progressively over time, and was greatest at 18 hpi when the virus-imposed suppression of host protein synthesis was strongest (Figure 2A). Analysis of these samples by immunoblotting indicated that phosphorylated eIF2α was more abundant in Xrn1-depleted cultures at all time points tested, and the inhibition of protein synthesis correlated precisely with (i) phosphorylated eIF2α levels and (ii) activation of the dsRNA-dependent eIF2α kinase PKR by phosphorylation (Figure 2B).
Figure 2.

Phosphorylated eIF2α Accumulation in Xrn1-Depleted Cells Correlates with Global Protein Synthesis Inhibition Late in the VacV Lifecycle
NHDFs transfected with ns control or Xrn1-specific siRNAs were infected with VacV (MOI = 5). At the indicated times (hpi) cells were metabolically pulse-labeled with [35S]Met-Cys for 30 min. Uninfected cells (UI) were harvested in parallel with 18 hpi samples.
(A) Total protein was isolated and separated by SDS-PAGE, and the fixed, dried gel exposed to X-ray film. Molecular mass standards (in kDa) are shown to the right.
(B) The same lysates were immunoblotted with the indicated antibodies. Tubulin served as a loading control.
(C) RNA from NHDFs treated with the indicated siRNAs and infected as in (A) was harvested at 6 hpi and subject to RT-qPCR using primers specific for K3L, E3L, or Xrn1 mRNAs. Each reaction product was normalized to the signal obtained using primers specific for 18S rRNA and expressed as the fold change relative to control siRNA-treated cells. Means of three independent experiments are plotted ± SEM.
As vaccinia encodes two proteins to antagonize eIF2α phosphorylation (Chang et al., 1992, Seo et al., 2008), one of which is a dsRNA-binding protein that prevents PKR activation (E3L), while the other is an eIF2α kinase pseudosubstrate (K3L), it was puzzling that robust eIF2α phosphorylation was observed in Xrn1-depleted cells. One possible explanation for this was a failure to produce E3L and K3L. Figure 2B shows that E3L is expressed at similar levels in control and Xrn1 knockdown cells at 6 and 9 hpi, when eIF2α becomes noticeably more phosphorylated in knockdown cells (Figure 2B). It is therefore not an insufficiency of this protein that is responsible for the phenotype. In the absence of suitable antisera available for K3L, qPCR was performed and showed that E3L and K3L mRNAs were expressed in Xrn1-depleted cells, exceeding those detected in control ns siRNA-treated cultures (Figure 2C).
Having shown that infection with UV-inactivated VacV, which delivers virion cargo into infected cells, but cannot express viral genes, is insufficient to inhibit protein synthesis in Xrn1-depleted cells (Figure 1C), infected NHDFs were treated with phosphonoacetic acid (PAA) to inhibit the viral DNA-dependent DNA polymerase. Importantly, PAA treatment prevented the cessation of protein synthesis observed in Xrn1-depleted, VacV-infected NHDFs, suggesting that viral DNA synthesis or an event closely associated with viral DNA synthesis like intermediate/late gene expression was required to trigger this phenotype (Figure 3 A). As VacV-induced inhibition of host protein synthesis is typically visible late in infection and is suppressed by PAA, expression of the viral early/intermediate gene product I3L was used to verify that the PAA-treated cells were indeed infected and had advanced to a stage preceding viral DNA replication (Figure 3B). To further parse the requirements to trigger the inhibition of VacV-infected cell protein synthesis by Xrn1 depletion, NHDFs were infected with a virus deficient in the intermediate transcription factor A23 (ΔA23) (Warren et al., 2012). While ΔA23-infected cells replicate viral DNA, they do not express intermediate or late genes whose transcription follows viral DNA synthesis and is absolutely dependent upon the A23 protein. Unlike WT VacV-infected cultures, protein synthesis proceeds and is not detectably impaired in Xrn1-depleted NHDFs infected with ΔA23 (Figures 3C and 3D). This indicates that the global inhibition of translation in Xrn1-depleted, VacV-infected cells is not triggered by viral DNA synthesis, but is instead dependent upon a specific virus transcription factor required for the biogenesis of discrete intermediate and late populations of VacV mRNAs. Moreover, it is consistent with a model positing that intermediate/late mRNAs or their protein products somehow promote eIF2α phosphorylation and globally inhibit translation in VacV-infected, Xrn1-depleted NHDFs.
Figure 3.

Inhibition of Protein Synthesis following Xrn1 Depletion Requires a VacV-Specific Late Gene Transcription Factor
(A) NHDFs transfected with ns control or Xrn1-specific siRNAs (−1 and −2) were mock-infected or infected with VacV (MOI = 5) in the presence or absence of PAA. At 18 hpi, cells were metabolically pulse-labeled with [35S]Met-Cys for 30 min. Total protein was collected and separated by SDS-PAGE, and 35S-labeled proteins visualized by exposing the fixed, dried gel to X-ray film. Molecular mass standards (in kDa) are shown to the right.
(B) The same lysates were also immunoblotted with the indicated antibodies. Tubulin was used as a loading control. VacV I3 (early expressed) serves as an infection control.
(C) As in (A), except NHDFs were infected with WT VacV or an A23-deficient virus (ΔA23).
(D) Lysates from (C) were immunoblotted with the indicated antibodies. eIF2α was used as a loading control.
Activation of PKR and RNase L Is Restricted by Xrn1 in VacV-Infected Cells
Of the four known mammalian eIF2α kinases that control translation in response to discrete stress, PKR, PERK, and GCN2 possess documented anti-viral activity, while only one, PKR, is encoded by an interferon-induced gene (Walsh et al., 2013). Importantly, although the inhibition of translation in Xrn1-depleted NHDFs infected with VacV correlated with PKR activation (Figure 2B), both PKR and PERK activities are antagonized by VacV-encoded effectors (Walsh et al., 2013). To determine which of these kinases might be required for eIF2α phosphorylation in Xrn1-depleted, VacV-infected NHDFs, each was depleted by RNAi in NHDFs treated with control ns or Xrn1-specific siRNA. While little detectable change in eIF2α phosphorylation was observed in mock-infected cells, only depleting the dsRNA-activated eIF2α kinase PKR reduced phosphorylated eIF2α levels in Xrn1-depleted, VacV-infected cells and basal phosphorylated eIF2α levels observed in VacV-infected, control ns siRNA-treated NHDFs (Figure 4 A). In addition, PKR activation above basal levels present in control ns siRNA-treated cultures was readily detected in Xrn1-depleted NHDFs (Figure 4A). In contrast, depletion of PERK, a distinct eIF2α kinase activated in response to unfolded protein accumulation in the ER, did not detectably reduce eIF2α phosphorylation in Xrn1-depleted cells and acted as a negative control (Figure 4A).
Figure 4.

PKR-Dependent eIF2α Phosphorylation and RNase L-Mediated rRNA Degradation in Xrn1-Depleted Cells Infected with VacV
(A) NHDFs transfected with the indicated siRNAs were mock-infected or infected with VacV (MOI = 5). Total protein was collected at 18 hpi and analyzed by immunoblotting with the indicated antibodies. Tubulin served as a loading control.
(B) NHDFs transfected with the indicated siRNAs were mock-infected or infected with VacV (MOI = 5). At 18 hpi, total RNA was isolated and analyzed using a Bioanalyzer Nano LabChip. 28S and 18S rRNA bands are indicated.
(C) NHDFs transfected with the indicated siRNAs were infected as in (A). At 18 hpi, cells were metabolically pulse-labeled with [35S]Met-Cys for 30 min. Total protein was collected and separated by SDS-PAGE, and 35S-labeled proteins visualized by exposing the fixed, dried gel to X-ray film. Molecular mass standards (in KDa) are shown to the right (upper panel). The same lysates were also immunoblotted (IB) with the indicated antibodies (lower panel). The RNase L-specific immunoreactive band is indicated by an arrow. Tubulin served as a loading control.
(D) Metabolically radiolabelled samples from (C) together with two additional independent replicate experiments were TCA precipitated. 35S incorporation into newly synthesized proteins was quantified by liquid scintillation counting. Means are plotted ± SEM. ∗ indicates p ≤ 0.05 by paired Student’s t test compared to Xrn1 siRNA-treated cells. See also Figures S2 and S3.
Besides PKR, 2′-5′ OASs are also encoded by a family of interferon-stimulated genes and are components of a separate arm of host dsRNA-dependent innate defenses. The resulting 2′-5′ oligoadenylate chains produced by OAS in response to dsRNA in turn selectively activate RNase L, an endonuclease that indiscriminately cleaves mRNAs and rRNA to inactivate ribosomes and inhibit protein synthesis (Sadler and Williams, 2008). To determine if RNase L was activated in Xrn1-depleted NHDFs infected with VacV, total RNA isolated from mock- versus VacV-infected NHDFs treated with control ns or either Xrn1 siRNA was analyzed using a Bioanalyzer Nano LabChip. While Xrn1 depletion resulted in little detectable difference in rRNA abundance in mock-infected cells, 28S and 18S rRNA breakdown products were only detected in VacV-infected NHDFs treated with Xrn1-specific compared to control ns siRNA (Figures 4B and S2). Thus, two dsRNA-activated innate immune defense pathways are specifically stimulated upon VacV infection of Xrn1-depleted cells. This suggests that Xrn1 is required to restrict the activity of both the eIF2α kinase PKR and RNase L, which is activated by the dsRNA-responsive OAS.
To determine the relative contribution of RNase L and/or PKR to the global inhibition of translation in Xrn1-depleted NHDFs upon VacV infection, the capacity of RNase L or PKR knockdown to prevent the inhibition of protein synthesis in Xrn1-depleted cells infected with VacV was evaluated. While infected cell protein synthesis was similar in cultures treated with control ns siRNA or siRNAs specific for PKR or RNase L, the inhibition of translation associated with Xrn1 knockdown was most effectively suppressed by co-depletion of RNase L (Figures 4C and 4D). Furthermore, knockdown of both Xrn1 and RNase L reduced phosphorylated eIF2α abundance compared to cultures treated with Xrn1 siRNA alone (Figures 4C and 4D). Triple depletion of Xrn1, RNase L, and PKR reduced phosphorylated eIF2α levels to below those observed in cultures treated with control ns siRNA, demonstrating the involvement of the eIF2α kinase PKR (Figures 4C and 4D). This did not, however, detectably augment protein synthesis beyond levels observed in Xrn1-RNase L doubly depleted cultures. Surprisingly, co-depletion of Xrn1 and PKR at best only modestly increased 35S-amino acid incorporation into protein, despite its efficacy at reducing phosphorylated eIF2α abundance below levels observed in VacV-infected cultures treated with control ns siRNA (Figure 4C). This likely is a consequence of sustained RNase L activation, which destroys both mRNA and rRNA and would be expected to restrict protein synthesis even though eIF2 activity is preserved. Equivalent results were obtained using a different Xrn1-specific siRNA (Figure S3). Taken together, these results show that the observed inhibition of protein synthesis in Xrn-depleted cells results primarily from activation of RNase L.
Control of dsRNA Accumulation in VacV-Infected Cells by Xrn1
Since dsRNA-responsive host defense proteins were activated in Xrn1-depleted, VacV-infected NHDFs; this suggested that overall steady-state dsRNA levels might be greater in VacV-infected cells. To test this possibility, control ns siRNA-treated and Xrn1-depleted NHDFs were mock-infected or infected with VacV. At different times post-infection, cultures were fixed and processed for indirect immunofluorescence using a monoclonal antibody that specifically detects dsRNA. Unlike earlier studies that detected dsRNA in VacV-infected cells with this antibody (Weber et al., 2006), we visualized the fluorophore signal without using tyramide signal amplification. By 12 hpi, cells containing elevated levels of dsRNA were readily detected in Xrn1-depleted NHDFs infected with VacV versus NHDFs treated with control ns siRNA (Figure 5 A). The fraction of dsRNA-containing VacV-infected cells increased through 18 hpi for cultures treated with Xrn1 siRNA compared to control ns siRNA (Figure 5A). This was confirmed by immuno-dot blotting on immobilized cytoplasmic extracts (Figure 5B). Furthermore, dsRNA-specific staining was consistently most intense coincident with DAPI-staining cytoplasmic compartments (Figure 5C), suggesting that dsRNA accumulation is occurring specifically within viral replication compartments.
Figure 5.

Massive dsRNA Accumulation in Xrn1-Depleted Cells Infected with VacV
NHDFs transfected with the indicated siRNAs were mock-infected or infected with VacV (MOI = 5). Cells were fixed at 6, 12, and 18 hpi and stained for immunofluorescence with J2 anti-dsRNA antibody (green). DNA was stained using DAPI (blue).
(A) Cells were visualized using fluorescence microscopy with a 20× objective.
(B) NHDFs treated with siRNAs and infected as in (A) were harvested and cell-free lysates prepared at 18 hpi. Equal volumes of lysates were dotted onto membrane and dsRNA detected by immunoblotting (upper panel). The dsRNA signal from (B) together with two independent replicates was quantified and the means plotted ± SEM (lower panel).
(C) Confocal image of Xrn1 siRNA-treated infected cells from (A), fixed at 18 hpi using 63× objective.
(D) Xrn1 siRNA-treated infected cells fixed at 18 hpi were treated with a mixture of single strand-specific RNase A/T1, dsRNA-specific RNase III, or buffer alone prior to immunostaining of dsRNA.
To verify that the immunoreactive signal was indeed due to dsRNA, fixed permeabilized cells were treated with either the single-strand-specific ribonucleases A and T1 or the dsRNA-specific RNase III. Figure 5D shows that dsRNA immunoreactivity in Xrn1-depleted NHDFs infected with VacV was insensitive to RNase A/T1 digestion, but abolished by pre-treatment with RNase III, indicating that this immunostaining is in fact specific for dsRNA. Immunopurification of dsRNA revealed enrichment for selected viral early and late mRNAs, but not two representative host mRNAs (actin, GAPDH). While this enrichment was readily observed in preparations treated with single-strand ribonucleases A and T1, it was eliminated upon treatment with the dsRNA-specific nuclease RNase III (Figures 6 A and 6B). This shows that dsRNA accumulating in VacV replication compartments contains virus-encoded mRNAs, although we cannot exclude the possibility that cellular mRNAs may also be represented. While overall actin and GAPDH mRNA levels remain relatively constant in Xrn1-depleted, VacV-infected NHDFs, the abundance of representative viral early mRNAs increased significantly (Figures 6C and 6D). Activation of RNase L late in infection (Figure S2) precluded analysis of how late VacV mRNA abundance is influenced by Xrn1 depletion. This is consistent with (i) a mechanism whereby the host 5′-3′ mRNA exoribonuclease Xrn1 restricts dsRNA accumulation by accelerating viral mRNA turnover and (ii) the possibility that the dsRNA originates from the products of overlapping VacV transcription units on opposite DNA strands.
Figure 6.

Elevated Viral mRNA Abundance and Their Enrichment in dsRNA Isolated from Xrn1-Depleted, VacV- Infected Cells
(A) Cell-free lysates from NHDFs transfected with Xrn1–1 siRNA and infected with VacV (MOI = 5) were prepared at 22 hpi and immunoprecipitated using J2 anti-dsRNA antibody. After treating with RNase A/T1 or RNase III, isolated RNA was analyzed by RT-qPCR using the indicated viral or cellular mRNA primers. mRNA abundances were normalized to actin and calculated relative to input (set to 1). The means of three independent experiments are plotted ± SEM.
(B) Equal volumes of buffer, input lysate (IP input), or the unbound fraction (IP unbound) were dotted onto a membrane and dsRNA detected by immunoblotting to demonstrate dsRNA depletion in the unbound fraction.
(C and D) NHDFs were treated with the indicated siRNAs and RNA isolated from uninfected cells (C) or 3 hpi with VACV (MOI = 5) (D). RNA was subject to RT-qPCR analysis for the indicated cellular or early viral mRNAs and each reaction product normalized to 18S rRNA and presented as the fold change relative to control siRNA-treated cells. The means of three independent experiments are plotted ± SEM. A significant difference by paired Student’s t test compared to control siRNA-treated cells is indicated by ∗ (p ≤ 0.05) or ∗∗ (p ≤ 0.01).
Regulation of the Response to Exogenous dsRNA in Uninfected Cells by Xrn1
The accumulation of dsRNA in Xrn1-depleted NHDFs infected with VacV raised the possibility that Xrn1 might regulate dsRNA responsiveness in uninfected cells and naturally buffer dsRNA accumulation. To investigate the impact of Xrn1 depletion on dsRNA responsiveness of uninfected cells, NHDFs treated with control ns or Xrn1-specific siRNA were transfected with increasing amounts of synthetic poly(I:C) dsRNA, and rates of protein synthesis were evaluated. While reductions in the intensity of individual protein bands were detected in Xrn1-depleted NHDFs exposed to 0.01–0.05 μg/ml poly(I:C) compared to NHDFs treated with control ns siRNA, global protein synthesis in Xrn1-depleted NHDFs exposed to poly(I:C) concentrations of 0.125 μg/ml and greater was significantly inhibited by poly(I:C) compared to corresponding controls (Figure 7 A). The reduction in ongoing mRNA translation in Xrn1-depleted NHDFs in response to poly(I:C) was mirrored by greater amounts of activated PKR and phosphorylated eIF2α compared to cultures treated with control ns siRNA (Figure 7B). However, while rRNA breakdown products were observed in poly(I:C)-treated cultures, further increase upon Xrn1 depletion was not detected (Figure S4). This could indicate differences in how infected versus uninfected NHDFs respond to Xrn1 depletion or reflect technical limitations in the assay. Nevertheless, the cellular 5′-3′ mRNA exonuclease unexpectedly can control the host response to exogenous dsRNA, an important pathogen-associated molecular pattern capable of activating potent innate defenses, in uninfected primary human cells.
Figure 7.

Increased Sensitivity to the dsRNA Analog Poly(I:C) in Response to Xrn1 Depletion in Uninfected Cells
(A and B) NHDFs treated with the indicated siRNAs were mock-transfected or transfected with increasing amounts of poly(I:C). After 3 hr, cells were metabolically pulse-labeled with [35S]Met-Cys for 30 min. Total protein was collected and separated by SDS-PAGE, and 35S-labeled proteins directly visualized by exposing the fixed, dried gel to X-ray film. Molecular mass standards (in KDa) are shown to the left (A). The same lysates were also immunoblotted with the indicated antibodies (B). Tubulin served as a loading control. See also Figure S4.
Discussion
By encoding two nudix hydrolase domain-containing proteins with enzymatic decapping activity in vitro that share homology to the host protein Dcp2, VacV accelerates global mRNA decay in infected cells (Parrish and Moss, 2006, Parrish and Moss, 2007, Parrish et al., 2007). In uninfected cells, cap removal results in mRNAs harboring a 5′ monophosphate mRNA terminus that are subsequently degraded by the mRNA 5′-3′ exonuclease Xrn1 (Arribas-Layton et al., 2013, Jonas and Izaurralde, 2013, Nagarajan et al., 2013). Here, we show that VacV growth is severely restricted in Xrn1-depleted primary human cells, as ongoing virus and host protein synthesis is inhibited. The global repression of protein synthesis was augmented in cells infected with a D9-deficient virus and dependent upon a specific transcription factor required for production of VacV intermediate/late mRNA. Significantly, substantial dsRNA accumulation and activation of host dsRNA-responsive innate defenses was observed, which resulted in inactivation of the translation initiation factor eIF2 and RNase L-mediated rRNA cleavage. Furthermore, Xrn1 depletion unexpectedly heightened uninfected cell sensitivity to dsRNA. This establishes a role for the host mRNA exonuclease Xrn1 as a potent effector limiting activation of host dsRNA-responsive innate immune defenses by regulating dsRNA abundance in uninfected and virus-infected cells.
The impressive increase in dsRNA accumulation in Xrn1-depleted, VacV-infected cells was unforeseen, as Xrn1 is a processive exonuclease whose substrate is single-stranded mRNA with exposed 5′-phosphate termini. Previous studies, however, have shown that it is capable of processing RNA stem loops (Decker and Parker, 1993, Poole and Stevens, 1997) and RNA:DNA duplexes (Jinek et al., 2011) provided that the 5′ RNA overhang is of sufficient length. Therefore Xrn1 may function not only to control VacV mRNA turnover, but also destabilize RNA duplexes and directly degrade imperfectly annealed strands. This could require or be stimulated by an ancillary RNA helicase to perhaps accelerate duplex unwinding. Our finding that Xrn1-depleted cells were more sensitive to poly(I:C)-induced PKR activation, eIF2α phosphorylation, and inhibition of translation is consistent with this notion, as poly(I:C) is a heterogeneous, synthetic dsRNA analog comprised of poly-nucleotide duplexes of varied lengths that likely contain single-stranded overhangs capable of being processed directly by Xrn1.
It has been argued for decades that dsRNA accumulation in DNA virus-infected cells arises via mRNAs produced from transcription units on opposing DNA strands, although direct evidence for this has been scant (Aloni, 1972, Aloni and Locker, 1973, Boone et al., 1979, Jacobs and Langland, 1996, Jacquemont and Roizman, 1975, Lucas and Ginsberg, 1972). While dsRNA has been isolated from infected cells and shown to activate PKR in vitro, it remained possible that such RNA structures formed as a result of the purification process and did not naturally exist as bona fide viral mRNA duplexes in infected cells (Maran and Mathews, 1988). Furthermore, although dsRNA has been detected by immunostaining in situ, its precise origin remained to be determined (Weber et al., 2006). Our data that dsRNA accumulation and activation of dsRNA-responsive host defenses in VacV-infected cells (i) are controlled by the major cellular mRNA exonuclease and (ii) are dependent upon a viral transcription factor required for the biogenesis of a specific class of mRNAs in Xrn1-depleted cells provide key biological support for the hypothesis that dsRNA comprised of viral mRNA duplexes activates PKR and RNase L.
In addition to E3L, a dsRNA binding protein that also inhibits RNase L (Chang et al., 1992, Rivas et al., 1998), and the eIF2α pseudosubstrate K3L (Seo et al., 2008), we show that VacV-encoded decapping enzymes required to accelerate mRNA turnover in infected cells also individually control eIF2α phosphorylation in infected primary human cells. In a parallel investigation, B. Moss and colleagues show that the combined deletion of D9 and D10 decapping enzymes results in dsRNA production, PKR activation, eIF2α phosphorylation, and RNase L activation similar to our findings in Xrn1-depleted cells (Liu et al., 2015 [this issue of Cell Host & Microbe]). Thus, increasing mRNA half-life may increase the probability that a specific set of mRNAs forms dsRNA, even with normal Xrn1 levels. A synthetic genetic interaction was detected involving D9 deficiency and Xrn1 depletion in that inhibition of translation in VacV-infected cells was more severe in the absence of this viral decapping enzyme. When Xrn1 levels are reduced, decapped mRNAs with the potential to form dsRNA are likely further stabilized, but cannot be efficiently utilized by the translation machinery as they lack an m7GTP cap. Interestingly, other examples indicating a powerful relationship between stress-induced anti-viral responses, including eIF2α phosphorylation, and mRNA decay have emerged from other systems. The HSV1-encoded endoribonuclease vhs stimulates mRNA decay and acts together with at least two additional viral factors to antagonize PKR in infected cells (He et al., 1997, Mulvey et al., 2003, Read, 2013, Sciortino et al., 2013). In contrast, overexpression of the cellular decapping complex component Dcp1a induced eIF2α phosphorylation mediated by the protein activator PACT and restricted poliovirus infection (Dougherty et al., 2011). Finally, cellular Dcp2 modulates IRF7 mRNA stability, and Dcp2-deficient cells exhibit sustained type I IFN production, which has an anti-viral effect (Li et al., 2012).
It is remarkable that the depletion of a single host gene has such an unprecedented impact on dsRNA accumulation and activation of host dsRNA-responsive innate immune defenses. While many viruses including VacV encode dsRNA binding proteins or other functions to limit eIF2α phosphorylation, prevent RNase L activation, and preserve the capacity of the cellular translation machinery to produce viral proteins (Walsh et al., 2013), Xrn1 represents a host factor that plays a significant role in allowing the virus to effectively antagonize host defenses. Since Xrn1 depletion does not prevent K3L and E3L expression, the resulting inhibition of protein synthesis in VacV-infected cells implies that the capacity of E3L and K3L to effectively antagonize host defenses is dependent upon preset levels of host factors including the cellular mRNA exonuclease Xrn1. Perturbing the natural homeostasis of host factors like Xrn1, which restrict dsRNA accumulation, can overwhelm the capacity of viral functions to antagonize host dsRNA-responsive innate defenses. This directly shows that viral antagonists of PKR and RNase L evolved to function within an environmental host context that regulates dsRNA accumulation, one component of which is Xrn1. Somewhat paradoxically, the effectiveness with which virus-encoded factors succeed in an “arms race” to antagonize host dsRNA-responsive defenses (Daugherty and Malik, 2012) is therefore reliant upon host functions that likewise limit dsRNA accumulation. Indeed, Xrn1 was among genes differentially expressed in high versus low responders to primary smallpox vaccination, perhaps reflecting, in part, its role in innate responses to dsRNA (Haralambieva et al., 2012). Moreover, it might be possible to exploit Xrn1 as a druggable target to selectively induce dsRNA production in acutely infected cells so that virus-encoded antagonists are overwhelmed, and the virus is revealed to the innate immune system.
Experimental Procedures
Antibodies and Chemicals
Monoclonal E3L antiserum was a kind gift from S. Isaacs (University of Pennsylvania). Polyclonal PABP1 antiserum was a kind gift of S. Morley (University of Sussex). I3 antiserum was a kind gift of D. Evans (University of Alberta). All other antibodies were purchased commercially as follows: Xrn1 (A300-443A; Bethyl Laboratories), α tubulin (6074; Sigma), eIF2α (5324; Cell Signaling Technology), phospho(ser51)-eIF2α (3398; Cell Signaling Technology), PKR (12297; Cell Signaling Technology), phospho(T446)-PKR (32036; Abcam), PERK (5683; Cell Signaling Technology), Hsc70 (10011384; Cayman Chemical), RNase L (13825; Abcam), dsRNA (J2; SciCons). PAA was from Sigma.
Cells and Viruses
Primary normal human diploid fibroblasts (NHDFs; Clonetics) were propagated in DMEM supplemented with 5% fetal bovine serum (FBS) plus1% penicillin/streptomycin (v/v), routinely sub-cultured 1:3 and maintained until passage 24. VacV (Western Reserve strain) was propagated in BSC40 cells, and virus stocks were prepared and titered as described (Walsh et al., 2008). UV-inactivated virus was prepared by exposing 1 ml of virus stock in a six-well dish to six pulses of 0.12 J/cm2 UV light in a Stratalinker (Stratagene). A23, D9, and D10 viruses were supplied by B. Moss (NIH NIAD, Bethesda).
Transfections and Infections
One day prior to siRNA transfection, NHDF cells were seeded in a 6-well or 12-well dish at 1.6 × 105 or 8 × 104 cells per well, respectively. Cells were transfected with siRNAs (see below) at a final concentration of 20 nM each using Lipofectamine RNAiMax (Life Technologies) at 1.25 μl/ml according to the manufacturer’s instructions. For double/triple combination siRNA experiments the negative control siRNA was used to ensure all cells received the same final concentration of siRNAs. Poly(I:C) transfections were conducted 3 days after siRNA transfection using poly(I:C) (Cat. No. P9582; Sigma) diluted in Opti-MEM and Lipofectamine 2000 (Life Technologies) according to manufacturer’s instructions. Prior to infection of siRNA-transfected cells the media was changed at 3 days post-transfection. Mock infections were performed with the same media used to dilute virus stocks. Following multicycle growth experiments, NHDF cultures were frozen at −80°C, and after three freeze-thaw cycles the amount of infectious virus was quantified by plaque assay in BSC40 cells.
siRNAs
Xrn1 siRNA-1 was custom-generated by Genelink as a duplex with UU 3′ overhangs with the following sequence: 5′−AGAUGAACUUACCGUAGAA−3′ (taken from Damgaard et al., 2008). All other siRNAs were commercially sourced as follows: AllStars Negative Control (SI03650318; QIAGEN), Xrn1–2 (SI00764127; QIAGEN), PKR (SI00042819; QIAGEN), PERK (SI02223725; QIAGEN), RNase L (SASI_Hs01_00081000; Sigma).
Immunofluorescence
Cells were seeded onto glass coverslips, and transfected/infected cells were fixed with 4% formaldehyde for 15 min and permeabilized with 0.5% Triton X-100. For RNase treatments cells were incubated in RNase buffer (10 mM Tris:HCl [pH 8.3], 10 mM MgCl2, 1 mM DTT, 60 mM NaCl) containing 50 U/ml RNase III (E6146S; New England Biolabs) or 50 μl/ml RNaseA/T1 mix (AM2286; Ambion), reflecting 25 U/ml and 1,000 U/ml, respectively, for 15 min at 37°C. Samples were then blocked in 4% FBS, incubated with primary antisera, and incubated with anti-mouse AlexaFluor 488 (A11029; Life Technologies) secondary antibody for 1 hr at room temperature. DNA was stained with 4′,6′-diamidino-2-phenylindole (DAPI). The fluorescent images were collected with a Zeiss LSM710 confocal microscope or a Zeiss Axiovert fluorescence microscope, using Zen 2008 software (Zeiss).
Immuno-Dot Blotting of dsRNA
NHDFs were lysed in cytoplasmic lysis buffer (15 mM Tris [pH 7.5], 0.3 M NaCl, 15 mM MgCl2, 1% Triton X-100, 100 U/ml RNase inhibitor [Fermentas]) containing complete EDTA-free protease inhibitors (Roche) for 15 min on ice. Samples were centrifuged for 1 min at 12,000 RCF at 4°C and supernatants collected. Extracts (5 μl) were dotted on to PVDF membrane and allowed to dry. RNA was then cross-linked using two pulses of 0.125 J/cm2 UV light in a Stratalinker (Stratagene). The membrane was then processed as for an immunoblot. Dot blots were quantified from film using Li-Cor Image Studio software to calculate signal intensity adjusted for background.
dsRNA Immunoprecipitation
Approximately 4 × 106 siRNA-transfected VacV-infected (MOI = 5) NHDFs were washed twice with cold PBS on ice and lysed in 1 ml IP buffer (15 mM Tris [pH 7.5], 0.1 M NaCl, 5 mM MgCl2, 0.5% Triton X-100, 1 mM dithiothreitol, 100 U/ml RNase inhibitor [Fermentas]) containing complete EDTA-free protease inhibitors (Roche) for 10 min on ice. Lysates were centrifuged for 1 min at 12,000 RCF at 4°C and supernatant collected. This was pre-cleared with 40 μl of protein-G+ agarose beads (25% slurry; Santa Cruz Biotechnology; SC-2002) for 1 hr at 4°C before incubation of lysate with 7 μg J2 dsRNA antibody overnight at 4°C on a rotating wheel. Lysate was then incubated with 40 μl of protein-G+ agarose beads for 3 hr at 4°C. Beads were then washed with IP buffer four times then treated with either 20 U/ml RNase III or 20 μl/ml RNaseA/T1 (25 U/ml/1,000 U/ml, respectively) for 15 min at 37°C. Beads were then washed three times and resuspended in 1 ml Trizol for RNA isolation.
Bioanalyzer RNA Analysis
RNA samples were analyzed on an Agilent 2200 Bioanalyzer using RNA Nano LabChips.
Acknowledgments
We thank members of the I.M. lab, D. Walsh, M. Garabedian, and A. Wilson for many discussions, and R.E. Rigby for helpful suggestions regarding use of the anti-dsRNA antibody. We are most grateful to B. Moss for his many helpful suggestions, generously providing A23, D9, and D10 mutant virus strains and for discussing unpublished data. This work was supported by NIH Grants AI073898 and GM056927 (to I.M.) and a Vilcek Fellowship awarded to H.M.B.
Published: March 11, 2015
Footnotes
Supplemental Information includes four figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2015.02.003.
Supplemental Information
References
- Aloni Y. Extensive symmetrical transcription of simian virus 40 DNA in virus-yielding cells. Proc. Natl. Acad. Sci. USA. 1972;69:2404–2409. doi: 10.1073/pnas.69.9.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloni Y., Locker H. Symmetrical in vivo transcription of polyoma DNA and the separation of self-complementary viral and cell RNA. Virology. 1973;54:495–505. doi: 10.1016/0042-6822(73)90159-1. [DOI] [PubMed] [Google Scholar]
- Arribas-Layton M., Wu D., Lykke-Andersen J., Song H. Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim. Biophys. Acta. 2013;1829:580–589. doi: 10.1016/j.bbagrm.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham C.J., Parker R. P bodies, stress granules, and viral life cycles. Cell Host Microbe. 2008;3:206–212. doi: 10.1016/j.chom.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone R.F., Parr R.P., Moss B. Intermolecular duplexes formed from polyadenylylated vaccinia virus RNA. J. Virol. 1979;30:365–374. doi: 10.1128/jvi.30.1.365-374.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J.E., Truffault V., Boland A., Huntzinger E., Chang C.T., Haas G., Weichenrieder O., Coles M., Izaurralde E. A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5′ exonucleolytic degradation. Nat. Struct. Mol. Biol. 2012;19:1324–1331. doi: 10.1038/nsmb.2413. [DOI] [PubMed] [Google Scholar]
- Chang H.W., Watson J.C., Jacobs B.L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E.G., Costantino D.A., Rabe J.L., Moon S.L., Wilusz J., Nix J.C., Kieft J.S. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science. 2014;344:307–310. doi: 10.1126/science.1250897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias S., Gaglia M.M., Kumar G.R., Wong W., Jackson A.O., Glaunsinger B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease Xrn1. PLoS Pathog. 2011;7:e1002339. doi: 10.1371/journal.ppat.1002339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard C.K., Kahns S., Lykke-Andersen S., Nielsen A.L., Jensen T.H., Kjems J. A 5′ splice site enhances the recruitment of basal transcription initiation factors in vivo. Mol. Cell. 2008;29:271–278. doi: 10.1016/j.molcel.2007.11.035. [DOI] [PubMed] [Google Scholar]
- Daugherty M.D., Malik H.S. Rules of engagement: molecular insights from host-virus arms races. Annu. Rev. Genet. 2012;46:677–700. doi: 10.1146/annurev-genet-110711-155522. [DOI] [PubMed] [Google Scholar]
- Decker C.J., Parker R. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- Dougherty J.D., White J.P., Lloyd R.E. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 2011;85:64–75. doi: 10.1128/JVI.01657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgadi M.M., Hayes C.E., Smiley J.R. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 1999;73:7153–7164. doi: 10.1128/jvi.73.9.7153-7164.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everly D.N., Jr., Feng P., Mian I.S., Read G.S. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J. Virol. 2002;76:8560–8571. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia M.M., Glaunsinger B.A. Viruses and the cellular RNA decay machinery. Wiley Interdiscip. Rev. RNA. 2010;1:47–59. doi: 10.1002/wrna.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia M.M., Covarrubias S., Wong W., Glaunsinger B.A. A common strategy for host RNA degradation by divergent viruses. J. Virol. 2012;86:9527–9530. doi: 10.1128/JVI.01230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau N.L., Wilusz J., Wilusz C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Gershon P.D., Ahn B.Y., Garfield M., Moss B. Poly(A) polymerase and a dissociable polyadenylation stimulatory factor encoded by vaccinia virus. Cell. 1991;66:1269–1278. doi: 10.1016/0092-8674(91)90048-4. [DOI] [PubMed] [Google Scholar]
- Haralambieva I.H., Oberg A.L., Dhiman N., Ovsyannikova I.G., Kennedy R.B., Grill D.E., Jacobson R.M., Poland G.A. High-dimensional gene expression profiling studies in high and low responders to primary smallpox vaccination. J. Infect. Dis. 2012;206:1512–1520. doi: 10.1093/infdis/jis546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Gross M., Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs B.L., Langland J.O. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- Jacquemont B., Roizman B. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 1975;15:707–713. doi: 10.1128/jvi.15.4.707-713.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagger B.W., Wise H.M., Kash J.C., Walters K.A., Wills N.M., Xiao Y.L., Dunfee R.L., Schwartzman L.M., Ozinsky A., Bell G.L. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science. 2012;337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Coyle S.M., Doudna J.A. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol. Cell. 2011;41:600–608. doi: 10.1016/j.molcel.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas S., Izaurralde E. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013;27:2628–2641. doi: 10.1101/gad.227843.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsafanas G.C., Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe. 2007;2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A.D., Frenkel N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA. 1987;84:1926–1930. doi: 10.1073/pnas.84.7.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Dai J., Song M., Fitzgerald-Bocarsly P., Kiledjian M. Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Mol. Cell. Biol. 2012;32:1164–1172. doi: 10.1128/MCB.06328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.W., Wyatt L.S., Orandle M.S., Minai M., Moss B. The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. J. Virol. 2014;88:202–211. doi: 10.1128/JVI.02426-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-W., Katsafanas G.C., Liu R., Wyatt L.S., Moss B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe. 2015;17:320–331. doi: 10.1016/j.chom.2015.02.002. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas J.J., Ginsberg H.S. Identification of double-stranded virus-specific ribonucleic acid in KB cells infected with type 2 adenovirus. Biochem. Biophys. Res. Commun. 1972;49:39–44. doi: 10.1016/0006-291x(72)90006-x. [DOI] [PubMed] [Google Scholar]
- Maran A., Mathews M.B. Characterization of the double-stranded RNA implicated in the inhibition of protein synthesis in cells infected with a mutant adenovirus defective for VA RNA. Virology. 1988;164:106–113. doi: 10.1016/0042-6822(88)90625-3. [DOI] [PubMed] [Google Scholar]
- Mohr I., Sonenberg N. Host translation at the nexus of infection and immunity. Cell Host Microbe. 2012;12:470–483. doi: 10.1016/j.chom.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan J.R., Cohen L.K., Roberts B.E. Identification of the DNA sequences encoding the large subunit of the mRNA-capping enzyme of vaccinia virus. J. Virol. 1984;52:206–214. doi: 10.1128/jvi.52.1.206-214.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013;5:a010199. doi: 10.1101/cshperspect.a010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B., Rosenblum E.N., Gershowitz A. Characterization of a polyriboadenylate polymerase from vaccinia virions. J. Biol. Chem. 1975;250:4722–4729. [PubMed] [Google Scholar]
- Mulvey M., Poppers J., Sternberg D., Mohr I. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 2003;77:10917–10928. doi: 10.1128/JVI.77.20.10917-10928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan V.K., Jones C.I., Newbury S.F., Green P.J. XRN 5′→3′ exoribonucleases: structure, mechanisms and functions. Biochim. Biophys. Acta. 2013;1829:590–603. doi: 10.1016/j.bbagrm.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins J.R., Joklik W.K. Isolation and partial characterization of the poly(A) polymerases from HeLa cells infected with vaccinia virus. J. Biol. Chem. 1977;252:6939–6947. [PubMed] [Google Scholar]
- Niles E.G., Lee-Chen G.J., Shuman S., Moss B., Broyles S.S. Vaccinia virus gene D12L encodes the small subunit of the viral mRNA capping enzyme. Virology. 1989;172:513–522. doi: 10.1016/0042-6822(89)90194-3. [DOI] [PubMed] [Google Scholar]
- Orban T.I., Izaurralde E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA. 2005;11:459–469. doi: 10.1261/rna.7231505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R., Song H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- Parrish S., Moss B. Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression. J. Virol. 2006;80:553–561. doi: 10.1128/JVI.80.2.553-561.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S., Moss B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 2007;81:12973–12978. doi: 10.1128/JVI.01668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S., Resch W., Moss B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. USA. 2007;104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotch S.J., Bouloy M., Ulmanen I., Krug R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- Poole T.L., Stevens A. Structural modifications of RNA influence the 5′ exoribonucleolytic hydrolysis by XRN1 and HKE1 of Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1997;235:799–805. doi: 10.1006/bbrc.1997.6877. [DOI] [PubMed] [Google Scholar]
- Read G.S. Virus-encoded endonucleases: expected and novel functions. Wiley Interdiscip. Rev. RNA. 2013;4:693–708. doi: 10.1002/wrna.1188. [DOI] [PubMed] [Google Scholar]
- Read G.S., Frenkel N. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J. Virol. 1983;46:498–512. doi: 10.1128/jvi.46.2.498-512.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas C., Gil J., Mĕlková Z., Esteban M., Díaz-Guerra M. Vaccinia virus E3L protein is an inhibitor of the interferon (i.f.n.)-induced 2-5A synthetase enzyme. Virology. 1998;243:406–414. doi: 10.1006/viro.1998.9072. [DOI] [PubMed] [Google Scholar]
- Sadler A.J., Williams B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciortino M.T., Parisi T., Siracusano G., Mastino A., Taddeo B., Roizman B. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J. Virol. 2013;87:3271–3276. doi: 10.1128/JVI.03049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo E.J., Liu F., Kawagishi-Kobayashi M., Ung T.L., Cao C., Dar A.C., Sicheri F., Dever T.E. Protein kinase PKR mutants resistant to the poxvirus pseudosubstrate K3L protein. Proc. Natl. Acad. Sci. USA. 2008;105:16894–16899. doi: 10.1073/pnas.0805524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S., Surks M., Furneaux H., Hurwitz J. Purification and characterization of a GTP-pyrophosphate exchange activity from vaccinia virions. Association of the GTP-pyrophosphate exchange activity with vaccinia mRNA guanylyltransferase. RNA (guanine-7-)methyltransferase complex (capping enzyme) J. Biol. Chem. 1980;255:11588–11598. [PubMed] [Google Scholar]
- Silva P.A., Pereira C.F., Dalebout T.J., Spaan W.J., Bredenbeek P.J. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J. Virol. 2010;84:11395–11406. doi: 10.1128/JVI.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan G., Martin S.E., Myers T.G., Buehler E., Szymczyk K.H., Ormanoglu P., Moss B. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc. Natl. Acad. Sci. USA. 2013;110:3519–3524. doi: 10.1073/pnas.1300708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G., Mayo T., Anderson P. ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep. 2006;7:72–77. doi: 10.1038/sj.embor.7400572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S., Gershowitz A., Moss B. Modification of the 5′ end of mRNA. Association of RNA triphosphatase with the RNA guanylyltransferase-RNA (guanine-7-)methyltransferase complex from vaccinia virus. J. Biol. Chem. 1980;255:903–908. [PubMed] [Google Scholar]
- Walsh D., Mohr I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011;9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D., Arias C., Perez C., Halladin D., Escandon M., Ueda T., Watanabe-Fukunaga R., Fukunaga R., Mohr I. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 2008;28:2648–2658. doi: 10.1128/MCB.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D., Mathews M.B., Mohr I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013;5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R.D., Cotter C.A., Moss B. Reverse genetics analysis of poxvirus intermediate transcription factors. J. Virol. 2012;86:9514–9519. doi: 10.1128/JVI.06902-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.