

Here, Li et al. investigate the mechanisms underlying αE-catenin inhibition of skin squamous cell carcinoma (SCC) development. They found that αE-catenin inhibits β4 integrin-mediated activation of SRC tyrosine kinase and that YAP1, an effector of the Hippo signaling pathway, is a direct SRC phosphorylation target, thereby providing new insights into the regulation of SRC–YAP1-mediated transformation.
Keywords: αE-catenin, SRC, YAP1, SCC, keratinocytes, skin
Abstract
Cell–cell adhesion protein αE-catenin inhibits skin squamous cell carcinoma (SCC) development; however, the mechanisms responsible for this function are not completely understood. We report here that αE-catenin inhibits β4 integrin-mediated activation of SRC tyrosine kinase. SRC is the first discovered oncogene, but the protein substrate critical for SRC-mediated transformation has not been identified. We found that YAP1, the pivotal effector of the Hippo signaling pathway, is a direct SRC phosphorylation target, and YAP1 phosphorylation at three sites in its transcription activation domain is necessary for SRC–YAP1-mediated transformation. We uncovered a marked increase in this YAP1 phosphorylation in human and mouse SCC tumors with low/negative expression of αE-catenin. We demonstrate that the tumor suppressor function of αE-catenin involves negative regulation of the β4 integrin–SRC signaling pathway and that SRC-mediated phosphorylation and activation of YAP1 are an alternative to the canonical Hippo signaling pathway that directly connect oncogenic tyrosine kinase signaling with YAP1.
Adherens junctions (AJs) are the cadherin–catenin protein complexes that are essential for proper intercellular adhesion. In addition to their structural function, these proteins have an important role in regulating intracellular signaling pathways, which help to maintain normal tissue homeostasis and protect from abnormal cellular proliferation and cancer (Klezovitch and Vasioukhin 2015). AJs play a critical role in the regulation of contact inhibition of cellular proliferation. They can sense an increase in local cell density and respond by signaling to halt proliferation (Stepniak et al. 2009; Vasioukhin 2012). The molecular mechanisms responsible for the signaling function of the AJ proteins are not fully understood. Epithelial α-catenin (encoded by Ctnna1) is an essential AJ protein that links cadherin–catenin protein complexes at the membrane to the actin cytoskeleton (Buckley et al. 2014). αE-catenin also has AJ-independent functions in the regulation of intracellular trafficking events (Lien et al. 2008a). It is frequently missing or down-regulated in various human epithelial cancers, and re-expression of αE-catenin in cancer cells has a prominent negative impact on tumor growth (Ewing et al. 1995; Bullions et al. 1997; Benjamin and Nelson 2008; Vasioukhin 2012). Genetic experiments in mice demonstrated the tumor suppressor function of αE-catenin in skin squamous cell carcinoma (SCC) (Kobielak and Fuchs 2006; Silvis et al. 2011). The analysis of mechanisms responsible for this function identified a functional connection between αE-catenin and YAP1, a pivotal transcriptional coactivator of the Hippo signaling pathway (Schlegelmilch et al. 2011; Silvis et al. 2011).
YAP1 binds numerous transcription factors and potently stimulates transcription (Yagi et al. 1999; Basu et al. 2003; Komuro et al. 2003; Zaidi et al. 2004; Zhao et al. 2008; Rosenbluh et al. 2012). YAP1 is a critical downstream target of the Hippo signaling pathway, which is necessary for the proper regulation of contact inhibition and tumor suppression in mammalian organisms (Barry and Camargo 2013; Yu and Guan 2013). At the core of the Hippo pathway is a kinase cascade consisting of the serine/threonine kinases MST1/2, which activate LATS1/2, which in turn phosphorylate and inactivate YAP1 (Enderle and McNeill 2013; Yu and Guan 2013).
We found previously that YAP1 is constitutively nuclear and is necessary for the hyperproliferative phenotype in αE-catenin−/− keratinocytes (Schlegelmilch et al. 2011; Silvis et al. 2011). Furthermore, analysis of the Hippo signaling pathway demonstrated that the activity of LATS1/2 and phosphorylation of YAP1 at S127 (LATS1/2 phosphorylation site) were not affected by αE-catenin, suggesting that a novel regulatory pathway may be responsible for constitutive nuclear localization of YAP1 in αE-catenin−/− keratinocytes (Silvis et al. 2011). We report here that αE-catenin regulates a Hippo-independent signaling pathway responsible for YAP1 activation. We found that αE-catenin attenuates β4 integrin-mediated activation of SRC. SRC is hyperactive in αE-catenin−/− keratinocytes; it phosphorylates YAP1 at three sites in its transcriptional activation domain, and this phosphorylation not only is required for transformation of αE-catenin−/− keratinocytes but is also necessary for general SRC-mediated oncogenic transformation.
Results
αE-catenin negatively regulates a β4 integrin–SRC signaling pathway, which is required for hyperproliferation of αE-catenin−/− keratinocytes
Our siRNA screen previously identified Yap1 as a gene necessary for the hyperproliferative and contact inhibition-defective phenotype of αE-catenin−/− mouse keratinocytes, and subsequent analysis demonstrated that YAP1 is constitutively active in these cells (Silvis et al. 2011). Since we did not observe changes in LATS1/2-mediated phosphorylation of YAP1 that could account for its constitutive activation, we extended our siRNA screen to examine genes necessary for hyperproliferation of αE-catenin−/− keratinocytes. We found that, in addition to Yap1, Src and Itgb4 were necessary for this phenotype (Fig. 1A,A′; Supplemental Fig. S1A,B). Moreover, αE-catenin−/− cells were also hypersensitive to PP2, a specific Src family kinase (SFK) inhibitor (Supplemental Fig. S1C). Thus, Src expression and SFK activity are essential for hyperproliferation of αE-catenin−/− keratinocytes. We next explored whether loss of αE-catenin expression affects SFK activity. Western blot analysis revealed increased SFK activity in αE-catenin−/− keratinocytes, and the difference in SFK activity between wild-type and αE-catenin−/− cells was erased after the knockdown of Src (Fig. 1B,B′,C).
Figure 1.

αE-catenin negatively regulates the β4 integrin–SRC signaling module, which is required for hyperproliferation of αE-catenin−/− keratinocytes. (A) αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes were plated at high density in the indicated siRNA-Lipofectamine mixtures in triplicates and cultured for 5 d. Relative cell numbers were determined by cell counting. Bar graphs show mean values ± standard deviation (SD). (A′) Western blot analyses of proteins from αE-cateninfl/fl (C) or αE-catenin−/− (K) keratinocytes transfected with the indicated siRNA oligos. (B) Western (WB) blot analyses of proteins from αE-cateninfl/fl or αE-catenin−/− keratinocytes with anti-αE-catenin, anti-phospho-SFK (pY416-SFK), or anti-SRC antibodies. (B′) Quantitation of active SFKs (pY416) versus total SRC. n = 3. Bar graphs show mean values ± SD. Student's t-test. (C) Western blot analyses of proteins from αE-cateninfl/fl or αE-catenin−/− keratinocytes transfected with control (siCtrl) or siSrc oligos. (L.E.) Longer exposure. (D) Coimmunoprecipitations of endogenous YAP1 from αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes and subsequent Western blot analyses with the indicated antibodies. (E) Western blot analyses of proteins from αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes transfected with control (siCtrl) or si-integrin β4 oligos with anti-αE-catenin, anti-phospho-SFK (pY416-SFK), anti-SRC, and integrin β4 antibodies. (F) Western blot analyses of proteins from αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes cotransfected with control (siCtrl) or si-integrin β4 oligos together with expression constructs encoding si-resistant human wild-type or signaling-incompetent Y341/357/394F (3YF) mutant β4 integrin. (G) Coimmunoprecipitations of endogenous β4 integrin from control or α-cat knockout keratinocytes and subsequent Western blot analyses with the indicated antibodies.
YAP1 associates with SFKs (Sudol 1994; Zaidi et al. 2004; Taniguchi et al. 2015); therefore, we examined whether endogenous YAP1 and SRC form a protein complex in mouse keratinocytes. Coimmunoprecipitation experiments revealed the presence of SRC in YAP1 protein complexes (Fig. 1D). αE-catenin did not influence the interaction between SRC and YAP1; however, SFKs were more active in the YAP1-containing protein complexes isolated from αE-catenin−/− cells (Fig. 1D). These results demonstrate that SRC and YAP1 form protein complexes, and αE-catenin negatively regulates SFK activity in mouse keratinocytes.
In addition to Src, Itgb4 (encoding β4 integrin) was necessary for hyperproliferation of αE-catenin−/− cells (Fig. 1A). There is significant coordination between cadherin-based and integrin-based adhesion structures within the cell (Collins and Nelson 2015). Moreover, β4 integrin is known as a potent activator of SRC (Bertotti et al. 2006). Therefore, we analyzed whether β4 integrin is required for hyperactivation of SRC in αE-catenin−/− cells. Knockdown of Itgb4 eliminated the increased activity of SRC in αE-catenin−/− keratinocytes (Fig. 1E). Re-expression of siRNA-resistant human β4 integrin rescued this phenotype (Fig. 1F). Moreover, re-expression of the triple tyrosine to phenylalanine mutant and therefore signaling-incompetent β4 integrin (Bertotti et al. 2006) failed to rescue hyperactivation of SRC in αE-catenin−/− cells (Fig. 1F). Coimmunoprecipitation experiments revealed a prominent increase in the interaction between β4 integrin and SRC in αE-catenin−/− keratinocytes (Fig. 1G). These data demonstrate that β4 integrin interacts with SRC and is necessary for SRC hyperactivation in αE-catenin−/− keratinocytes.
β4 integrin is involved in the formation of adhesive hemidesmosomes in wild-type keratinocytes. To determine whether αE-catenin regulates localization of β4 integrin, we performed immunofluorescent stainings of keratinocytes with anti-β4 integrin antibodies. As expected, β4 integrin was present at cell–substratum adhesion structures in wild-type cells; however, it was localized to E-cadherin-positive cell–cell adhesion structures in αE-catenin−/− cells (Fig. 2A,B). In addition, while SRC displayed diffuse cytoplasmic localization in wild-type cells, it was localized to cell–cell junctions in αE-catenin−/− keratinocytes (Fig. 2B).
Figure 2.
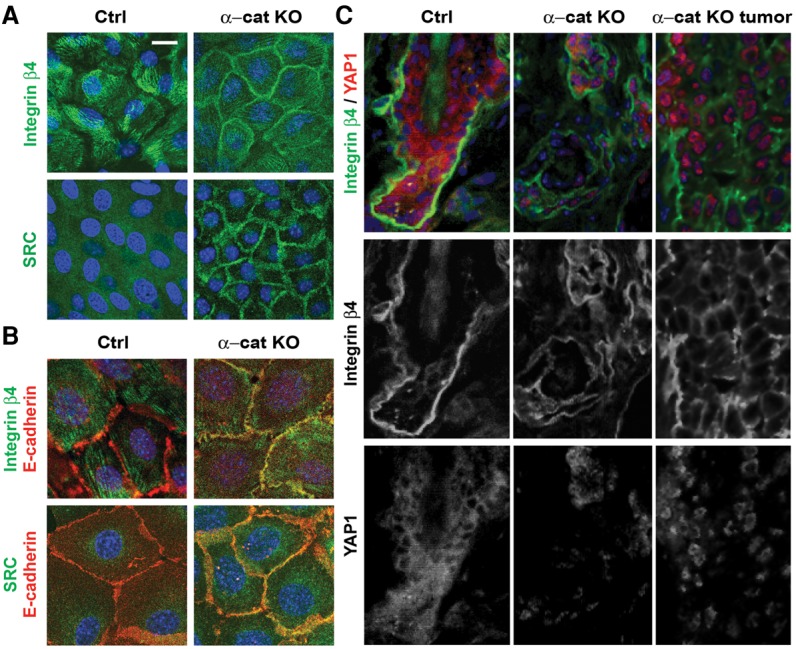
Cell–cell junctional localization of SRC and β4 integrin in αE-catenin−/− keratinocytes. (A) Immunofluorescent staining of αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes with anti-SRC and anti-integrin β4 antibodies (green). (B) Immunofluorescent staining of αE-cateninfl/fl or α-cat knockout mouse keratinocytes with anti-SRC, anti-integrinβ4 (green), and anti-E-cadherin (red) antibodies. DAPI indicates nuclear counterstain (blue). (C) Immunofluorescent staining of skin sections from 22-wk-old GFAP-Cre (Ctrl) and GFAP-Cre/αE-cateninfl/fl (α-cat KO and α-cat KO tumor) mice with anti-integrin β4 (green) and anti-YAP1 (red) antibodies. (Blue) Nuclear DAPI staining. Bars: A, 20 µm; B, 10 µm; C, 12 µm.
Immunofluorescent stainings of skin and tumor sections from control and GFAP-Cre/αE-cateninfl/fl mice revealed mislocalization of β4 integrin and nuclear localization of YAP1 in αE-catenin−/− keratinocytes in vivo (Fig. 2C). Overall, we conclude that αE-catenin prevents junctional localization of β4 integrin and SRC and that β4 integrin targeted to cell–cell junctions activates SRC, which drives hyperproliferation of αE-catenin−/− keratinocytes.
Active SRC promotes nuclear localization and activation of YAP1 in confluent keratinocytes independently from the canonical Hippo signaling pathway
Since YAP1 is constitutively nuclear in αE-catenin−/− keratinocytes (Silvis et al. 2011) and since we found increased activation of SFKs associated with YAP1 in αE-catenin−/− cells (Fig. 1D), we next examined whether activation of SRC in normal keratinocytes impacts YAP1 localization and transcriptional activity. In confluent cell monolayers, the active Hippo signaling pathway leads to cytoplasmic retention and inactivation of YAP1 (Zhao et al. 2007). Consistent with these findings, YAP1 was localized to the cytoplasm in confluent wild-type keratinocytes. However, expression of the constitutively active form of SRC-Y529F (CA-SRC) (Piwnica-Worms et al. 1987) resulted in nuclear-localized YAP1 in confluent cells (Fig. 3A,B). Treatment with an inhibitor of ABL/SFKs, dasatinib (DAS), rescued this phenotype, indicating that tyrosine kinase activity was required for the constitutive nuclear localization of YAP1 in confluent cells (Fig. 3A-A′). In addition, siRNA-mediated knockdown of Src resulted in a decrease in the levels of nuclear YAP1 in αE-catenin−/− keratinocytes (Supplemental Fig. S2A).
Figure 3.

Active SRC promotes Hippo pathway-independent nuclear localization and activation of YAP1 in confluent keratinocytes. (A) Immunofluorescent staining of keratinocytes stably transduced with vector or CA-SRC and treated with DMSO or DAS with anti-αE-catenin (α-cat; red) and anti-YAP1 (green) antibodies. DAPI indicates nuclear counterstain (blue). Cells were treated with either vehicle (DMSO) or 5 nM DAS for 30 min at 37°C. Bar, 15 µm. (A′) Quantitation of YAP1 localization in A. Bars represent the average of YAP1 localization between the cytoplasm, the nucleus, or both ±SD for >150 cells for each condition. (B) Western blot (WB) analyses of proteins from nuclear and cytoplasmic fractions of control (Ctrl) or α-cat knockout (KO) mouse keratinocytes transduced with vector (−) or CA-SRC (+). (C) Luciferase assay using empty control (Ctrl-LUC) or TEAD/YAP1 8xGTIIC reporter in keratinocytes stably transduced with empty vector or CA-SRC. DAS indicated 0.2 nM DAS. Bars represent means ± SD. n = 3. Student's t-test. (C′) Western blot analyses of total proteins from keratinocytes expressing empty control or CA-SRC constructs and treated with DMSO (−) or DAS using the indicated antibodies. (D) Luciferase assay using control (Ctrl-LUC) or the TEAD/YAP1 8xGTIIC reporter in keratinocytes stably transduced with empty vector or CA-SRC and transiently transfected with siCtrl or siLATS1/2 oligos. Bars represent means ± SD. n = 3. Student's t-test. (D′) Western blot analyses of total proteins from keratinocytes expressing empty control or CA-SRC constructs and transiently transfected with siCtrl or siLATS1/2 oligos using the indicated antibodies.
Luciferase assays with YAP1-regulated reporter constructs and quantitative RT–PCR (qRT–PCR) analysis of endogenous gene targets of YAP1 demonstrated that the subcellular relocalization of YAP1 to the nucleus induced by CA-SRC was accompanied by an increase in YAP1-mediated transcription, which was erased by DAS (Fig. 3C,C′; Supplemental Fig. S2B,C).
It has been reported that CA-SRC can negatively regulate the canonical Hippo signaling pathway (Enomoto and Igaki 2013; Kim and Gumbiner 2015; Kwon et al. 2015). To determine whether CA-SRC regulates YAP1 activity in keratinocytes via the canonical Hippo signaling pathway, we performed luciferase assays with the YAP1-regulated reporter in cells with knockdown of endogenous LATS1/2 (Fig. 3D,D′). While ablation of LATS1/2 resulted in a detectable increase in YAP1 transcriptional activity, CA-SRC retained its ability to activate endogenous YAP1 even in LATS1/2 knockdown cells, indicating that CA-SRC activated YAP1 in confluent keratinocytes through a Hippo pathway-independent mechanism. Consistent with this finding, phosphorylation of YAP1 at S127/S397, which is mediated by LATS1/2 kinases (Zhao et al. 2007), was not affected in keratinocytes expressing CA-SRC, indicating that SRC-mediated activation of YAP1 in confluent keratinocytes does not involve changes in the canonical Hippo phosphorylation cascade (Fig. 3C′). Interestingly, a similar analysis of sparse keratinocytes revealed a prominent SRC-mediated dephosphorylation of YAP1 at S127, indicating the potential involvement of SFKs in the regulation of the canonical Hippo pathway in non-contact-inhibited cells (Supplemental Fig. S2D). Thus, we conclude that activation of SRC is sufficient for constitutive nuclear localization and transcriptional activation of YAP1 in confluent keratinocytes through a mechanism that is independent of LATS1/2-mediated YAP1 phosphorylation.
Active SRC prominently cooperates with YAP1 to malignantly transform epidermal keratinocytes
To determine the functional significance of SRC and YAP1 in malignant transformation, we overexpressed CA-SRC and/or wild-type or S127/397A (2SA) mutant (constitutively active in the Hippo pathway) YAP1 in primary mouse keratinocytes. Expression of CA-SRC or YAP1 alone increased the saturation density of normal keratinocytes, and these genes displayed a significant additive effect when expressed together in the same cells (Fig. 4A,A′). Interestingly, this was also observed when CA-SRC was expressed together with the 2SA YAP1 mutant, which is constitutively active in the Hippo pathway (Fig. 4A,A′). In addition, endogenous YAP1 was necessary for a CA-SRC-mediated increase in the saturation density of normal keratinocytes (Supplemental Fig. S3).
Figure 4.
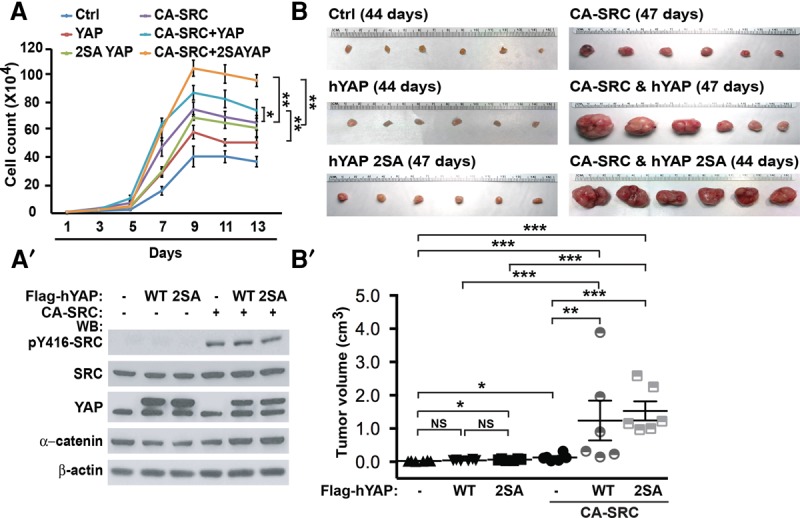
Active SRC prominently cooperates with YAP1 in the malignant transformation of primary keratinocytes. (A) Growth curve analysis of keratinocytes stably transduced with empty vectors (Ctrl), wild-type hYAP1 (YAP), 2SA YAP1 (2SA YAP), and CA-SRC. Values represent mean ± SD. n = 3. Student's t-test. (A′) Western blot (WB) analyses of total proteins from the cells in A using the indicated antibodies. (B) Orthotopic allograft tumor formation assays in NOD-SCID mice using primary keratinocytes stably transduced with the expression constructs described in A. (B′) Quantitation of tumor size differences in B. Data represent mean ± SE. P-values were calculated by unpaired t-test.
We also analyzed whether CA-SRC and YAP1 can synergize in the malignant transformation of nontumorigenic mouse keratinocytes using subcutaneous allograft tumor formation assays. CA-SRC strongly cooperated with both wild-type and 2SA mutant YAP1 proteins in driving tumor formation (Fig. 4B,B′). These results demonstrate that CA-SRC prominently synergizes with YAP1 in malignant transformation.
SRC directly phosphorylates YAP1 at three tyrosines (Y341, Y357, and Y394) in the transcriptional activation domain, and these sites are hyperphosphorylated in αE-catenin−/− keratinocytes
Since we found that SRC binds to YAP1 and that constitutive activation of SRC in confluent keratinocytes results in activation of YAP1 independently of LATS1/2-mediated phosphorylation, we hypothesized that SRC directly phosphorylates YAP1 in keratinocytes and that this phosphorylation is responsible for SRC-mediated YAP1 activation. Indeed, it has been reported previously that YAP1 can be phosphorylated at Y357 by ABL and YES1 tyrosine kinases (Levy et al. 2008; Tamm et al. 2011; Rosenbluh et al. 2012). Moreover, an increase in Y357 YAP1 phosphorylation was previously documented in intestinal epithelial cells with an active gp130–SFK pathway (Taniguchi et al. 2015). Here, we found that YAP1 was prominently tyrosine phosphorylated in keratinocytes expressing CA-SRC, and this phosphorylation was lost in cells exposed to the ABL/SFK inhibitor DAS (Fig. 5A). YAP1 was also hyperphosphorylated on tyrosine residues in αE-catenin−/− keratinocytes, and this phosphorylation was erased in DAS-treated cells (Fig. 5B).
Figure 5.

SRC directly phosphorylates YAP1 at Y341/357/394, and these sites are hyperphosphorylated in αE-catenin−/− keratinocytes. (A) Western blot (WB) analyses of total (input) or anti-Flag immunoprecipitated (Flag IP) proteins from αE-cateninfl/fl mouse keratinocytes stably transduced with Flag-YAP1 and empty vector or CA-SRC constructs using the indicated antibodies. Cells were treated with 5 nM DMSO or DAS for 30 min and then with 10 µM pervanadate for 5 min before lysis. (pTyr) Anti-phospho-tyrosine. (B) Western blot analyses of proteins immunoprecipitated with anti-YAP1 antibodies (YAP IP) from αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes using anti-phospho-tyrosine (pTyr) and anti-YAP1 antibodies. (C) SRC phosphorylation consensus motifs in the YAP1-1β and YAP1-2γ isoforms of YAP1. Note that the previously identified ABL phosphorylation site corresponds to Y357 in YAP1-1β and to Y407 in YAP1-2γ. (C′) Evolutionary conservation of Y341/Y357/Y394 (YAP1-1β) sites in YAP1 proteins. (D) Western blot analyses of total (input) or anti-Flag immunoprecipitated (Flag IP) proteins from HEK293 cells transiently transfected with empty vector (−) or CA-SRC (+) and wild-type (WT) or tyrosine mutant Flag-YAP1 constructs. (3YF) Y341/357/394F. (E) Western blot analyses of total (Input) or anti-Flag immunoprecipitated (Flag IP) proteins from keratinocytes stably transduced with empty vector (Ctrl), wild-type (WT), or tyrosine mutant Flag-YAP1 constructs using the indicated antibodies. (F) In vitro kinase assay using purified YAP1 and CA-SRC proteins. In vitro translated HALO-tagged wild-type (WT), Y341F, Y357F, Y394F, or Y341/357/394F (3YF) YAP1 proteins were purified using HALO resin and incubated with or without ATP and purified CA-SRC for 30 min at 30°C. YAP1 proteins were cleaved off from HALO resin with TEV protease and analyzed by Western blot with anti-phospho-tyrosine (pTyr), anti-YAP, anti-pY341-YAP, pY357-YAP, and anti-pY394 antibodies. (G) Western blot analyses of total proteins from αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes at 50% or 100% confluency treated with DMSO (−) or DAS.
Since Y357 is a known YAP1 tyrosine phosphorylation site, we analyzed whether it is hyperphosphorylated in αE-catenin−/− keratinocytes. Western blotting with phospho-Y357-specific YAP1 antibodies demonstrated a marked increase in Y357 phosphorylation in αE-catenin−/− keratinocytes (Supplemental Fig. S4A). As YAP1 and SRC are necessary for hyperproliferation of αE-catenin−/− keratinocytes (Fig. 1A), we hypothesized that SRC activates YAP1 in αE-catenin−/− cells by interacting with and phosphorylating YAP1 at Y357. To analyze whether phosphorylation of YAP1 at Y357 is necessary for hyperproliferation of αE-catenin−/− cells, we performed YAP1 knockdown rescue experiments (Supplemental Fig. S4B,B′). As expected, the knockdown of endogenous Yap1 prominently decreased accumulation of αE-catenin−/− keratinocytes, and this was rescued by overexpression of siRNA-resistant human YAP1; however, overexpression of the nonphosphorylatable Y357F YAP1 mutant was as efficient as re-expression of the wild-type YAP1 (Supplemental Fig. S4B,B′). We conclude that phosphorylation at Y357 is not responsible for SRC-mediated activation of YAP1 and hyperproliferation of αE-catenin−/− cells.
Since phosphorylation at Y357 cannot explain SRC-mediated activation of YAP1, we explored the possibility that SRC may directly phosphorylate YAP1 at other sites. To identify the SRC-dependent phosphorylation sites in YAP1, we analyzed the sequences of short and long isoforms of YAP1 (YAP1-1β and YAP1-2γ) for SRC consensus phosphorylation motifs and identified five potential sites (Fig. 5C,C′). We then generated Flag-tagged Y-to-F YAP1 mutants for each potential site and analyzed their tyrosine phosphorylation in HEK293 and mouse keratinocytes overexpressing CA-SRC (Fig. 5D,E). Mutation of Y341, Y357, or Y394 (Y391, Y407, and Y444 in the longer YAP1-2γ isoform) significantly decreased SRC-mediated tyrosine phosphorylation of YAP1, and the triple mutation Y341/357/394F (3YF) completely erased tyrosine phosphorylation of YAP1 in CA-SRC-expressing cells (Fig. 5D,E).
We next analyzed whether SRC can directly phosphorylate Y341/357/394 in YAP1. An in vitro kinase assay using purified CA-SRC and wild-type, Y341F, Y357F, Y394F, or triple-mutant 3YF YAP1 revealed a prominent SRC-dependent tyrosine phosphorylation of wild-type and, to a lesser degree, single-mutant YAP1 proteins but not the triple mutant 3YF YAP1 (Fig. 5F). Since phosphorylation of YAP1 at Y341 and Y394 has not been previously analyzed, we generated phospho-specific Y341 and Y394 YAP1 antibodies. These antibodies were highly specific for phospho-Y341 and phospho-Y394 because they recognized SRC phosphorylated wild-type but not the nonphosphorylated wild-type, the Y341F mutated, or the Y394F mutated YAP1 proteins (Fig. 5F). Western blot analysis of total protein lysates revealed a marked increase in Y341/357/394 YAP1 phosphorylation in confluent αE-catenin−/− keratinocytes, which is consistent with increased SRC activity in these cells (Fig. 5G). Overall, we conclude that SRC directly phosphorylates YAP1 at three sites (Y341/357/394) in its transcription activation domain, and YAP1 is hyperphosphorylated at these sites in αE-catenin−/− keratinocytes.
Phosphorylation of Y341/357/394 is necessary for YAP1 transcriptional activity, nuclear localization, and interaction with TEAD
To analyze the potential role of Y341/357/394 phosphorylation in the regulation of YAP1 transcriptional activity, we used luciferase assays using a YAP1/TEAD-responsive promoter. As expected, expression of wild-type YAP1 increased the activity of the promoter; however, expression of the 3YF mutant significantly attenuated YAP1 activity (Fig. 6A). In addition to the YAP1 3YF mutant, which cannot be phosphorylated by SRC, we also generated and analyzed a YAP1 Y341/357/394E (3YE) mutant, where substitution of Y to E may mimic the negative charge that is normally introduced by phosphorylation of Y341/357/394. It had been demonstrated previously that such substitution in other proteins can partially simulate Y phosphorylation, particularly when this phosphorylation alters the conformation of the target protein (Roura et al. 1999; Kassenbrock and Anderson 2004). The transcriptional activity of the YAP1 3YE mutant was similar to wild-type YAP1 (Fig. 6A). To determine whether Y341/357/394 phosphorylation is also necessary for the transcriptional activity of the 2SA YAP1 mutant, which is constitutively active in the canonical Hippo pathway, we generated 2SA+3YF and 2SA+3YE YAP1 mutants. We found that Y341/357/394 phosphorylation was necessary for transcriptional activity of the 2SA YAP1 mutant, indicating that tyrosine phosphorylation regulates YAP1 activity independently of the canonical Hippo pathway (Fig. 6A). S94 of YAP1 is necessary for interaction between YAP1 and TEAD, and the S94A mutant of YAP1 cannot bind TEAD (Li et al. 2010). As expected, the 2SA+S94A mutant of YAP1 completely failed to activate the reporter, confirming YAP1–TEAD complex specificity of our luciferase reporter assays. Similar data were obtained using qRT–PCR analyses of endogenous gene targets of YAP1 (Supplemental Fig. S5A). Overall, we conclude that phosphorylation of the transcriptional activation domain of YAP1 at Y341/357/394 is crucial for YAP1 transcriptional activity.
Figure 6.

Functional significance and mechanisms of Y341/357/394 YAP1 phosphorylation. (A,B) Y341/357/394 (3Y) YAP1 phosphorylation is necessary for YAP1 transcriptional activity. Luciferase assay using empty control (Ctrl-LUC) or the TEAD/YAP1 8xGTIIC reporter in αE-catenin−/− keratinocytes stably transduced with empty vector (Ctrl) or the indicated human YAP1 (Flag-hYAP) constructs and also transfected with control (siCtrl) or mouse YAP1 (simYAP1) siRNA oligos. The graph shows mean values ± SD. Student's t-test. (B) Y341/357/394 (3Y) YAP1 phosphorylation is necessary for increased YAP1 activity in αE-catenin−/− keratinocytes. Luciferase assay similar to A using αE-cateninfl/fl (Ctrl) or αE-catenin−/− (α-cat KO) keratinocytes stably expressing the indicated human YAP1 constructs and transiently transfected with negative control (siCtrl) or mouse YAP1-specific (si-YAP) oligos. The graph shows mean values ± SD. Student's t-test. (C,C′) Y341/357/394 (3Y) YAP1 phosphorylation is necessary for nuclear localization of wild-type (WT) but not 2SA mutant YAP1. Western blot analyses and quantitation of proteins from nuclear and cytoplasmic fractions of αE-catenin−/− keratinocytes stably expressing indicated YAP1 constructs. Bar graph, mean values ± SD. n = 3. Student's t-test. (D,D′) Y341/357/394 (3Y) YAP1 phosphorylation is necessary for interaction with TEAD of both wild-type and 2SA mutant YAP1. Western blot analyses and quantitation of total (Input) or anti-Flag antibody immunoprecipitated (Flag IP) proteins from αE-catenin−/− keratinocytes stably expressing the indicated YAP1 constructs. The bar graph shows mean values ± SD. n = 3. Student's t-test. (E) Y341/357/394 (3Y) YAP1 phosphorylation is necessary for hyperproliferation of αE-catenin−/− keratinocytes. Cell counts of αE-catenin−/− keratinocytes stably expressing vector control (Ctrl), wild-type (WT), or the indicated Y and/or S mutant human YAP1 proteins transfected with control (siCtrl) or mouse YAP1-specific (siYAP1) oligos, plated at high confluency, and cultured for 5 d. The bar graph shows mean values ± SD. Student's t-test. (E′) Western blot (WB) analyses of total proteins from cells described in E. (F,G′) Y341/357/394 (3Y) YAP1 phosphorylation is necessary for CA-SRC–YAP1-mediated transformation of primary keratinocytes. Orthotopic allograft tumors generated in Nude mice by keratinocytes stably expressing the indicated YAP1 and CA-SRC constructs. Graphs in F′ and G′ show quantitation of tumor volumes in F and G, respectively. Tumors were analyzed 6 wk after cell injection. Data represent means ± SE. Unpaired t-test.
To determine whether Y341/357/394 phosphorylation is responsible for increased YAP1 activity in αE-catenin−/− keratinocytes, we performed luciferase transcriptional activity assays using YAP1 knockdown rescue experiments in both αE-cateninfl/fl and αE-catenin−/− keratinocytes. As expected, the activity of the YAP1 reporter was significantly higher in αE-catenin−/− cells than in αE-cateninfl/fl cells (Fig. 6B). The knockdown of endogenous Yap1 erased the differences in YAP1 transcriptional activity between these cells, and this was rescued by overexpression of siRNA-resistant human YAP1; however, overexpression of the nonphosphorylatable 3YF YAP1 mutant failed to rescue the differences in YAP1 transcriptional activity between the αE-catenin−/− and αE-cateninfl/fl keratinocytes (Fig. 6B). Thus, we conclude that YAP1 Y341/357/394 phosphorylation is necessary for the increased transcriptional activity of YAP1 in αE-catenin−/− cells.
We found previously that activation of SRC increases nuclear localization of YAP1 (Fig. 3A,B). Thus, we analyzed the role of Y341/357/394 phosphorylation in YAP1 localization. Expression of 3YF mutant YAP1 significantly decreased YAP1 nuclear localization (Fig. 6C-C′). In contrast, the 3YF mutation had no effect on nuclear localization of the 2SA mutant, which is consistent with the known function of S127 phosphorylation in YAP1 interaction with 14-3-3 and its cytoplasmic retention (Zhao et al. 2007). Since TEAD proteins play a critical role in the transcriptional output of the Hippo pathway (Wu et al. 2008; Zhao et al. 2008), we analyzed whether Y341/357/394 phosphorylation has an impact on the interaction between YAP1 and TEAD. As expected, coimmunoprecipitation experiments revealed interaction between wild-type YAP1 and TEAD4; however, this interaction was significantly attenuated in the 3YF YAP1 mutant (Fig. 6D,D′; Supplemental Fig. S5B). A similar effect was observed in experiments with the 2SA YAP1 mutant, which is constitutively active in the canonical Hippo pathway (Fig. 6D,D′; Supplemental Fig. S5C). Overall, we conclude that Y341/357/394 phosphorylation promotes nuclear localization of YAP1 and enhances the interaction between YAP1 and TEAD proteins.
Phosphorylation of Y341/357/394 of YAP1 is necessary for hyperproliferation of αE-catenin−/− cells and SRC–YAP1-mediated transformation of normal keratinocytes
To analyze whether phosphorylation of YAP1 at Y341/357/394 is necessary for hyperproliferation of αE-catenin−/− cells, we performed YAP1 knockdown rescue experiments. The knockdown of endogenous Yap1 decreased accumulation of αE-catenin−/− keratinocytes, and this phenotype was rescued by overexpression of siRNA-resistant human YAP1; however, overexpression of the nonphosphorylatable 3YF YAP1 mutant failed to completely rescue the endogenous Yap1 knockdown phenotype, while the 3YE mutant was as efficient as the wild-type YAP1 (Fig. 6E,E′; Supplemental Fig. S5D,D′). Similar results were observed in experiments with 2SA YAP1 mutants that are constitutively active in the Hippo pathway (Fig. 6E,E′). The 2SA+S94A YAP1 mutant failed to rescue the endogenous Yap1 knockdown phenotype, indicating a critical role of the YAP1–TEAD interaction for hyperproliferation of αE-catenin−/− cells. We conclude that phosphorylation of the transcriptional activation domain of YAP1 at Y341/357/394 is functionally important for the hyperproliferative phenotype of αE-catenin−/− keratinocytes.
SRC is the first discovered oncogene, and it functions as a tyrosine kinase (Collett et al. 1980); however, while many important SRC substrates have been found throughout the last two and a half decades, a SRC phosphorylation substrate that is pivotal for cellular transformation has not been identified. We hypothesized that SRC-mediated phosphorylation and activation of YAP1 may directly connect SRC with gene transcription and play an important role in SRC-mediated transformation. We used the orthotopic allograft SCC tumor formation model to investigate the role of YAP1 tyrosine phosphorylation in the SRC–YAP1-mediated transformation of primary keratinocytes. Consistent with our previous findings, the expression of CA-SRC and wild-type YAP1 transformed keratinocytes; however, the expression of the 3YF YAP1 mutant significantly attenuated the tumor formation phenotype (Fig. 6F-F′; Supplemental Fig. S5E). The 3YE YAP1 mutant was as efficient as wild-type YAP1. We conclude that tyrosine phosphorylation of YAP1 at Y341/357/394 plays an important role in SRC–YAP1-mediated cellular transformation.
To analyze whether tyrosine phosphorylation of YAP1 at Y341/357/394 continues to play an important role in SRC-mediated transformation when YAP1 is already fully activated in the canonical Hippo pathway, we repeated experiments with CA-SRC+YAP1-mediated transformation but used the 2SA mutant YAP1, which cannot be negatively regulated by Hippo signaling. As expected, the coexpression of CA-SRC with 2SA YAP1 transformed primary keratinocytes (Fig. 6G). Importantly, the expression of 2SA–3YF YAP1 was significantly less efficient in transformation than the 2SA YAP1 mutant (Fig. 6G-G′). These findings indicate that phosphorylation of Y341/357/394 plays an important role in SRC-mediated transformation even when YAP1 cannot be attenuated by the canonical Hippo signaling pathway.
Prominent tyrosine phosphorylation of YAP1 in mouse and human SCC and the potential therapeutic significance of SRC–YAP1 inhibition in these tumors
Since we found that αE-catenin negatively regulates the ITGB4–SRC–YAP1(pY341/357/394) oncogenic pathway in keratinocytes, we decided to analyze tyrosine phosphorylation of YAP1 in SCC tumors and adjacent noninvolved skin in GFAP-Cre/αE-cateninfl/fl/TP53fl/fl mice (Silvis et al. 2011). Western blot analysis revealed a marked increase in the activity of SRC kinases (pY416-SRC) and Y341/357/394 phosphorylation of YAP1 in these tumors (Fig. 7A,A′). Similar analysis of human skin SCC tumors revealed a significant correlation between low αE-catenin expression and tyrosine phosphorylation of YAP1 in human cancer (Fig. 7B,B′). We conclude that extensive phosphorylation of YAP1 at Y341/357/394 takes place in both human and mouse autochthonous SCC tumors with low or absent αE-catenin.
Figure 7.

Prominent Y341/357/394 YAP1 phosphorylation in mouse and human SCCs and the potential therapeutic significance of SRC–YAP1 pathway inhibition in these tumors. (A) Western blot (WB) analyses of total proteins extracted from primary SCC tumors and control uninvolved skin of GFAP/αE-cateninfl/fl/P53fl/fl mice using the indicated antibodies. Note that αE-catenin is deleted in hair follicle stem cells but is present in the epidermis of GFAP/αE-cateninfl/fl/P53fl/fl mice. All tumors lose αE-catenin; however, excised skin tumors were often contaminated by skin epidermis expressing αE-catenin. (A′) Quantitation of the data in A. The bar graphs show mean values ± SE. Student's t-test. (B) Western blot analyses for total proteins extracted from human normal skin (Ctrl) and primary skin SCC tumor samples using the indicated antibodies. (B′) Quantitation of data in B. Human skin SCC samples were separated into two groups with low (4–6) and high (7–10) levels of αE-catenin expression. The graphs show mean values ± SE. Student's t-test. (C) MTT cell number assay of SCC cells from GFAP/αE-cateninfl/fl/P53fl/fl mice stably transduced with doxycycline (DOX)-inducible control shLUC or shYAP1 constructs, plated at high density, and cultured for 5 d with and without doxycycline. The graph shows mean values ± SD. Student's t-test. (C′) Western blot analyses of total proteins from the cells used in C with the indicated antibodies. (C′′) Orthotopic tumor formation assay of the SCC cells described in C using NOD-SCID mice. Mice were exposed to doxycycline-containing food for 45 d, and the resulting tumors were excised and analyzed. (C′′′) Quantitation of tumor volumes in C′′. Data represent means ± SE. Unpaired t-test. (D) Western blot analyses of total proteins from SCC cells isolated from tumors in GFAP/αE-cateninfl/fl/P53fl/fl mice incubated with indicated concentrations of DAS. (D′) Orthotopic tumor formation assay of the SCC cells described in D using NOD-SCID mice. Mice were exposed to food with or without 70 mg/kg DAS for 45 d, and the resulting tumors were excised and analyzed. (D′′) Quantitation of tumor volumes in D′. Data represent means ± SE. Unpaired t-test. (E) Model of canonical Hippo and inhibited by αE-catenin SRC–YAP1 signal transduction pathways.
To explore the potential therapeutic significance of the connection between loss of αE-catenin, Y341/357/394 YAP1 phosphorylation, and the development of SCC tumors, we established an orthotopic allograft SCC tumor model. For this purpose, we generated cells line from primary SCC tumors that formed in GFAP-Cre/αE-cateninfl/fl/p53fl/fl mice. To determine whether YAP1 is a valid therapeutic target in these tumors, SCC cells were transduced with either a doxycycline-inducible shLUC control or a shYAP1 retroviral construct and analyzed in orthotopic allograft tumor formation assays. While shLUC control cells injected intradermally into NOD-SCID mice generated large SCC tumors, the knockdown of endogenous YAP1 in these SCC cells strongly inhibited tumor growth, indicating that YAP1 is necessary for the survival of SCC cells (Fig. 7C–C′′′; Supplemental Fig. S6).
Since we previously found that the ABL/SFK inhibitor DAS completely blocked Y341/357/394 phosphorylation of YAP1, which is necessary for YAP1 transforming activity, we analyzed whether αE-catenin−/−/p53−/− SCC cells are sensitive to DAS treatment. We found that a dose-dependent decrease in SRC activity in DAS-treated cells coincided with a decrease in tyrosine phosphorylation of YAP1 (Fig. 7D). To analyze whether αE-catenin−/−/p53−/− SCC tumors are sensitive to DAS treatment in vivo, we performed orthotopic allograft experiments. Mice injected intradermally with αE-catenin−/−/p53−/− SCC keratinocytes were randomly separated into two groups and fed with either control or DAS-containing food. We observed prominent inhibition of tumor growth in mice treated with DAS (Fig. 7D′,D′′). Thus, SRC family tyrosine kinase activity is required for tumorigenesis of αE-catenin−/−/p53−/− SCC cells, and DAS treatment may be a very effective tool in the treatment of SCC tumors displaying down-regulation of endogenous αE-catenin.
Discussion
In this study, we demonstrate that αE-catenin negatively regulates the signaling activity of β4 integrin, which in turn activates SRC, leading to Y341/357/394 phosphorylation and activation of YAP1 in αE-catenin−/− cells. These data provide a molecular mechanism for the tumor-suppressing function of αE-catenin and reveal an alternative to the canonical Hippo signaling pathway that connects oncogenic tyrosine kinase signaling with the transcriptional output of the Hippo pathway (Fig. 7E).
Molecular mechanism of αE-catenin-mediated tumor suppression
αE-catenin is frequently down-regulated in human skin SCC tumors, and conditional deletion of αE-catenin in mouse hair follicle stem cells promotes the development of skin SCC, strongly implicating αE-catenin as a tumor suppressor in this cancer type (Silvis et al. 2011).
αE-catenin may have multiple mechanisms responsible for its tumor suppressor function. They include negative regulation of β-catenin signaling (Gottardi and Gumbiner 2004), YAP1 signaling (Varelas et al. 2010; Kim et al. 2011; Schlegelmilch et al. 2011; Silvis et al. 2011; Benham-Pyle et al. 2015), FAK–PAK signaling (Livshits et al. 2012), and the NFkB pathway (Kobielak and Fuchs 2006; Piao et al. 2014). While attenuation of β-catenin signaling is a plausible mechanism of αE-catenin tumor suppressor activity, the in vivo analyses of potential changes in β-catenin transcriptional activity did not identify a significant impact on β-catenin signaling in αE-catenin−/− tissues (Vasioukhin et al. 2001; Lien et al. 2008b). Since genetic analysis revealed the tumor suppressor function of αE-catenin in skin SCC, we previously focused on the role of αE-catenin in keratinocytes. Unlike normal primary keratinocytes, αE-catenin−/− keratinocytes do not withdraw from the cell cycle upon reaching confluency (Vasioukhin et al. 2001). The siRNA screen demonstrated that β-catenin is not required for hyperproliferation of αE-catenin−/− cells, indicating the involvement of a β-catenin-independent mechanism (Silvis et al. 2011). Subsequent analysis identified Yap1 as a gene necessary for hyperproliferation of αE-catenin−/− cells and demonstrated an αE-catenin-dependent inhibition of nuclear YAP1 localization in confluent epithelial cells. Knockdown-mediated depletion of αE-catenin revealed a similar connection between αE-catenin and YAP1 in a variety of cell lines (Varelas et al. 2010; Kim et al. 2011; Schlegelmilch et al. 2011).
Analysis of the mechanisms responsible for αE-catenin-dependent regulation of YAP1 uncovered significant differences between cell types. In the mammary epithelial cell line MCF10A, αE-catenin as well as both β-catenin and E-cadherin prominently regulated MST1/2-LATS1/2 kinases, indicating that αE-catenin affects the canonical Hippo signaling pathway (Kim et al. 2011). In contrast, while αE-catenin was a prominent regulator of YAP1, it did not affect MST1/2 or LATS1/2 activities in epidermal keratinocytes (Schlegelmilch et al. 2011; Silvis et al. 2011). In HaCaT keratinocytes, αE-catenin binds to YAP1 and prevents its interaction with PP2A, which can dephosphorylate S127 and therefore activate YAP1 (Schlegelmilch et al. 2011). In contrast, loss of αE-catenin from primary human and mouse keratinocytes not only failed to cause inactivation of MST1/2-LATS1/2 kinases but also did not affect YAP1 phosphorylation at LATS1/2 sites, indicating that an alternative mechanism must be involved in YAP1 activation in these cells (Silvis et al. 2011). In this study, we reveal a distinct mechanism of YAP1 regulation that involves SRC-mediated phosphorylation. We show that this mechanism is primarily responsible for αE-catenin-mediated control of YAP1 localization and activity in keratinocytes and skin SCC cells. Identification of SRC–YAP1 complexes as a critical growth-promoting and potentially oncogenic module negatively regulated by αE-catenin has not only a general scientific interest but also a substantial therapeutic significance. Our findings suggest that DAS administration as well as other modalities inhibiting SFKs may prove to be very effective interventions for the treatment of SCC tumors displaying inactivation of αE-catenin.
While our results demonstrate a negative role of αE-catenin in the regulation of β4 integrin signaling to SRC, it is presently not clear whether there is a direct connection between αE-catenin and β4 integrin or this is an indirect route of activation due to general disruption of cell polarity in αE-catenin−/− cells (Vasioukhin et al. 2001; Nemade et al. 2004). Unlike MCF10A cells, which do not form tight junctions and completely depend on E-cadherin/β-catenin protein complexes for cell–cell adhesion, epidermal keratinocytes maintain adhesion upon the knockdown of E-cadherin and β-catenin. Therefore, the lack of YAP1 activation in E-cadherin and β-catenin knockdown keratinocytes does not necessarily indicate that αE-catenin-mediated regulation of YAP1 is independent from its function in the AJs. Significant literature indicates that loss of proper cell–cell adhesion results in abnormal activation of various receptor type tyrosine kinases (RTKs), and the inactivation of RTK signaling in confluent cells is primarily responsible for contact inhibition of cell proliferation (Vermeer et al. 2003; Qian et al. 2004; McClatchey and Yap 2012). SFKs are important downstream targets of various oncogenic RTKs (Muthuswamy 2011), and an increase in RTK signaling mediated by the loss of cell–cell adhesion may be responsible for hyperactivation of SRC and subsequent phosphorylation and activation of YAP1 in αE-catenin−/− keratinocytes.
The SRC–YAP1 oncogenic module and the noncanonical YAP1 signal transduction pathway
We show here that SRC is a potent activator of YAP1. In the canonical Hippo pathway, MST1/2 kinases phosphorylate and activate LATS1/2, which in turn phosphorylate and inactivate YAP1 (Pan 2010; Yu and Guan 2013). This pathway is activated in confluent cells, where it suppresses YAP1 nuclear localization and thereby inhibits its transcriptional activity (Zhao et al. 2007). It had been demonstrated previously that SRC and other SFKs can positively (Shanzer et al. 2015) and negatively regulate canonical Hippo signaling pathway (Enomoto and Igaki 2013; Pijuan-Galito et al. 2014; Kim and Gumbiner 2015; Kwon et al. 2015). In accordance with these findings, we observed negative regulation of S127 YAP1 phosphorylation in sparse keratinocytes (Supplemental Fig. S2D). In contrast, in confluent keratinocytes, when Hippo signaling is activated, SRC promotes nuclear YAP1 localization without impacting the canonical Hippo pathway (Fig. 3C,D). Instead, it functions by directly phosphorylating YAP1 at three sites in its transcription activation domain, and this is necessary for YAP1 activation. Prominent phosphorylation of endogenous YAP1 at these sites in primary SCC tumors strongly suggests an important role of this event in tumorigenesis.
We found that SRC is the principal SFK that phosphorylates YAP1 in αE-catenin−/− keratinocytes; however, it is likely that other SFKs are also involved in a similar regulation of YAP1. Indeed, YAP1 was cloned as an interacting partner of the SRC family tyrosine kinase YES1 (Sudol et al. 1995). In colon cancer cells driven by constitutively active β-catenin, YES1 phosphorylates YAP1 at Y357, and this phosphorylation uses an unknown mechanism to promote the transcriptional activity of YES1–YAP1–β-catenin–Tbx5 protein complexes without impacting their integrity (Rosenbluh et al. 2012). Tbx5 is not expressed in keratinocytes, and the knockdown of both Tbx5 and β-catenin does not impact hyperproliferation of αE-catenin−/− keratinocytes, indicating that SRC-mediated YAP1 phosphorylation promotes YAP1 activity via a distinct mechanism.
Activation of the gp130–SFK pathway in the intestinal epithelium results in increased tyrosine phosphorylation and activation of YAP1, and treatment with the SFK-specific inhibitor erases YAP1 phosphorylation and suppresses its nuclear localization in intestinal organoids with constitutively active gp130 (Taniguchi et al. 2015). While the significance of YAP1 tyrosine phosphorylation was not directly analyzed in that study, it is likely that, similar to our findings in keratinocytes, the direct phosphorylation of YAP1 by SFK at Y341/357/394 is responsible for YAP1 activation in this model system.
SRC is the first cloned oncogene, and significant literature implicates SRC in human cancer, especially in skin SCC (Matsumoto et al. 2003; Lee et al. 2010; Sudol 2011). Nevertheless, the mechanisms of SRC oncogenic activity are still poorly understood, as the essential SRC phosphorylation targets responsible for cellular transformation have not been identified. Our findings suggest that YAP1 is a strong candidate for such a downstream phosphorylation target. Additional work will be necessary to determine whether the Y341/357/394 YAP1 phosphorylation identified here is responsible for cellular transformation induced by constitutive activation of other oncogenic tyrosine kinases.
Materials and methods
Cell culture, siRNA transfections, luciferase assays, and cell fractionation
Primary mouse keratinocytes were isolated and cultured as described (Wang et al. 1997). Cells were first established in low-calcium E-medium and later adapted and maintained in regular-calcium E-medium (Rheinwald and Green 1975). αE-catenin−/− cells were generated as described (Silvis et al. 2011). αE-catenin−/−/p53−/− mouse SCC cells were isolated from GFAP-Cre/αE-cateninfl/fl/p53fl/fl mouse tumors as described above. HEK293FT cells were maintained in DMEM (Gibco) supplemented with glutamine, sodium pyruvate (HyClone), nonessential amino acids, penicillin–streptomycin, and 10% FBS. For siRNA experiments, 1 × 104 cells were seeded directly into a siRNA-Lipofectamine 2000 (Invitrogen) mixture on a 96-well plate and cultured for 5 d. The medium was changed daily, and the relative cell number was determined by MTT assay (Promega) or direct cell count. siRNA oligos were from Qiagen (5 pmol per well). Keratinocytes were selected in 200 µg/mL G418, 150 µg/mL hygromycin B, or 2 µg/mL puromycin. DAS was purchased from LC Laboratories. Luciferase assays were performed as described (Silvis et al. 2011). Cytoplasmic and nuclear fractionations were obtained using NE-PER nuclear and cytoplasmic extraction (Pierce).
Allograft tumor formation assays
All procedures involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) of the Fred Hutchinson Cancer Research Center. For the tumor formation assays in NOD-SCID mice (Jackson Laboratories, catalog no. 001303), 2 × 106 keratinocytes in PBS were injected intradermally or subcutaneously into anesthetized mice. To determine the sensitivity of tumors to DAS, mice with injected SCC cells were exposed for 45 d to a soft transgenic dough diet (Bio-Serv, S3472) or the same food mixed with 70 mg/kg DAS (LC Laboratories). For the tumor formation assays in Nude mice (Jackson Laboratories, catalog no. 007850), 2 × 106 keratinocytes were resuspended in F medium (DMEM with 10% FBS) and mixed 1:1 with Matrigel (BD Biosciences, catalog no. 356237), and 50 µL of the mixture was injected intradermally into anesthetized mice.
Data analysis
Statistical significance was determined by the unpaired Student's t-test. The P-value is indicated by asterisks in the figures (P < 0.05 [*], P < 0.01 [**], and P < 0.001 [***]). Differences of P < 0.05 and lower were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. J.A. Cooper and Dr. L. Trusolino for the gift of CA-SRC and β4 integrin plasmids, and Dr. Slobodan Beronja for help with intradermal cell injections. This work was supported by National Cancer Institute grants CA188452 and CA179914 (to V.V.) and National Institutes of Health training fellowship grants from T32 CA09657 (to M.R.S. and Y.H.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.274951.115.
References
- Barry ER, Camargo FD. 2013. The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr Opin Cell Biol 25: 247–253. [DOI] [PubMed] [Google Scholar]
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J. 2003. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell 11: 11–23. [DOI] [PubMed] [Google Scholar]
- Benham-Pyle BW, Pruitt BL, Nelson WJ. 2015. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and β-catenin activation to drive cell cycle entry. Science 348: 1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin JM, Nelson WJ. 2008. Bench to bedside and back again: molecular mechanisms of α-catenin function and roles in tumorigenesis. Semin Cancer Biol 18: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A, Comoglio PM, Trusolino L. 2006. β4 integrin activates a Shp2–Src signaling pathway that sustains HGF-induced anchorage-independent growth. J Cell Biol 175: 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ, Dunn AR. 2014. Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science 346: 1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullions LC, Notterman DA, Chung LS, Levine AJ. 1997. Expression of wild-type α-catenin protein in cells with a mutant α-catenin gene restores both growth regulation and tumor suppressor activities. Mol Cell Biol 17: 4501–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett MS, Purchio AF, Erikson RL. 1980. Avian sarcoma virus-transforming protein, pp60src shows protein kinase activity specific for tyrosine. Nature 285: 167–169. [DOI] [PubMed] [Google Scholar]
- Collins C, Nelson WJ. 2015. Running with neighbors: coordinating cell migration and cell–cell adhesion. Curr Opin Cell Biol 36: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enderle L, McNeill H. 2013. Hippo gains weight: added insights and complexity to pathway control. Sci Signal 6: re7. [DOI] [PubMed] [Google Scholar]
- Enomoto M, Igaki T. 2013. Src controls tumorigenesis via JNK-dependent regulation of the Hippo pathway in Drosophila. EMBO Rep 14: 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing CM, Ru N, Morton RA, Robinson JC, Wheelock MJ, Johnson KR, Barrett JC, Isaacs WB. 1995. Chromosome 5 suppresses tumorigenicity of PC3 prostate cancer cells: correlation with re-expression of α-catenin and restoration of E-cadherin function. Cancer Res 55: 4813–4817. [PubMed] [Google Scholar]
- Gottardi CJ, Gumbiner BM. 2004. Distinct molecular forms of β-catenin are targeted to adhesive or transcriptional complexes. J Cell Biol 167: 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassenbrock CK, Anderson SM. 2004. Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J Biol Chem 279: 28017–28027. [DOI] [PubMed] [Google Scholar]
- Kim NG, Gumbiner BM. 2015. Adhesion to fibronectin regulates Hippo signaling via the FAK–Src–PI3K pathway. J Cell Biol 210: 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NG, Koh E, Chen X, Gumbiner BM. 2011. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci 108: 11930–11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klezovitch O, Vasioukhin V. 2015. Cadherin signaling: keeping cells in touch. F1000Res 4: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak A, Fuchs E. 2006. Links between α-catenin, NF-κB, and squamous cell carcinoma in skin. Proc Natl Acad Sci 103: 2322–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro A, Nagai M, Navin NE, Sudol M. 2003. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem 278: 33334–33341. [DOI] [PubMed] [Google Scholar]
- Kwon HJ, Waghmare I, Verghese S, Singh A, Singh A, Kango-Singh M. 2015. Drosophila C-terminal Src kinase regulates growth via the Hippo signaling pathway. Dev Biol 397: 67–76. [DOI] [PubMed] [Google Scholar]
- Lee JH, Pyon JK, Kim DW, Lee SH, Nam HS, Kim CH, Kang SG, Lee YJ, Park MY, Jeong DJ, et al. 2010. Elevated c-Src and c-Yes expression in malignant skin cancers. J Exp Clin Cancer Res 29: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Adamovich Y, Reuven N, Shaul Y. 2008. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell 29: 350–361. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, Xu Y. 2010. Structural insights into the YAP and TEAD complex. Genes Dev 24: 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Gelfand VI, Vasioukhin V. 2008a. αE-catenin binds to dynamitin and regulates dynactin-mediated intracellular traffic. J Cell Biol 183: 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Null M, Vasioukhin V. 2008b. αE-catenin is not a significant regulator of β-catenin signaling in the developing mammalian brain. J Cell Sci 121: 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livshits G, Kobielak A, Fuchs E. 2012. Governing epidermal homeostasis by coupling cell–cell adhesion to integrin and growth factor signaling, proliferation, and apoptosis. Proc Natl Acad Sci 109: 4886–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Jiang J, Kiguchi K, Ruffino L, Carbajal S, Beltran L, Bol DK, Rosenberg MP, DiGiovanni J. 2003. Targeted expression of c-Src in epidermal basal cells leads to enhanced skin tumor promotion, malignant progression, and metastasis. Cancer Res 63: 4819–4828. [PubMed] [Google Scholar]
- McClatchey AI, Yap AS. 2012. Contact inhibition (of proliferation) redux. Curr Opin Cell Biol 24: 685–694. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK. 2011. Trastuzumab resistance: all roads lead to SRC. Nat Med 17: 416–418. [DOI] [PubMed] [Google Scholar]
- Nemade RV, Bierie B, Nozawa M, Bry C, Smith GH, Vasioukhin V, Fuchs E, Hennighausen L. 2004. Biogenesis and function of mouse mammary epithelium depends on the presence of functional α-catenin. Mech Dev 121: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. 2010. The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao HL, Yuan Y, Wang M, Sun Y, Liang H, Ma L. 2014. α-Catenin acts as a tumour suppressor in E-cadherin-negative basal-like breast cancer by inhibiting NF-κB signalling. Nat Cell Biol 16: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Galito S, Tamm C, Anneren C. 2014. Serum inter-α-inhibitor activates the Yes tyrosine kinase and YAP/TEAD transcriptional complex in mouse embryonic stem cells. J Biol Chem 289: 33492–33502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwnica-Worms H, Saunders KB, Roberts TM, Smith AE, Cheng SH. 1987. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell 49: 75–82. [DOI] [PubMed] [Google Scholar]
- Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. 2004. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J 23: 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinwald JG, Green H. 1975. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6: 331–343. [DOI] [PubMed] [Google Scholar]
- Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, et al. 2012. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151: 1457–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. 1999. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J Biol Chem 274: 36734–36740. [DOI] [PubMed] [Google Scholar]
- Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, et al. 2011. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 144: 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanzer M, Ricardo-Lax I, Keshet R, Reuven N, Shaul Y. 2015. The polyomavirus middle T-antigen oncogene activates the Hippo pathway tumor suppressor Lats in a Src-dependent manner. Oncogene 34: 4190–4198. [DOI] [PubMed] [Google Scholar]
- Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V. 2011. α-Catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal 4: ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniak E, Radice GL, Vasioukhin V. 2009. Adhesive and signaling functions of cadherins and catenins in vertebrate development. Cold Spring Harb Perspect Biol 1: a002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudol M. 1994. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 9: 2145–2152. [PubMed] [Google Scholar]
- Sudol M. 2011. From Rous sarcoma virus to plasminogen activator, src oncogene and cancer management. Oncogene 30: 3003–3010. [DOI] [PubMed] [Google Scholar]
- Sudol M, Bork P, Einbond A, Kastury K, Druck T, Negrini M, Huebner K, Lehman D. 1995. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem 270: 14733–14741. [DOI] [PubMed] [Google Scholar]
- Tamm C, Bower N, Anneren C. 2011. Regulation of mouse embryonic stem cell self-renewal by a Yes–YAP–TEAD2 signaling pathway downstream of LIF. J Cell Sci 124: 1136–1144. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. 2015. A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 519: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL. 2010. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β–SMAD pathway. Dev Cell 19: 831–844. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V. 2012. Adherens junctions and cancer. Subcell Biochem 60: 379–414. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. 2001. Hyperproliferation and defects in epithelial polarity upon conditional ablation of α-catenin in skin. Cell 104: 605–617. [DOI] [PubMed] [Google Scholar]
- Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, Welsh MJ. 2003. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 422: 322–326. [DOI] [PubMed] [Google Scholar]
- Wang X, Zinkel S, Polonsky K, Fuchs E. 1997. Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proc Natl Acad Sci 94: 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Liu Y, Zheng Y, Dong J, Pan D. 2008. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell 14: 388–398. [DOI] [PubMed] [Google Scholar]
- Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. 1999. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J 18: 2551–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Guan KL. 2013. The Hippo pathway: regulators and regulations. Genes Dev 27: 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Sullivan AJ, Medina R, Ito Y, van Wijnen AJ, Stein JL, Lian JB, Stein GS. 2004. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J 23: 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22: 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.