

SUMMARY
The hair follicle (HF) is a complex miniorgan that serves as an ideal model system to study stem cell (SC) interactions with the niche during growth and regeneration. Dermal papilla (DP) cells are required for SC activation during the adult hair cycle, but signal exchange between niche and SC precursors/transit amplifying progenitor cells (TACs) that regulates HF morphogenetic growth is less explored. Here we use six transgenic reporters to isolate 14 major skin and HF cell populations. With next-generation RNA sequencing we characterize their transcriptomes and define unique molecular signatures. SC precursors, TACs and the DP niche express a plethora of ligands and receptors. Signaling interaction network analysis reveals a birds-eye view of pathways implicated in epithelial-mesenchymal interactions. Using a systematic tissue-wide approach this work provides a comprehensive platform, linked to an interactive online database, to identify and further explore the SC/TAC/niche crosstalk regulating HF growth.
Keywords: hair follicle, hair growth, progenitors, dermal papilla, stem cells, stem cell niche, epithelial-mesenchymal interactions, transcriptomics, systems biology, big data, signaling networks
Graphical Abstract
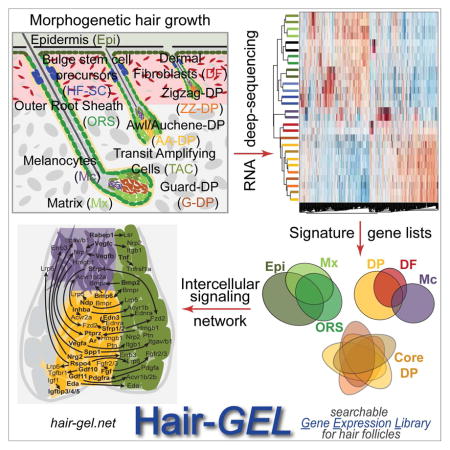
INTRODUCTION
Embryonic hair follicle (HF) formation, hair growth after birth, and regulation of the adult hair cycle involve complex signaling interactions between epithelial stem cells (SCs) and progenitors, and a dermal specialized niche compartment, the dermal papilla (DP) (Lee and Tumbar, 2012; Rezza et al., 2014; Rompolas and Greco, 2014; Sennett and Rendl, 2012). At the end of the resting phase of the hair cycle, DP cells signal to bulge/germ SCs to activate new HF growth, and recent ligand supplementation experiments and receptor ablation studies in the SCs indicate an important role for DP-derived TGFB2, FGF7 and inhibitory BMP signals (Greco et al., 2009; Kobielak et al., 2003; Oshimori and Fuchs, 2012). Laser-mediated ablation established the absolute requirement of these cells for SC activation during the hair cycle (Rompolas et al., 2012).
Similarly, during hair growth, DP cells are thought to act as a core signaling center for surrounding epithelial progenitors (transit amplifying cells or TACs) within the wider matrix (Mx) compartment that proliferate, migrate upwards, and differentiate into the multiple layers of the hair shaft and the inner root sheath channel (Hsu et al., 2014a). The outer root sheath (ORS) lines the epithelial HF compartment, is contiguous with the Mx and epidermis, and contains the SC precursors of the adult HF bulge during early hair growth (Schlake, 2007). FGF signals from the DP have been implicated in controlling hair growth (Petiot et al., 2003) while BMP and WNT signaling play an important role in hair shaft progenitor differentiation, but the precise source of BMP and WNT ligands is unclear (DasGupta and Fuchs, 1999; Kobielak et al., 2003). SHH is produced by a subpopulation of TAC progenitors that reside right next to the DP compartment (Gambardella et al., 2000; Hsu et al., 2014b). It is still unclear if a broader requirement for TAC-derived signals interacting with the DP niche exists, as pure TAC cells of growing HFs have not been isolated and characterized. Finally, the third major cellular component in the HF bulb are melanocytes (Mc) that provide pigment to the epithelial cells and are thought to receive regulatory signals from the DP niche (Enshell-Seijffers et al., 2008; Enshell-Seijffers et al., 2010). Whether Mc signal with TAC progenitors and Mx cells is currently unclear.
Previous studies have tried to identify signals involved in driving HF growth using a global transcriptomic approach (Driskell et al., 2009; Rendl et al., 2005). Isolation of HF cell populations during the morphogenetic growth phase and subsequent gene expression analysis with early microarrays identified enriched genes for the DP, as well as for Mc, Mx and ORS cells (Rendl et al., 2005). Although this study provided important insights into the molecular composition of major HF cell types, it was not yet possible to distinguish TAC progenitors from Mx, HFSC precursors from the remaining ORS, or hair type-specific DP subpopulations. Indeed, during early postnatal hair growth four hair types can be recognized that form in three embryonic developmental waves and have different HF lengths within the skin (Figure 1A) as well as different hair shaft sizes, kinks and bends externally (Schlake, 2007). Guard (G) HFs which develop first, are the largest, and extend deepest in the dermis. Awl and Auchene (AA) follicles develop subsequently, are thinner and reach the lower part of the reticular dermis. Zigzag (ZZ) HFs appear last and are ultimately the shortest. While these morphological and developmental idiosyncrasies have been noted for years, it is unclear if hair type-specific DP subpopulations are molecularly distinct and control regulation of hair type sizes and shapes. In previous work, ZZ-enriched DP cells were isolated and ZZ-enriched DP genes were described with a possible role in controlling ZZ hair type (Driskell et al., 2009). Conversely, another recent study manipulated the total number of DP cells per HF resulting in hair type switching, suggesting that the cumulative signaling output from the niche determines hair type rather than its intrinsic molecular features (Chi et al., 2013).
Figure 1. Multicolor cell sorting of stem cell precursors, transit amplifying progenitors, and their niche from developing hair follicles.

(A) Schematic of postnatal day (P)5 back skin with hair follicles (HF) from the three HF developmental waves. Right: list of cell populations isolated from six different fluorescent reporter mouse lines in four double-transgenic combinations.
(B) Isolation of eight main skin/HF populations from K14-H2BGFP;Lef1-RFP back skin. Top: P5 skin section shows strong H2BGFP expression in epithelial Epi and ORS cells, and RFP expression in upper DFs, the DP and Mc. Mx expresses low levels of H2BGFP. The Shh expressing subpopulation of TAC progenitors and few differentiating cells co-express H2BGFP and RFP. Bottom: FACS plots and gates for cell sorting from HF-enriched dermal preparations. Seven gates mark Mx, ORS, TAC, Mc, and DP from HFs, and DF and a mixture of negative cells (Neg) from the upper dermis. Right: qRT-PCR of known markers confirms TAC and DP enrichment. Data are mean ± SD from two measurements.
(C) Isolation of HFSC precursors from K14-RFP;Sox9-GFP P5 back skin. Top: P5 skin section shows GFP expression in the upper ORS of the future bulge area. All epithelial cells are RFP. Bottom: FACS plots and gates for isolation of HFSC precursors and the remaining HF-ORS, and HF-Mx.
(D,E) Isolation of pure DP subpopulations from P5 back skin. (D) Top: section of P5 Sox2GFP;Lef1-RFP back skin and GFP quantification shows GFP expression in G-DP and AA-DP cells, compared to ZZ-DP. (E) Top: section of P5 Crabp1-GFP;Lef1-RFP back skin and GFP quantification shows GFP expression in AA-DP and ZZ-DP cells, but not in G-DP. Bottom: FACS plots and gates for sorting. Note that all DP subpopulations are highly enriched as RFP+ and ITGA9+ cells.
Scale bars are 100 μm (B, C), 20 μm (D, E). See also Figure S1.
Here, we comprehensively define the molecular traits of all DP subpopulations, SHH expressing TAC progenitors, and HFSC precursors from growing HFs, in conjunction with other major skin/HF cell types, and identifiy signaling interactions potentially involved in HF growth. For this we utilized six different fluorescent transgenic mouse reporter lines combined with immunofluorescence to isolate a total of 14 distinct skin/HF populations from postnatal day 5 back skin and performed genome-wide transcriptome analysis by multiplexed RNA deep-sequencing. We defined molecular signatures of uniquely enriched genes for each population, establishing a comprehensive set of markers and identifying interacting ligand/receptor combinations for key HF cell types during hair growth. Molecular characterization of hair type-specific DP subpopulations showed only few specific signature genes, revealing a remarkable molecular relatedness at the mRNA level. We further defined a core DP molecular signature of genes uniquely enriched and expressed by all DP subpopulations. HFSC precursors from growing HFs showed common features with adult HFSCs, but mostly expressed unique signature genes as they mature during development. TAC progenitors expressed numerous uniquely enriched genes, including many signaling factors, as was the case for DP, suggesting a rich crosstalk between these populations. Finally, our global unbiased analysis of intercellular signaling interaction revealed a network of multiple ligand/receptor interaction pairs involving all cell types during HF growth, with a particular density in the HF bulb. With this study we establish a comprehensive birds-eye-view of the complex signaling interactions in growing HFs of developing skin, and share it with the community on the Hair-GEL online database for further validation and investigation.
RESULTS
Isolation of key cell populations from growing skin and hair follicles
To purify and molecularly characterize all major cellular constituents of developing HF during the first hair growth phase, we devised an integrated approach that utilized pairwise combinations of six different transgenic reporter mouse lines together with three specific immunofluorescence stainings. In this manner we were able to isolate by fluorescence activated cell sorting (FACS) of postnatal day (P)5 back skins a total of 14 distinct skin/HF cell populations and subpopulations including SC precursors and TAC progenitors, as well as hair type-specific DP niche cells (Figure 1A).
First, to purify seven core skin and hair cell types we revisited, improved and expanded cell isolations from K14-H2BGFP;Lef1-RFP transgenic mice previously utilized to obtain HF matrix (Mx), outer root sheath (ORS), dermal papilla (DP) cells, and melanocytes (Mc) (Rendl et al., 2005). In these reporters nuclear GFP is expressed in all epithelial cells of the epidermis and HFs under the keratin-14 promoter, while RFP is present in DP, Mc, and upper dermal fibroblasts (Figure 1B) driven by a Lef1 promoter fragment. P5 back skins were harvested, and epidermis and dermis were enzymatically separated and processed to obtain epidermal and HF-enriched dermal preparations of single cells. From the epidermal sample, we selected basal epidermal cells (Epi) as the K14-H2BGFP+ population (81% of live cells) (Figure S1A). The dermal sample was subjected to further immunofluorescence marker stainings (Figure 1B). Based on GFP expression alone we selected Mx (35%) and ORS (21%) cells as K14-H2BGFPLow and K14-H2BGFPHigh populations, respectively, as previously described (Rendl et al., 2005). The RFP+ population was subdivided to obtain CD117+ Mc (2.24%), and DP cells (1.1%) that express ITGA9, a marker of these cells at P5 (Rendl et al., 2005). Quantification of immunofluorescence stainings of back skin sections demonstrated that all DP cells from all HF types express ITGA9 (Figure S1B). As RFP is also strongly expressed in the papillary dermis of Lef1-RFP mice, we selected RFP+CD117−ITGA9− cells to enrich for dermal fibroblasts (DF; 0.6%). RFP+ cells of the arrector pili muscle (APM) are also contained within the DF population. We also gated for negative cells (GFP−RFP−CD117−ITGA9−) representing an unlabeled mixture of residual dermal cells that include endothelial, smooth muscle and immune cells (Neg; 9%).
Finally, with detailed analysis of K14-H2BGFP;Lef1-RFP P5 back skin sections, we identified GFPLowRFP+ cells in the anterior side or on both sides within the matrix compartment that surrounds the DP (Figure 1B, Figure S1C), strongly resembling the expression pattern of Shh in a subpopulation of TAC progenitors (Gambardella et al., 2000; Hsu et al., 2014b). GFPLowRFP+ cells could sometimes also be found in the most proximal cells of the inner root sheath. These cells enriched in Shh expressing TACs were clearly distinguishable by FACS analysis (Figure 1B; 2%), allowing the isolation of this subpopulation of specialized signaling progenitors from growing HFs.
We next isolated all populations by FACS, extracted RNAs and performed Real-Time Quantitative Reverse Transcription PCR (qRT-PCR) for known marker genes to confirm the correct isolation of all cell types (Figures 1B and S1D). Epithelial markers K14 and K5 were strongly enriched in Epi, whereas Vdr and Lgr5 were most expressed in ORS. Matrix markers Msx2 and Shh were present in the Mx population and TAC cells. Melanocyte markers Dct and Mitf were highest in Mc, Col1a1 was strongly enriched in DF, whereas endothelial markers Tie2 and CD31 were almost exclusively expressed in the Neg population. Interestingly, Acta2 was strongly expressed in Neg and DF, confirming the presence of smooth muscle cells or APM cells in the DF population. DP markers Wif1 and Sox2 were strongly expressed in DP cells. Finally, Shh was highest in TAC cells - at even higher levels compared to Mx cells - confirming the enrichment of an epithelial progenitor subpopulation within this compartment. Taken together, our qRT-PCR analyses of marker gene expression confirmed the accuracy of our sorting strategy for concomitant isolation of epidermal cells, dermal fibroblasts, melanocytes, ORS, DP, matrix and TAC from growing HFs.
To additionally purify the precursors of future bulge SCs from within the ORS compartment of same stage growing HFs (HFSC), we used a GFP reporter line for the SC marker Sox9 crossed with an RFP reporter for all skin epithelial cells under the K14 promoter (Figure 1C), as previously described (Wang et al., 2013). From K14-RFP;Sox9-GFP transgenic P5 back skins we prepared single cells, stained for basement membrane marker ITGA6 and sorted by FACS the HFSCs as the RFPHighITGA6HighGFP+ population. We further co-isolated the remainder of the ORS (HF-ORS; RFPHighITGA6HighGFP−), matrix cells (HF-Mx; RFPLow) and basal epidermal cells (EpiT; RFPHighITGA6HighGFP− from the epidermal fraction). We then confirmed the purity of HFSC isolation by qRT-PCR (Figure S1E). Expression of the HFSC markers Sox9, Lhx2 and Nfatc1 was strongly enriched in the HFSC population, with Lhx2 also present in HF-Mx cells, as previously described (Wang et al., 2013), whereas matrix markers Shh and Msx2 were only detected in HF-Mx.
Finally, to isolate and explore potential subtypes of DP niche cells from the three major back skin hair formation waves, we utilized two different transgenic reporters together with ITGA9 immunofluorescence. With a Sox2GFP knock-in reporter line crossed with Lef1-RFP, we selected pure DPs from zigzag hairs (ZZ) and distinguished a complementary mixture of guard/awl/auchene hair (G/AA) DPs (Figure 1D), similarly to the previously described use of Sox2-GFP transgenic mice (Driskell et al., 2009). In this configuration, total DPs were selected as RFP+ITGA9+ cells (Figure 1D, 1%), from which we could distinguish GFP− ZZ-DP (15%) and GFP+ G/AA-DP cells (80%). Conversely, to isolate pure guard (G) DPs as well as complimentary mixtures of zigzag/awl/auchene hair (ZZ/AA) DPs we utilized Crabp1-GFP transgenic reporter mice (Figure 1E) that we identified in a screening effort for hair type-specific fluorescent DP reporter lines (not shown). In P5 back skins of this line, close to 100% of AA-DP and ZZ-DP cells express GFP, whereas no GFP was found in G-DPs, although endogenous Crabp1 is expressed in these cells (Figure S1F). Using the same Lef1-RFP, ITGA9 strategy we could then isolate from Crabp1-GFP;Lef1-RFP P5 pups pure G-DP cells as GFP− (2%) and a mixture of AA-DPs and ZZ-DPs (AA/ZZ-DP) as GFP+ (90%) DP cells. Together with the four different DP subpopulations we further isolated RFP+ dermal fibroblasts and the remaining mixed negative population (Neg) to confirm cell enrichment and sorting efficiencies. To determine initial sort purity we next performed enzymatic reactions for DP marker Alkaline Phosphatase on isolated and cytospun DP populations demonstrating >90% enrichment, compared to 1% in RFP+ and 0% in Neg cells. We then extracted RNA and performed qRT-PCR analyses to confirm expression of known markers (Figure S1E). All tested DP markers were found enriched in both DP subpopulations of both isolation strategies, with the exception of barely detectable Sox2 in ZZ-DP. GFP mRNA was only present in GFP+ populations (G/AA-DP and AA/ZZ-DP).
In summary, with combinations of six different mouse transgenic reporter lines together with immunofluorescence we successfully isolated 14 epidermal, dermal and HF cell types for further molecular characterization, including for the first time the SC precursors, TAC progenitors, and several DP niche subpopulations from different hair types.
Transcriptome profiles of the major HF populations by RNA deep sequencing
For all isolated populations we performed transcriptome profiling with RNA sequencing from low RNA amounts (6ng) in two biological replicates, as previously described (Sennett et al., 2015) (Table S1).
We then established global lineage relationships by identifying differentially expressed genes between all populations. Principal component analysis (PCA) of all mapped genes showed that dermal (DF, DP and the 4 DP subpopulations) and epithelial populations (Epi, ORS, Mx, TAC, HF-SC and HF-ORS) each group together, whereas Mc cells represented a distinct expression profile (Figure 2A). Total DP and all DP subpopulations clustered together, as did Mx and TAC cells. In order to interrogate the global relatedness of gene expression profiles between the developing HF and skin populations, we calculated coefficients of determination (r2) for all expressed genes (Figure 2B). As expected all biological replicate samples displayed high correlation (r2>0.85). Interestingly, all DP populations (DP, ZZ-DP, G/AA-DP, G-DP and AA/ZZ-DP) showed highly similar gene expression profiles with r2>0.83. They also all exhibited a high correlation with DF (r2>0.81), but low similarity to every other population (r2<0.71). Apart from these, the two closest populations were Mx and TAC (r2>0.91), and both were quite similar to the other epithelial populations (Epi, ORS; r2>0.77). Mc showed a very unique expression profile with plot-wide low r2 values (r2<0.75), as they are specialized cells of neural crest origin. HF-SC and HF-ORS were highly correlated to each other (r2>0.85) and similar to ORS and Epi (r2>0.80).
Figure 2. Transcriptome analyses of 14 distinct skin/hair follicle cell populations with next-generation RNA-sequencing.

(A) Principal component analysis of all expressed genes with PC1 (42.07% variance captured), PC2 (11.64% variance captured), and PC3 (7.97% variance captured).
(B) Heat map of coefficient of determination (r2) for gene expression profiles of all skin/HF populations.
(C) Hierarchical clustering of differentially expressed genes (DEG, p-value < 0.05). Note that DP populations are highly similar with replicates not clustering together. DEGs are listed in Table S2.
To determine the enriched genes for each population, we first compared the FPKMs of all genes that are differentially expressed in at least one population (CuffDiff; p < 0.05). Hierarchical clustering identified a total of 10,627 differentially expressed genes (Figure 2C; Table S2). The main populations clustered together in correlation with their epithelial, mesenchymal and neural crest origins. Total DP and all DP subpopulations clustered together, but they appeared to exhibit extremely closely related molecular profiles as biological replicates themselves were unclustered (Figure 2C). All other biological replicates clustered together. Next, we established gene signatures identifying uniquely enriched genes in each population compared to all relevant others by analyses of variance calculations (ANOVA; FDR < 0.05) for each of the four cell sorting strategies. We defined molecular signatures for the main skin and HF populations including TAC progenitors, for the four subpopulations of DP, and the HFSC precursors as subpopulation of the ORS. In total, 3,015 signature genes were identified for all 14 key cell populations of growing HFs that we discuss in detail below.
Identification of unique molecular characteristics of the major HF populations
A first ANOVA included the main eight populations from the K14-H2BGFP;Lef1-RFP sort, i.e. Epi, ORS, Mx, TAC, Mc, DF, DP and Neg, identifying 5,807 genes (Figure 3A). The epithelial populations clustered on one arm, apart from the dermal populations, while Mc stood alone. Mx and TAC grouped together, as did DF and DPs, illustrating their close lineage relationships. Enriched signature genes for each population were then selected from the Cuffdiff/ANOVA analyses as genes with FPKM > 1 and an expression fold change (FC) ≥ 2 compared to all other populations (Figure 3B; Table S3). Neg were excluded for comparisons with the epithelial populations (Epi, ORS, Mx, TAC). With this approach, we identified a total of 2,049 signature genes for all seven populations, with 414 for Epi and 145 for ORS populations. Only 21 signature genes were found for Mx compared to 128 for TAC, illustrating their highly overlapping expression profiles. Considering the high similarity and lineage relationship, Mx and TAC were not compared to each other and expanded overlapping signature lists contained 96 and 234 genes for Mx and TAC respectively. The TAC progenitor population will be discussed in detail below. 604 and 124 signature genes defined the Mc and DF populations, respectively. Finally, the total DP population showed a signature of 578 genes.
Figure 3. Molecular signatures of eight main skin/HF cell populations.

(A) Hierarchical clustering of significantly enriched genes identified by ANOVA of the main eight skin/HF populations from the K14-H2BGFP;Lef1-RFP sort.
(B) Venn diagrams of cell-type specific signatures. The overlaps represent commonly enriched genes in corresponding populations compared to all others. Overlapping gene lists are in Table S4.
(C) Gene ontology analysis of molecular signatures. Notable terms are highlighted; all terms are listed in Table S5.
(D) Total number of signature genes with listed representative cell-type specific markers organized according to functional categories. Fold-change enrichment is indicated. FPKM fold changes are between Epi vs. ORS, ORS vs. Mx, Mx vs. ORS, Mc vs. DF, DF vs. DP, and DP vs. DF. FDR of q < 0.05; p-value < 0.05. Gene lists are provided in Table S3.
(E) qRT-PCR validation of selected signature genes, relative expression is to dermis. Data are mean ± SD from two measurements. See also Figure S2.
(F) Immunofluorescence staining for DP marker MC3R on K14-H2BGFP P5 back skin section. Scale bar is 100 μm.
(G) Comparison of P5 signatures with embryonic signatures from Sennett et al., 2015. Overlapping genes are listed in Table S6. Statistically significant overlap was calculated with Fisher’s exact test.
Considering the hierarchical and/or developmental relationships between these six cell types (Epi, ORS, Mx, Mc, DF, DP) we next inspected the shared expressed genes in these populations (Figure 3B; Table S4). Enriched epithelial-type genes expressed in Epi, ORS and Mx were selected as genes with a FC ≥ 2 compared to every non-epithelial population (Mc, DF, DP). These 3 populations shared 156 genes in common. 46.7% (234/501) of ORS and 51.8% (235/453) of Mx genes are epithelial-enriched and overlap with the Epi population. ORS and Mx share 278 genes, 55.5% and 61.4% of expressed epithelial genes, respectively. Similarly, common dermal genes were identified as genes with a FC ≥ 2 in DF and DP compared to epithelial populations (Epi, ORS, Mx and TAC). DP and DF cells have a large overlap sharing 422 dermal-type genes, 72% and 40% of DF and DP genes, respectively, that are enriched compared to epithelial cells.
To gain insights into the biological significance of our gene signatures, we next analyzed functional categories of gene ontologies (GO) using EnrichR (Figure 3C; Table S5) (Chen et al., 2013). Genes related to epidermis development or epithelial differentiation were represented well in all epithelial populations (Epi, ORS, Mx), and categories involving cell adhesion and ECM were associated with Epi and ORS, as well as with DF and DP in the dermal compartment. The specialized Mc population showed distinct GO terms, such as pigment cell differentiation and melanosome localization. Finally, neural categories and signaling pathway regulation (Wnt, BMP, Notch) were particularly enriched in the DP niche, as previously described (Rendl et al., 2005).
To focus on specific genes within the signatures, we first corroborated that previously known markers described in expression and/or functional studies of single genes were found within the signature lists of each population (Figure 3D; Table S3). Epidermal markers Cdh1, Klf5, Itga6 and Krtdap were most enriched in Epi, whereas SC markers Nfatc1, Sox9, Lgr5 were found in the ORS signature, as was Krt19 and Vdr. Note that Krt14 and Krt5 are more highly expressed in Epi than ORS. Matrix markers Dlx3, Foxn1, Fbp1 and Ovol1 were enriched in Mx. Markers for Mc (Sox10, Pax3, Kit, Mc1r, Mlph), and DF (CD34, Itga8) were present in their respective signatures, as well as Crabp1, Enpp2, Hhip, Sox2, Sostdc1 and many others for total DP. Several known marker genes were independently confirmed by qRT-PCR (Figure S2A). We also identified many signature genes for all cell types (Figure 3D, Table S3). Genes involved in signaling were identified in all populations such as the Rho guanine exchange factor Net1 and the growth factor Figf (Vegfd) in Epi, the TNF-induced metabolic regulator Steap4 and the connective tissue growth factor (Ctgf) in ORS, and the frizzled ligand Wnt8a, the hydrolase Liph and the placode marker Edar in Mx. Similarly, the inducer of Pi3k/Akt pathway Igf1 and the adrenoreceptors Adra1a and Adra1d were present in DF signature, the activin receptor Acvr1c and the chemokine Ccl25 in Mc and the activator of canonical Wnt pathway Rspo3 and the BMP ligand Gdf11 in DP. Identities of transcription factors were also uncovered in multiple populations, including the bHLH family member Ascl4 in Mx and the silencing factor Satb1 in Epi. The DP niche was further characterized by a number of previously undescribed neural-associated genes, such as the receptors Mc3r and Trpm3, the cellular migration regulator Slit2, and the Wnt antagonist guidance protein Draxin. We validated expression of numerous signature genes by qRT-PCR (Figures 3E, S2B). Immunofluorescence staining for the DP signature gene MC3R confirmed its strong expression in DP cells (Figure 3F), although it also appeared to be expressed in fibroblasts and/or adipocytes of the lower dermis and the dermal sheath. Insights into several signature genes are described below in detail.
We recently isolated the earliest HF progenitors, dermal condensate niche, and other distinct skin populations during embryonic HF formation at E14.5 and defined their gene signatures (Sennett et al., 2015). In order to relate these characterized populations from developing skin after birth with their embryonic precursors, we compared the gene signatures for Mc, Epi, DF and DP with their corresponding embryonic counterparts (Figure 3G; Table S6). Embryonic and postnatal Mc showed a large overlap of signature genes (168; 27.8% of P5 Mc), mostly associated with pigmentation. 23 DF genes at P5 were found in common with embryonic DF (18.5% of P5 DF), mostly related to ECM organization. Lipid processing categories were well represented in the 77 overlapping Epi signature genes (414; 18.6% of P5 Epi). Finally, only 12.3 % (71/578) of the total DP gene signature at P5 were common with the E14.5 condensate niche signature, and were highly associated with cell adhesion and nervous system development. This suggests a conserved role of these functional categories in basic DP biology, while otherwise widespread transcriptional shifts occur as the cells mature. A comparison with our previously published P5 molecular signatures (Rendl et al., 2005) showed a majority of Mc (55.2%) and DP (51.9%) signature genes in both signatures, whereas 25.8% and 13.1% of previous ORS and Mx signatures overlapped with our gene lists (Figure S2C; Table S6). We also compared our total DP signature to the DP markers identified by Driskell and coworkers at P2 (Driskell et al., 2009). Our total DP signature of 578 genes shared only 82 of 364 genes (22.5%) from that study (Figure S2D; Table S6). This apparent low overlap is most likely due to the use of different labeling and sorting methods, as we employed the stringent combination of Lef1-RFP and ITGA9 for highly enriched DP isolation, whereas Driskell et al. used CD133 alone, which may also be expressed in other skin cell types. Finally, DP cells have also been molecularly characterized during the 2nd hair cycle and compared with bulge and hair germ SCs (Greco et al., 2009). We revisited this analysis and defined a total of 2,043 enriched DP genes for all three time points in that study as genes with a FC ≥ 2 compared to bulge and germ SCs. Interestingly, 55.8% (207/578) of our P5 DP signature overlapped with this very large DP genes list (Figure S2E; Table S6). The 207 common DP genes have a high association with tissue morphogenesis, ECM organization, neurogenesis and the hair cycle (data not shown).
In summary, using RNA sequencing we gained insights into the molecular lineage relationships and functional gene categories of the main skin and HF populations, their relatedness to embryonic precursors and adult counterparts, and we uncovered a plethora of signature genes that likely play important roles in the function of all analyzed cell types.
Hair type-specific DP cells are highly similar and exhibit only few signature genes
To gain insights into the similarities and differences between individual DP subpopulations, we next performed two separate ANOVAs for the four DP subtypes. ZZ-DP and G/AA-DP from the Sox2GFP;Lef1-RFP sorts (Figure 4A), and G-DP and AA/ZZ-DP from the Crabp1-GFP;Lef1-RFP isolations (Figure 4B) were compared along with seven of the eight main populations, i.e. Epi, ORS, Mx, TAC, Mc, DF, Neg. In order to uncover hair type-enriched DP genes, we computed gene expression comparisons between both GFP+ and GFP− DP subpopulations for significantly changed genes (FPKM ≥ 1; FC ≥ 1.4x). GFP− G-DP and ZZ-DP populations were compared to each other as well. To our surprise but consistent with the high similarity of the DP subpopulations in the PCA and clustering analyses (Figure 2), only 31 and 74 signature genes were identified for the G-DP and ZZ-DP subpopulations, respectively (Figure 4C, Table S3). 351 and 224 genes were highly enriched in the AA/ZZ-DP and G/AA-DP signatures, respectively. We then calculated AA-DP signature genes that were present in both G/AA-DP and AA/ZZ-DP signatures, and had an expression FC ≥ 1.4 compared to both ZZ-DP and G-DP (Figure 4D; Table S3). We identified only 76 AA-DP enriched genes, a similarly low number as for G-DP and ZZ-DP (Figure 4D). In addition to identification of few signature genes in each DP subpopulation, the level of enrichment was typically modest. For example, qRT-PCR demonstrated the previously described ZZ-DP-associated transcription factor Sox18 as highly expressed in ZZ-DP, although it was also enriched in G/AA-DP compared to all other populations (Figure 4E). We confirmed a similar pattern for several other DP subpopulation-enriched genes, such as the frizzled ligand Wnt5b and the enzyme Man1c1 for G-DP, the modulator of Wnt signaling Frzb (Sfrp3) and the lipid transporter Abcg1 for ZZ-DP and the glutathione peroxidase Gpx3 and the transcriptional regulator Plagl1 for AA-DP (Figures 4E and S3A). At the protein level, immunofluorescence staining for WNT5B and GPX3 protein confirmed their presence in G-DP and AA-DP, respectively, but the detectable staining of both proteins in DPs of the other hair types did not confirm a hair type-specific expression pattern (Figure 4F). Only very few genes showed high FC enrichment in only one DP subpopulation including Sox2, Sostdc1, and interestingly several ncRNAs, suggesting DP subpopulations exhibit very limited hair type-specific signals at the transcriptomic level.
Figure 4. Hair type-specific DP subpopulations are highly similar and express only few signature genes.

(A, B) Hierarchical clustering of significantly enriched genes identified by ANOVA for hair type-specific DP subpopulations from the (A) Sox2GFP;Lef1-RFP and (B) Crabp1-GFP;Lef1-RFP sorts.
(C, D) DP subpopulation-specific molecular signatures. Signature genes are organized according to functional categories. Indicated FPKM fold changes are between (C) G-DP vs. AA/ZZ-DP, ZZ-DP vs. G/AA-DP, and (D) AA/ZZ-DP vs. G-DP, G/AA-DP vs. ZZ-DP. The AA-DP signature is a logical list of shared enriched genes in AA/ZZ-DP and G/AA- DP compared to each pure G-DP and ZZ-DP (fold change average). FDR of q < 0.05; p-value < 0.05. Gene lists are provided in Table S3.
(E) qRT-PCR validation of signature genes, relative expression is to dermis. Data are mean ± SD from two measurements. See also Figure S3.
(F) Immunofluorescence stainings for G-DP marker WNT5B and AA-DP marker GPX3 on K14-H2BGFP P5 back skin sections. Note that although these proteins are expressed in the DP subpopulation, they are also expressed in DPs of other hair types. Scale bar is 20 μm.
(G) Venn diagram of expanded and total DP gene lists. The expanded DP subpopulation signature includes enriched genes compared to all other cell types except DP. The overlap between the four expanded DP subpopulation and total DP signatures represents the core DP signature of shared genes in all five DP populations. Expanded DP gene lists are provided in Table S3.
(H) The core DP molecular signature. Signature genes are organized according to functional categories. Average FPKM fold changes are shown of all DP signatures vs. DF. FDR of q < 0.05; p-value < 0.05. The full core DP signature is listed in Table S3.
Comparison of our ZZ-DP and G/AA-DP signature to previously reported DP genes for the same DP subpopulations (Driskell et al., 2009) showed that our ZZ-DP signature only shared five genes (6.8%) with the previously published list, and G/AA-DP had 41 overlapping genes (18.3%) (Figure S3B; Table S6). The low overlap is most likely due to the difference of DP isolation methods, as CD133 might have been the best marker available at the time but stains only a subpopulation of DP cells and is also expressed in non-DP cells (Figure S3C).
As all DP subpopulations isolated in our study showed only few specific markers, we then defined a robust core DP signature, consisting of genes enriched in all DP subpopulations to document their similarity. We first established expanded molecular signatures for each DP subpopulation compared to all other skin/HF cell types. These were assessed for overlap with each other, and with the total DP signature. For this we calculated expression FC ≥ 2 by comparing FPKMs (≥1) of all significantly increased genes (ANOVA/CuffDiff) in each DP subpopulation with FPKMs of all other main cell types (Epi, ORS, Mx, Mc, DF, TAC and Neg). These expanded signatures ranged from 844 genes for G/AA-DP cells to 419 genes for G-DP cells (Figure 4G; Table S3). 289 ZZ-DP genes (47.9%), 404 G/AA-DP genes (47.8%), 247 G-DP genes (58.9%) and 364 AA/ZZ-DP genes (53.5%) were previously identified as signature genes in the total DP population (Figure 2). As predicted from the PCA and clustering analysis and the low signature gene numbers, all four DP subpopulations showed strikingly similar molecular profiles with most genes shared with at least one other DP subpopulation (90% of ZZ-DP, 78% of G/AA-DP, 93% of G-DP and 86% of AA/ZZ-DP). Importantly, 285 genes were common to all four DP subpopulations (not shown), and of those a total of 202 genes (70.9%) were also present in the total DP signature, establishing with stringent criteria a core DP signature of universally expressed DP genes (Table S3). As predicted this core signature contained all previously identified DP markers, such as Wif1, Corin, Lepr and Pbx1. Notably, it also identified many core signature genes such as the calcium channel Trpm3, the epoxyde hydroxylase Ephx2, the neuron navigator family member Nav3 and the cell morphology regulator Wasf3. Similar to previous GO analyses, the core signature was enriched for genes involved in epithelium and nervous system development, and regulation of Smoothened, Wnt and BMP signaling pathways (Figure S3D; Table S5).
Taken together, it appears that all DP subpopulations share most of their genes, and differentially express only very few markers, including several ncRNAs. A convergent analysis of all hair type-specific DP subpopulations together with total DP established a comprehensive and stringent core DP molecular signature that is shared by the DPs of all HF types.
Stem cell precursors from developing follicles express distinct signature genes and are molecularly more similar to adult bulge stem cells than embryonic placode progenitors
Many studies have focused on characterizing HFSCs at the key stages of HF development and cycling (Greco et al., 2009; Lien et al., 2011; Sennett et al., 2015), but no transcriptome-wide information is available for the precursors of adult bulge SCs during the early HF growth phase. By subdividing the ORS into the HFSC precursors and the remaining HF-ORS, we were able to define the HFSC precursor signature. With a fourth ANOVA (Figure 5A) we compared HFSC precursor and HF-ORS subpopulations (K14-RFP;Sox9-GFP sort), again in conjunction with the seven of the eight main populations from the K14-H2BGFP;Lef1-RFP sort, i.e. Epi, Mx, TAC, Mc, DF, DP, Neg.
Figure 5. Molecular characterization of bulge stem cell precursors.

(A) Hierarchical clustering of significantly enriched genes identified by ANOVA for HFSC precursors and the remaining ORS subpopulation from the K14-RFP;Sox9-GFP sort.
(B) HFSC precursor-specific molecular signature. Signature genes are organized according to functional categories. FPKM fold changes are between HF-SC vs. HF-ORS. FDR of q < 0.05; p-value < 0.05. Gene lists are provided in Table S3.
(C) Comparison of the HFSC precursor signature with previously published embryonic placode (Pc) progenitor and adult bulge gene signatures. Some overlapping genes are listed here; all are in Table S6. Statistically significant overlap was calculated with Fisher’s exact test.
(D) Gene ontology analysis of the HFSC precursor signature. Notable terms are highlighted; all terms are listed in Table S5.
(E) qRT-PCR validation of selected signature genes, relative expression is to dermis. Data are mean ± SD from two measurements. See also Figure S4.
(F) Immunofluorescence staining for HFSC precursor marker ITGB8 on K14-H2BGFP P5 back skin section. Scale bar is 100 μm.
By comparing FPKMs ≥ 1 of all differentially expressed genes by ANOVA and/or CuffDiff, we identified 127 HFSC precursor signature genes with an expression FC ≥ 2 compared to all other populations (Figure 5B, Table S3). Several of these markers were validated by qRT-PCR (Figure S4A). Only a few signature genes had been previously reported as HFSC precursor markers in developing HFs, such as Nfatc1, Sox9, and Lgr6. Several other signature genes uncovered by our analysis were adult SC markers, such as Lrig1, Tgfb2, Tgfb3 and Gata6, but previously not known to be expressed in precursors during early hair growth. Indeed, 21% (27/127) and 23% (29/127) of these genes were common with previously described HFSC signatures during the second hair cycle at P56 and P43-P69, respectively (Figure 5C; Table S6). Interestingly, only seven genes (5.5%) were commonly expressed with E14.5 placode progenitors, suggesting that HFSC precursors at P5 are molecularly closer already to adult HFSC.
GO analysis of the HFSC precursor molecular signature showed that enriched genes are highly associated with extracellular matrix organization, cell communication, epithelium development, regulation of cell adhesion, and stem cell development (Figure 5D; Table S5), whereas genes enriched in the rest of the ORS (HF-ORS) are closely associated with regulation of protein transport and regulation of cell activation (data not shown). Enrichment for signature genes including transcription factor Foxi3, adhesion molecule Itgb8, docking protein Nedd9 and Wnt receptor Fzd7 was validated by qRT-PCR (Figures 5E and S4B), and immunofluorescence staining for ITGB8 confirmed specific expression in the upper part of the ORS around the area of the future bulge.
Taken together, our analysis identified many signature genes as well as several previously described genes from adult bulge SCs that are already expressed in the HFSC precursors of early growing HFs. It also confirmed that HFSC precursors at this stage have continued to molecularly diverge from placode progenitors (Ouspenskaia et al., 2016).
Transit amplifying progenitors of early growing follicles have a unique signature enriched in signaling factors
TAC progenitors within the larger matrix compartment are thought to exchange signals with the DP at the base of growing HFs, and here we isolated and profiled the Shh expressing TAC subpopulation. As stated earlier, we identified only 21 Mx and 128 TAC signature genes (Figure 3A; Table S2), due to their close lineage relationship. When Mx and TAC were not directly compared to each other, 96 and 234 genes were compiled for each population, respectively, for an expanded signature including 61 significantly increased genes in both populations compared to all other cell types (Figure 6A; Table S6). Most previously described Mx genes, such as Msx2 and Ovol1, were enriched in both Mx and TAC progenitors, whereas other Mx markers were TAC specific genes and only enriched in this population, including Hoxc13 and Lef1 (Figure 6B; Table S3). The TAC signature also contained many enriched genes that belong to several important functional gene categories such as signaling (Bambi, Bmp2, Kitl, Tiam2), transcription (Egr3, Foxq1, Tob1, Zfhx3) or adhesion (Unc5b, Cdhr5, Dsg4, Fndc9) (Figure 6B). Expression of TAC signature genes was confirmed by qRT-PCR (Figure 6C). The signature genes identified here may play an important role in the signal exchange and cell fate changes that control HF progenitor proliferation, migration, and differentiation into the hair shaft and inner root sheath channel.
Figure 6. Molecular characterization of a transit amplifying progenitor TAC subpopulation in developing hair follicles.

(A) Overlapping expanded signatures of Mx and TAC. 61 genes were found commonly enriched in both populations and are listed in Table S6.
(B) TAC-specific molecular signature. Signature genes are organized according to functional categories. FPKM fold changes are between TAC vs. ORS. FDR of q < 0.05; p-value < 0.05. Gene lists are provided in Table S3.
(C) qRT-PCR validation of selected signature genes, relative expression is to dermis. Data are mean ± SD from two measurements.
(D) Gene ontology analysis of the TAC signature. Notable terms are highlighted; all terms are listed in Table S5.
(E) Comparison of P5 TAC signature with previously published adult TAC and embryonic placode (Pc) progenitor signatures. Some common factors are listed here; all are in Table S6. Statistically significant overlap was calculated with Fisher’s exact test.
Epithelial proliferating progenitors have previously been isolated and molecularly characterized from adult HFs during the anagen growth phase of the hair cycle (Lien et al., 2011). To explore the similarities and dynamic changes between HF development and adult re-growth, we directly compared the signatures (Figure 6D). Our TAC population shared 62 genes (26.5%) with adult TAC progenitors, many which have been implicated in HF morphogenesis/cycling and serine/threonine kinase signaling (Figure 6B; Table S6). Several GO categories involving signaling regulation were highly associated with TAC, such as BMP, Wnt and Smoothened pathways (Figure 6C; Table S5). This signaling pathway enrichment was not detected for other Mx cells (Figure 3B). Fittingly, similar signaling pathways are also well represented in the DP GO analysis (Figure 3B; Wnt, BMP, Notch) underlining the central importance of signaling crosstalk in these two neighboring populations. In summary, this analysis showcases the unique molecular profile of the specialized TAC progenitor subpopulation during early hair growth, with a significant enrichment of signaling genes and overlap with adult TAC progenitors.
Intercellular network analysis uncovers complex signaling crosstalk in the developing hair follicle
Our transcriptome-wide analyses of molecular features of the major HF cell types during early growth revealed in most populations expression of many signature genes involved in signaling pathways and cell communication (Figure 3B, D; Figure 6B–C). In order to systematically identify signals between these populations, we constructed expression heatmaps for the seven main populations covering known key signaling pathways (Figure S5). Genes were selected from the KEGG pathway database and included in this analysis when they were expressed by at least one population with an FPKM ≥ 1. For Wnt pathway-related genes, most Wnt ligands were expressed by the epithelial populations, whereas secreted Wnt-regulators were specifically expressed by dermal cells, most prominently DP cells. BMP ligands and receptors were expressed in both epithelial and dermal compartments, while BMP inhibitors were mostly expressed by DP and DF with the exception of highest Bambi expression by TAC progenitors. Shh was most strongly expressed by TAC, as previously shown (Hsu et al., 2014b), while Ptch1 receptor and downstream effectors were highest in the DP. Notch ligands were highly expressed in epithelial populations, and downstream effectors were highly expressed in DP, Mx and TAC progenitors.
Although this analysis strategy provided a birds-eye view of major signaling pathways, it could not reveal ligand/receptor interactions and did not include all known signaling pathways. To comprehensively interrogate the signal exchange between all key HF populations, we conducted an unbiased intercellular network analysis using all identified signature ligands and receptors for all HF cell types (Epi, ORS, Mx, Mc, DF, DP, TAC and HF-SC). We first identified 342 and 353 unique ligands and receptors, respectively, and described 878 unique interaction pairs (Figure 7A; Table S7). A similar approach was previously used to define a cell-cell communication network in the human hematopoietic system (Qiao et al., 2014). Several signature ligands and receptors were identified (Figure 7B). Mx cells uniquely expressed only one single receptor (Edar), while TAC progenitors produced several ligands and receptors, fitting for a cell population involved in signaling. Interestingly, the highest numbers of signature ligands and receptors was found in DP, characteristic for a signaling center. We then constructed an unbiased signaling network of 53 unique ligand/receptor interactions (Figure 7C; Table S7). Some signal/receptor pairs were between distant cells, such as Figf and Pdgfc from Epi to DP, but numerous interactions were revealed between neighboring cells. For example TAC progenitors appeared to signal to Mc (Kitl), ORS (Efnb2) and DP cells (Shh, Tnf). Interestingly, DP cells seemed to interact with all populations by secreting ligands, such as Bdnf and Edn3 to Mc, Efnb3, Rspo1 and Spp to ORS, Eda to Mx, and Fgf7 to DF. It is also interesting to note that the only populations exhibiting autocrine regulations were Mc (Npy), TAC progenitors (Shh) and the DP niche (Mdk, S100b, Spp1, Vegfa). Next we searched for potentially activated pathways by identifying intracellular effectors in target populations. For this, we performed a KEGG pathway analysis of signature receptors, and systematically identified intracellular KEGG pathway members expressed in the signatures of corresponding cell types (Figure 7D; Table S7). Several activated major pathways and their intracellular effectors were identified (Figure 7D) for numerous ligand/receptor pairs (Figure 7C, D, blue arrows). Fgfr1 and Pdgfra in DP may bind different ligands from other specialized cells such as Vegfc from Mc and Fgf5 from ORS, resulting in the potential activation of Pi3K-Akt, Ras, Rap1 and Mapk pathways, considering the presence of intracellular effectors Gng8, Mras, Magi2, Map2k6 of these pathways in the DP signature (Figure 7D, Table S7). Similarly, Edn3 from DP could activate receptor Ednrb on Mc to potentially activate the Calcium signaling pathway with intracellular effectors Adcy2, Adcy9, Slc25a4 present in the Mc signature. Interestingly, some of these receptors/pathways were already described as important regulators in the corresponding population such as Ednrb, Met or Kit (Yaar and Park, 2012) in Mc, but the precise source of their ligands has been unclear until now.
Figure 7. Signaling interaction network in growing hair follicles of developing skin.

(A) A ligand and receptor database containing 878 unique interaction pairs. See Table S7.
(B) Ligands and receptors present in the gene signatures identified in this study. Gene lists are in Table S7.
(C) Cell-cell communication network constructed from signature ligands and receptors of each cell population. Signature ligands are color-coded for the source population. Color-coded signature receptors in target cells are listed below. Blue arrows denote interactions also found in KEGG pathway analysis (Figure 7D). All interactions are listed in Table S7.
(D) KEGG pathway analysis of signatures. Color-coded signature receptors of enriched KEGG pathways are next to the arrows. The major pathways and color-coded intracellular signature effectors in the target cell population are listed below. Note that numerous other KEGG pathway members were found in the signatures (Table S7). All interactions are listed in Table S7.
(E) DP/TAC/Mc interaction network with all expressed ligands and receptors, independent of signature status. Left: 50 ligand/receptor interaction pairs with either ligand or receptor a signature gene. Signature genes are in bold. Right: 73 non-signature ligand/receptor interactions. Arrows connect source with target populations. All interactions are listed in Table S7.
Finally, as this network was exclusively built with signature ligands and receptors, it likely does not include important signal exchange where the ligand and/or receptor is not uniquely enriched as a signature gene in only one population. To uncover such interactions in the subsequent analysis, we included ligand/receptors from all expressed genes (FPKM ≥ 5) in neighboring Mc, TAC progenitors and DP niche (Figure 7E; Table S7). This analysis revealed a total of 137 interactions, of which 14 connected signature ligands and signature receptors (included in Figure 7C), 50 pairs involved a single signature ligand or receptor (Figure 7E, left) and 73 interactions associated non-signature ligand and receptor gene pairs (Figure 7E, right). Although most of these interactions are identified here and need further corroboration of ligand/receptor expression by immunofluorescence and functional studies to uncover their biological significance, known crosstalk was identified such as Bmp receptor activation in TAC keratinocytes (Kobielak et al., 2003), DP (Rendl et al., 2008) and Mc (Kawakami et al., 2008). Regardless, this comprehensive signaling interaction network provides numerous insights into the complexity of the signal exchange during skin and HF development.
DISCUSSION
Several studies have focused on identifying ligands, their receptors, and transcriptional/other regulators to gain insights into the signaling interactions and molecular controls of HFSCs and their dermal niche during HF formation, growth and regeneration (Driskell et al., 2009; Greco et al., 2009; Rendl et al., 2005; Sennett et al., 2015). More than six years ago, two studies molecularly characterized HF cell populations of the neonatal growth phase through a global transcriptome-wide approach. The earlier study isolated and characterized five major HF/skin populations, but did not include epidermis, and could at the time not distinguish Shh+ TAC progenitors from Mx, HFSC precursors from remaining ORS, or hair type-specific DP subpopulations (Rendl et al., 2005). Similarly, Driskell and coworkers molecularly defined for the first time the DP niche of zigzag HFs (ZZ-DP), but did not apply a stringent fold change threshold when defining signatures as many blood vessel and blood related genes (Flt1, Kdr, Lyve1, Bmx) can be found in their ZZ-DP gene list. This study also lacked characterization of other DP subpopulations and any epithelial populations (Driskell et al., 2009). Moreover Driskell et al. isolated DP using CD133 that on its own may not be specific to DP and may have labeled other skin cells as well (Figure S3C). Of note, both previous studies used microarrays for transcriptome analyses, which could contribute to the difference of signature genes numbers observed between these reports and our findings.
In the present study, we built on our previous molecular definition of nascent HFs during embryonic skin development (Sennett et al., 2015), and characterized for the first time bulge SC precursors (HFSC), Shh expressing transit amplifying matrix progenitors (TAC), and all three hair type-specific DPs from early growing HFs, in addition to eight main skin/HF populations. To accomplish this, we used four paired combinations of six fluorescent transgenic mouse lines and three specific antibody stainings to isolate a total of 14 cell populations from P5 back skins, characterized gene expression transcriptome-wide by RNA sequencing, and defined cell type-specific molecular signatures of more than 3,000 signature genes combined.
Overlapping these signatures with previously published gene lists during HF formation, growth or regeneration demonstrated that all populations shared a significant but varying number of genes, providing fresh insights into their molecular evolution as they mature during development. DP shared 12.3% (71/578) of genes with DC precursors at E14.5 (Sennett et al., 2015), 16.3% (94/578) with the previously published P5 signature (Rendl et al., 2005), and 35.8% (207/578) with adult DP during the second cycle (Greco et al., 2009), suggesting these cells express a constant subset of genes that defines their cellular uniqueness independently of their developmental stage. Regarding our unprecedented characterization of SC precursors from developing HFs, signature comparisons revealed that they are already molecularly closer to adult bulge SCs (Greco et al., 2009) than to embryonic placode progenitors (Sennett et al., 2015), yet express a distinct gene expression signature related to their developmental stage. Similarly, our molecular definition of TAC progenitors during morphogenetic HF growth identified a significant number of signature genes shared with previously described P28 TAC (Lien et al., 2011) and with E14.5 placode progenitors, but also a uniquely enriched set of genes that could play an important role in the signaling exchange with the DP niche during early HF growth after birth. Of note, E14.5 placode cells share several signature genes with both P5 HFSC precursors and TAC progenitors, indicative of the broader developmental potential of these embryonic progenitors.
A notable strength of this study is the molecular characterization of all three DP subpopulations: G-DP, AA-DP, and ZZ-DP. Our integrated comparative analysis uncovered highly similar molecular profiles for all DP subpopulations at the mRNA level, while identifying only few hair type-specific DP genes. This finding is in agreement with the previously proposed hypothesis that the total number of DP cells and their overall signaling output in differently sized niche compartments dictates hair type fate (Chi et al., 2013), rather than intrinsic molecular specificity in individual DP subpopulations (Driskell et al., 2009). On the other hand, some of the signature genes with the highest differential expression in DP subpopulations were miRNAs, suggesting that hair type-determination could be regulated at the posttranscriptional level. Also, it is interesting to note that AA genes were enriched in the total DP signature, most likely because the total sorted DP population contained a majority of DPs from AA HFs (80%), based on the HF enrichment and cell preparation method. This suggests that other studies using a similar dermal preparation method for DP isolation might show a bias towards AA DP cells as well. With this analysis, we also defined an original overarching core signature of 202 genes that are co-expressed and enriched by all DP subpopulations. This commonly expressed set of DP-enriched genes shares several signature genes with E14.5 DC and adult DP, suggesting that a subset of the core signature is developmentally conserved.
Finally, our intercellular signaling network analysis using transcriptome-wide gene expression information allowed comprehensive spatial mapping of ligands and receptors, downstream signaling pathways and corresponding intracellular effectors throughout HFs and the skin. It revealed a complex web of cell-cell communication through paracrine signals involving all populations included in our analysis. Notably, the DP appears to be a major signaling center as it exhibited the highest number of interacting ligands and receptors in its signature. Overall, only few signals identified in our study have been previously described, such as Shh secretion from TAC (Hsu et al., 2014b) or Ednrb activation in Mc (Pla and Larue, 2003), but for most of these identified signals, the respective source or target cell had not been previously identified. Of note, Epi seemed heavily involved in signaling crosstalk, primarily with DF and Neg (endothelial and immune cells). Our analysis identified interleukin 6 receptor alpha, Il6ra as a potential activating receptor of Pi3k-Akt signaling in Epi, with ligand Il6 secreted from Neg cells. As Il6 was recently implicated in regulation of keratinocyte terminal differentiation (Son et al., 2014) and wound induced hair neogenesis (Nelson et al., 2015), it will be interesting to confirm the precise source of Il6 and the involvement of Pi3k-Akt pathway in these processes.
Within the HF itself, ORS and Mc expressed many more receptors in their signatures compared to ligands. Interestingly, Mc appears to activate the cAMP signaling pathway through its receptor Npyr in an autocrine fashion. As Npyr ligand Npy has been associated with vitiligo in patients (Tu et al., 2001), our analysis provides a possible molecular insight into melanocyte dysfunction in the pathogenesis of vitiligo. Our focused signaling interaction analysis identified additional signals of particular interest: Nrg2 signaling from DP may activate Erbb3 in Mc, which is involved in Mc maturation and melanoma development (Buac et al., 2009). Similarly, DP-secreted Inhba could bind Acvr2a in Mc to modulate hair pigmentation, as Acvr2a and Bmpr2 ablation in Mc induces graying (Han et al., 2012). Also, Edar is known to activate Shh signaling through the NF-kB pathway (Schmidt-Ullrich et al., 2006), and could be the missing link between Eda secretion from DP and Shh production from TAC. Finally, as Fgfr2IIIb null skins display HF growth retardation and impairment of hair type specification and hair shaft formation (Petiot et al., 2003), Fgfr2 activation in TAC through binding of FGF ligands produced by DPs could be an important regulator of progenitor differentiation.
Overall, by isolating and characterizing transcriptome-wide all specialized key populations of developing HFs at an unprecedented level of refinement and sensitivity, we built a global glossary of specific signature genes and intercellular signaling interactions in the neonatal HF and skin. We share this set of signatures with the scientific community in our open access online database hair-gel.net enabling rapid interrogation of gene expression in all major skin/HF cell populations. It is our hope that promoting broad exchange of gene expression information in this resource will spark development of reporter systems, launch functional genetic studies, and provide a deeper understanding of these SC, progenitor and niche cell populations, and their complex signaling network controlling hair growth.
EXPERIMENTAL PROCEDURES
Mice
K14-H2BGFP, Lef1-RFP, Sox2GFP, K14-RFP and Sox9-GFP mice were described previously (Rendl et al., 2005; Sennett et al., 2015; Wang et al., 2013). The Crabp1-GFP was originally generated by the GENSAT program at the Rockefeller University (Gong et al., 2003). All animals were housed in facilities operated by the Center for Comparative Medicine and Surgery (CCMS) at Icahn School of Medicine. All animal experiments were conducted in accordance with the guidelines and approval of the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai.
Immunofluorescence staining
Back skins sections were fixed with 4% PFA, washed with PBS, then incubated with primary and secondary antibodies as described in more detail in the Supplemental Experimental Procedures.
Cell isolation by FACS
To isolate cells, K14-H2BGFP;Lef1-RFP, Sox2GFP;Lef1-RFP, Crabp1-GFP;Lef1-RFP and K14-RFP;Sox9-GFP P5 back skins were processed as previously described (Clavel et al., 2012; Rendl et al., 2005). Details are provided in Supplemental Experimental Procedures.
cDNA generation, library manufacture, and RNA-sequencing
RNA extraction, amplification and library production were performed as previously described (Sennett et al., 2015). Sequencing libraries were generated with 36 unique barcoded adapters (Biological duplicates for 8 samples from K14-H2BGFP;Lef1-RFP, 3 samples from Sox2GFP;Lef1-RFP, 3 samples from Crabp1-GFP;Lef1-RFP, 4 samples from K14-RFP;Sox9-GFP) and subsequently sequenced on the IlluminaHiSeq 2000 platform using a 100nt single-read setting. Reads were mapped, aligned and quantified to determine the Fragments Per Kilobase of transcript per Million mapped reads (FPKM). This approach resulted in a high-quality output, with a >30 Mean Quality Score (Q score), >90% Perfect Index Reads for all samples, a total of >1.3 billion reads representing on average 46.8 million reads with 31.1 million aligned reads and 8.2 million cDNA fragments per sample (Table S1). Details of library production and RNA-sequencing analysis are provided in Supplemental Experimental Procedures.
Real-Time Quantitative Reverse Transcription PCR
qRT-PCR was performed as previously described (Sennett et al., 2015). Differences between samples and controls were calculated based on the 2−ΔΔCt method and normalized to Gapdh. Measurements were recorded in duplicate. Details and primers used are provided in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
RNA-seq identifies transcriptomes of 14 skin populations during hair growth
SC precursors, progenitors and hair type-specific DP niche signatures are defined
Comparison with embryonic and adult signatures shows dynamic gene expression
Signaling interaction network reveals a complex web of intercellular exchanges
Acknowledgments
We thank Nicole Dubois for valuable comments on the manuscript and Joseph McCarty for providing the ITGB8 antibody. Many thanks to the personnel of the Flow Cytometry Core Facility, and the Microscopy Shared Resource Facility at ISMMS. A.R. was supported by a fellowship from the Fondation pour la Recherche Médicale and the Prix Claude Rozé/CECED. R.S. was supported by fellowship F30AR065847 from NIH/NIAMS. A.M. was supported by NIH grants R01GM098316, R01DK088541, U54CA189201 and U54HL127624. M.R. was supported by grants from the NIH/NIAMS (R01AR059143; R01AR063151) and New York State Department of Health (NYSTEM-C029574), and a fellowship from the Irma T. Hirschl Trust.
Footnotes
ACCESSION NUMBERS
RNA-seq data have been submitted to NCBI-GEO under the accession number GSE77197.
Supplemental Information includes Extended Experimental Procedures, five figures, and seven tables and can be found with this article online.
AUTHOR CONTRIBUTIONS
Conceptualization and Methodology, A.R. and M.R.; Investigation, A.R., Z.W., R.S., W.Q. and D.W.; Validation, N.H., K.W.M. and C.C.; Supervision, R.Y., P.Z. and A.M.; Writing, A.R. and M.R.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Buac K, Xu M, Cronin J, Weeraratna AT, Hewitt SM, Pavan WJ. NRG1 / ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res. 2009;22:773–784. doi: 10.1111/j.1755-148X.2009.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi W, Wu E, Morgan BA. Dermal papilla cell number specifies hair size, shape and cycling and its reduction causes follicular decline. Development. 2013;140:1676–1683. doi: 10.1242/dev.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel C, Grisanti L, Zemla R, Rezza A, Barros R, Sennett R, Mazloom AR, Chung CY, Cai X, Cai CL, et al. Sox2 in the dermal papilla niche controls hair growth by fine-tuning BMP signaling in differentiating hair shaft progenitors. Dev Cell. 2012;23:981–994. doi: 10.1016/j.devcel.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Driskell RR, Giangreco A, Jensen KB, Mulder KW, Watt FM. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development. 2009;136:2815–2823. doi: 10.1242/dev.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enshell-Seijffers D, Lindon C, Morgan BA. The serine protease Corin is a novel modifier of the Agouti pathway. Development. 2008;135:217–225. doi: 10.1242/dev.011031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enshell-Seijffers D, Lindon C, Wu E, Taketo MM, Morgan BA. Beta-catenin activity in the dermal papilla of the hair follicle regulates pigment-type switching. Proc Natl Acad Sci U S A. 2010;107:21564–21569. doi: 10.1073/pnas.1007326107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambardella L, Schneider-Maunoury S, Voiculescu O, Charnay P, Barrandon Y. Pattern of expression of the transcription factor Krox-20 in mouse hair follicle. Mech Dev. 2000;96:215–218. doi: 10.1016/s0925-4773(00)00398-1. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Greco V, Chen T, Rendl M, Schober M, Pasolli HA, Stokes N, Dela Cruz-Racelis J, Fuchs E. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell. 2009;4:155–169. doi: 10.1016/j.stem.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R, Beppu H, Lee YK, Georgopoulos K, Larue L, Li E, Weiner L, Brissette JL. A pair of transmembrane receptors essential for the retention and pigmentation of hair. Genesis. 2012;50:783–800. doi: 10.1002/dvg.22039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Li L, Fuchs E. Emerging interactions between skin stem cells and their niches. Nat Med. 2014a;20:847–856. doi: 10.1038/nm.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Li L, Fuchs E. Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell. 2014b;157:935–949. doi: 10.1016/j.cell.2014.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Kimura S, Kawa Y, Kato M, Mizoguchi M, Soma Y. BMP-4 upregulates Kit expression in mouse melanoblasts prior to the Kit-dependent cycle of melanogenesis. J Invest Dermatol. 2008;128:1220–1226. doi: 10.1038/sj.jid.5701136. [DOI] [PubMed] [Google Scholar]
- Kobielak K, Pasolli HA, Alonso L, Polak L, Fuchs E. Defining BMP functions in the hair follicle by conditional ablation of BMP receptor IA. Journal of Cell Biology. 2003;163:609–623. doi: 10.1083/jcb.200309042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Tumbar T. Hairy tale of signaling in hair follicle development and cycling. Semin Cell Dev Biol. 2012;23:906–916. doi: 10.1016/j.semcdb.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Guo X, Polak L, Lawton LN, Young RA, Zheng D, Fuchs E. Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell. 2011;9:219–232. doi: 10.1016/j.stem.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AM, Reddy SK, Ratliff TS, Hossain MZ, Katseff AS, Zhu AS, Chang E, Resnik SR, Page C, Kim D, et al. dsRNA Released by Tissue Damage Activates TLR3 to Drive Skin Regeneration. Cell Stem Cell. 2015;17:139–151. doi: 10.1016/j.stem.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimori N, Fuchs E. Paracrine TGF-beta signaling counterbalances BMP-mediated repression in hair follicle stem cell activation. Cell Stem Cell. 2012;10:63–75. doi: 10.1016/j.stem.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouspenskaia T, Matos I, Mertz AF, Fiore VF, Fuchs E. WNT-SHH Antagonism Specifies and Expands Stem Cells prior to Niche Formation. Cell. 2016;164:156–169. doi: 10.1016/j.cell.2015.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Conti FJ, Grose R, Revest JM, Hodivala-Dilke KM, Dickson C. A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development. 2003;130:5493–5501. doi: 10.1242/dev.00788. [DOI] [PubMed] [Google Scholar]
- Pla P, Larue L. Involvement of endothelin receptors in normal and pathological development of neural crest cells. Int J Dev Biol. 2003;47:315–325. [PubMed] [Google Scholar]
- Qiao W, Wang W, Laurenti E, Turinsky AL, Wodak SJ, Bader GD, Dick JE, Zandstra PW. Intercellular network structure and regulatory motifs in the human hematopoietic system. Mol Syst Biol. 2014;10:741. doi: 10.15252/msb.20145141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendl M, Lewis L, Fuchs E. Molecular dissection of mesenchymal-epithelial interactions in the hair follicle. Plos Biology. 2005;3:1910–1924. doi: 10.1371/journal.pbio.0030331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendl M, Polak L, Fuchs E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes & Development. 2008;22:543–557. doi: 10.1101/gad.1614408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezza A, Sennett R, Rendl M. Adult stem cell niches: cellular and molecular components. Curr Top Dev Biol. 2014;107:333–372. doi: 10.1016/B978-0-12-416022-4.00012-3. [DOI] [PubMed] [Google Scholar]
- Rompolas P, Deschene ER, Zito G, Gonzalez DG, Saotome I, Haberman AM, Greco V. Live imaging of stem cell and progeny behaviour in physiological hair-follicle regeneration. Nature. 2012;487:496–499. doi: 10.1038/nature11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Greco V. Stem cell dynamics in the hair follicle niche. Semin Cell Dev Biol. 2014;25–26:34–42. doi: 10.1016/j.semcdb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlake T. Determination of hair structure and shape. Semin Cell Dev Biol. 2007;18:267–273. doi: 10.1016/j.semcdb.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich R, Tobin DJ, Lenhard D, Schneider P, Paus R, Scheidereit C. NF-kappaB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post-initiation hair placode down growth. Development. 2006;133:1045–1057. doi: 10.1242/dev.02278. [DOI] [PubMed] [Google Scholar]
- Sennett R, Rendl M. Mesenchymal-epithelial interactions during hair follicle morphogenesis and cycling. Semin Cell Dev Biol. 2012:917–927. doi: 10.1016/j.semcdb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennett R, Wang Z, Rezza A, Grisanti L, Roitershtein N, Sicchio C, Mok KW, Heitman NJ, Clavel C, Ma’ayan A, et al. An Integrated Transcriptome Atlas of Embryonic Hair Follicle Progenitors, Their Niche, and the Developing Skin. Dev Cell. 2015;34:577–591. doi: 10.1016/j.devcel.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son ED, Kim HJ, Park T, Shin K, Bae IH, Lim KM, Cho EG, Lee TR. Staphylococcus aureus inhibits terminal differentiation of normal human keratinocytes by stimulating interleukin-6 secretion. J Dermatol Sci. 2014;74:64–71. doi: 10.1016/j.jdermsci.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Tu C, Zhao D, Lin X. Levels of neuropeptide-Y in the plasma and skin tissue fluids of patients with vitiligo. J Dermatol Sci. 2001;27:178–182. doi: 10.1016/s0923-1811(01)00134-7. [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang Z, O’Loughlin E, Wang L, Fan X, Lai EC, Yi R. MicroRNA-205 controls neonatal expansion of skin stem cells by modulating the PI(3)K pathway. Nat Cell Biol. 2013;15:1153–1163. doi: 10.1038/ncb2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Park HY. Melanocytes: a window into the nervous system. J Invest Dermatol. 2012;132:835–845. doi: 10.1038/jid.2011.386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.