

Abstract
Medication adherence and insufficient drug levels are central to HIV/AIDS disease progression. Recently, Fletcher et al. confirmed that HIV patients on oral antiretroviral therapy had lower intracellular drug concentrations in lymph nodes than in blood. For instance, in the same patient, multiple lymph node drug concentrations were as much as 99 % lower than in blood. This study built upon our previous finding that HIV patients taking oral indinavir had 3-fold lower mononuclear cell drug concentrations in lymph nodes than in blood. As a result, an association between insufficient lymph node drug concentrations in cells and persistent viral replication has now been validated. Lymph node cells, particularly CD4 T lymphocytes, host HIV infection and persistence; CD4 T cell depletion in blood correlates with AIDS progression. With established drug targets to overcome drug insufficiency in lymphoid cells and tissues, we have developed and employed a “Systems Approach” to engineer multi-drug-incorporated particles for HIV treatment. The goal is to improve lymphatic HIV drug exposure to eliminate HIV drug insufficiency and disease progression. We found that nano-particulate drugs that absorb, transit, and retain in the lymphatic system after subcutaneous dosing improve intracellular lymphatic drug exposure and overcome HIV lymphatic drug insufficiency. The composition, physical properties, and stability of the drug nanoparticles contribute to the prolonged and enhanced drug exposure in lymphoid cells and tissues. In addition to overcoming lymphatic drug insufficiency and potentially reversing HIV infection, targeted drug nanoparticle properties may extend drug concentrations and enable the development of long-acting HIV drug therapy for enhanced patient compliance.
Keywords: Lymphatic drug insufficiency, Long-acting, Targeted drug delivery, Systems approach, HIV
Introduction to Systems Approach in drug delivery and targeting
Advances in technology have provided an impetus for tremendous growth in the pharmaceutical industry. Novel drug delivery systems are believed to hold promising business opportunities and have attracted intense research interest. Drug delivery approaches could be defined broadly as physical dosage form, molecular design optimization and engineering, and other physiochemical approaches built on the understanding of drug metabolism and pharmacokinetics (DMPK) as well as therapeutic impact. With a more narrow definition, novel drug dosage forms, carriers, and formulations capture US$50–82 billion worth of revenue annually in the USA [1]. As will be discussed below, clinical translation of advances in medical sciences and drug discovery have been challenged by late-stage clinical failure due to unforeseen toxicity or lack of efficacy. To address these limitations and to accelerate drug development, this commentary focuses on a new strategy called a Systems Approach for drug delivery and targeting. As an example, this Systems Approach has been used to address drug insufficiency and residual virus in lymphoid tissues of HIV patients on chronic drug therapies. While we focus on HIV/AIDS treatment, the Systems Approach may be used to improve safe and effective exposure of drugs to a wide range of diseases including cancer, metabolic, cardiovascular, and neurological diseases and symptoms.
With completion of human genome mapping and characterization of candidate molecular mechanisms associated with human diseases, drug targets have continued to grow from about 500 in the 1990s to over 1700 validated targets for which chemical or biological compounds could be used to influence the course of human diseases. Depending on the stringency in validation and definition, the current drug target number has been estimated to be 2200 or more [2]. Yet, the growth in drug-target number has not been fully realized in numerical growth of new drugs approved for disease treatment in the past 25 years. The pharmaceutical industry that sponsors new drug development has invested in a number of approaches, particularly early attrition to shorten drug development time. This approach uses in vitro and in vivo tools to evaluate drug properties including absorption, distribution, metabolism, and elimination (ADME) and has helped eliminate drugs with liability in poor absorption, high metabolic rate, toxicity, and drug-drug interactions. However, many of the drugs that have passed the early screen still failed in late-stage clinical testing due to unanticipated toxicity or lack of efficacy [3]. These late-stage failures are costly to the pharmaceutical industry, as sponsors may spend up to several hundred million dollars in phase 2 and 3 human clinical tests [4]. While the role and quality of data linking drug target and disease symptoms leading to drug failure is being debated, it is clear that drug distribution to off-target tissues and cells, and related metabolic conversion, could pose significant impact on both efficacy and toxicity (or safety).
We propose a Systems Approach for drug delivery and targeting to improve on-target drug distribution into tissues and cells that mediate or are linked to a clinical syndrome. The Systems Approach incorporates a holistic approach to drug development that includes the biological and physiological context, drug distribution, drug action, and validated measures or surrogate markers of disease outcomes. With the understanding and definition of key or rate-limiting steps, an appropriate drug delivery system could be established to best deliver the drug to target tissues and cells while a maximal overlap between the distribution of drug molecule (and exposure) and that of drug targets (cells and tissues) throughout the body could be realized. Thus, based on the integration of molecular, cellular, and physiological knowledge, and an understanding of pharmacology and pharmacokinetics of drug-drug target interactions, the Systems Approach for drug delivery could be specifically designed to reduce off-target effects and improve on-target drug exposure in vivo. This concept includes consideration of the time-course of drug concentrations and the extent of drug exposure to target cells and tissues in the body in relation to blood levels, which are typically monitored by the classical pharmacokinetic parameters.
The emphasis of most drug delivery systems is on developing sustained drug release polymers or smart particles and demonstrating their capabilities in vitro as proof of concepts. In contrast, the Systems Approach provides guidance and specification on in vivo tissues and cell targets prior to initiating the engineering aspect of nanoparticles such as surface decoration with targeting moieties. For the Systems Approach, the simplest tissue and cell localization strategy would be favored. This minimalist approach is beneficial to develop a scalable drug delivery system requiring fewer steps and to ensure a stable pharmaceutical dosage or a particulate form to reach the target site of action. The overall strategy of the Systems Approach would include the drug formulation with a cost-effective and scalable preparation method to accelerate product development.
A major goal of the Systems Approach to drug delivery and targeting is to accelerate product development through reducing risks and improving success rates by considering the following issues early in the course of product development. A visual representation can be seen in Fig. 1.
The definition and characteristics of drug targets at molecular, intra/extra-cellular, and tissue levels
The distribution, uptake, metabolism, and elimination of a drug or drug candidate (with unique properties) in the tissues and cells within the body
Opportunity to integrate the physiochemical properties of a drug and the physiological/biological systems limitation to design the simplest and best drug delivery system
Design a simple and effective means to influence drug distribution and exposure in cells and tissues expressing high levels of drug targets
Validate the surrogate marker that represents disease outcome to assist in early decisions on drug development
Fig. 1.
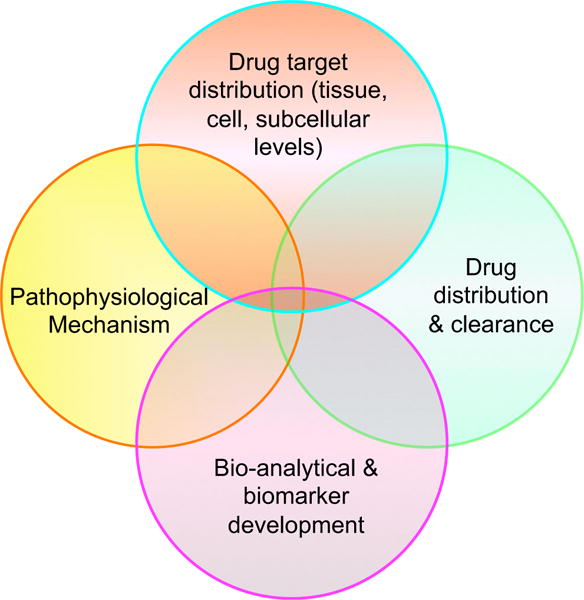
A graphical representation of the Systems Approach to drug delivery and targeting. The Systems Approach is based on integration of the intercepts among four areas of biomedical knowledge: (1) drug target localization information from an intracellular context to cells and tissues as well as organs in the body, (2) knowledge about the biodistribution and clearance of drug candidates in the body over time, (3) the physiological and pathological consequences of the aberration in drug targets that reflect disease symptoms, and (4) quantitative or bioanalytical markers that could be measured in blood or other samples for predicting drug levels and disease outcome. These overlapping themes and conditions should be taken into consideration when implementing the Systems Approach to drug delivery and targeting
Effectiveness of combination antiretroviral HIV therapies is limited by drug exposure and residual virus in tissues
To demonstrate the application of the Systems Approach, we will use the example of HIV and the development of novel therapeutics to target sites of viral persistence. HIV infection is now well-described, and surrogate markers of disease progression have already been validated. HIV infection in CD4 T cells leads to T cell decline that can be detected in blood, resulting in disease progression to AIDS opportunistic infections and eventual death [5]. While viremia is an important factor, a precipitous decline in CD4 T cells is among the best clinical hallmarks of disease progression. With research investment from the government, pharmaceutical industry, and academia, all HIV proteins have been identified and their molecular structures have been elucidated in intricate detail. These HIV molecules graphically presented in Fig. 2 form the basis of druggable targets that are validated to allow the development of multiple compounds with selective lethal efficacy against HIV but minimal cross activity against human host protein counterpart. Many of these HIV drugs, approved by the FDA for human use, are shown to suppress HIV in plasma/blood selectively and effectively. The FDA has approved 25 agents for the treatment of HIV, some of which are available as combination products. These compounds are listed below in Fig. 2 according to their targets.
Fig. 2.
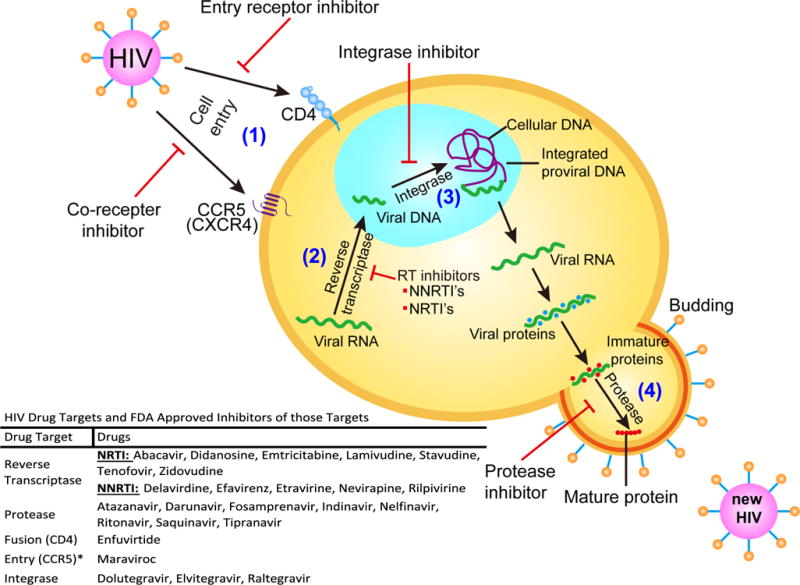
HIV infection, replication, key proteins, and drug targets. Shown are the protein targets that have been identified in HIV as well as the selective effects of HIV therapies. These targets are responsible for steps in viral replication such as (1) cell entry, (2) reverse transcription into viral DNA, (3) integration into the human genome, and (4) maturation of viral proteins into functional proteins. This figure also lists FDA-approved drugs that target specific proteins such as (1) CCR5 and CD4 inhibitors, (2) nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs), (3) integrase inhibitors, and (4) protease inhibitors. HIV regimens commonly include inhibitors of multiple steps in viral replication to reduce the incidence of viral drug resistance. *As a note, additional entry proteins have been characterized such as CXCR4, but there are currently no FDA-approved drug products targeting that protein
The number of drugs in the list approved for prevention and treatment of HIV infection continues to grow. Anti-HIV drugs are divided into different classes depending on which viral protein or enzyme they target, such as protease, reverse transcriptase, integrase, fusion proteins, and co-receptors [6]. These HIV proteins are well understood and have been validated to modify virus levels and rate of disease progression. Early studies with mono-drug therapies have exhibited high frequency in harboring drug-resistant viruses that are linked to therapeutic failure [7]. The combination of HIV drugs targeted to multiple HIV proteins or the same protein, i.e., reverse transcriptase at different druggable domains, has proven to keep the induction of drug-resistant virus at bay [8]. The combination of highly active antiretroviral therapies (often referred to as HAART) has also been proven to reduce the detectable viral load (typically based on a highly sensitive assay capable of detecting down to a single virus copy of HIV’s ribonucleotide sequences) in blood to nearly undetectable levels in HIV-infected patients [9]. However, patients with no detectable virus in the blood still carry virus in lymph nodes and tissues.
Despite the success in suppressing virus to undetectable levels in blood, the cure for HIV remains elusive as virus will rebound with stoppage of HAART [10]. Even prior to the cessation of HAART, virus can be isolated from the lymph nodes of patients. Additional analysis reveals that virus isolated from the lymph nodes of patients without detectable virus in blood remains sensitive to the drug combinations in the prescribed therapy. Therefore, drug resistance is not the major cause of inability to clear virus in tissues. While the fraction of residual virus between those latently hidden in host CD4 T cells and those accessible for HIV drug action is being debated, active replication of HIV (albeit at low levels) could be revealed by the synthesis of HIV RNA in infected cells and tissues.
Therefore, in 2003, we determined whether the residual HIV-infected cells in lymphoid tissues and cells could be a result of insufficient drug exposure in these tissues after oral HAART. To address this question, the lymph nodes of HIV patients on the oral HIV protease inhibitor indinavir were collected and peripheral blood mononuclear cells (which include CD4 T lymphocytes) in blood and mononuclear cells from lymph nodes were isolated [11]. At the time of tissue and cell collection, these patients were on oral indinavir therapy and had reached steady-state drug levels in blood after multiple doses of indinavir. The blood mononuclear cells and the equivalent cells from lymph nodes were collected at the same time during patients’ clinical visit. As shown in Table 1, we found that intracellular indinavir levels in the same cells from lymph nodes were about 3-fold lower than those found inside the cells from blood. This data validates the hypothesis that drug insufficiency is one of the factors likely to contribute to the residual virus in lymphoid tissues. In other words, limited drug exposure in tissue after oral administration is insufficient to provide viral control or clear HIV from the lymphoid tissues. Thus, our research team has not only reported the existence of drug insufficiency in lymphoid tissues after oral dosing but also highlighted the need to address this issue for improved effectiveness of HIV therapy.
Table 1.
Indinavir concentration (nM) in plasma, peripheral blood mononuclear cells (PBMCs), and lymph node mononuclear cells (LNMCs) in HIV-1-infected humans on oral indinavir therapya
| Plasma | PBMC | LNMC | LNMC/PBMC ratiob | |
|---|---|---|---|---|
| HIV-positive patients on oral indinavir therapy | ||||
| Patient A | 1004 | 1401 | 489 | 0.35 |
| Patient B | NA | 1531 | 432 | 0.28 |
| Patient C | NA | 432 | 101 | 0.23 |
Patients received oral indinavir 800 mg TID as part of combination therapy for at least 1 month prior to testing. PBMCs were isolated from blood and LNMCs were isolated from lymph node tissue for drug analysis as previously described [17]. Intracellular concentration (nM) was calculated assuming an average intracellular volume of 4×10−9 mL per cell [22]. Reported pharmacokinetic parameters of indinavir include a Cmax of 11000 nM and Ctrough of 211 nM
Ratio of intracellular drug concentration in LNMCs over intracellular concentration in PBMCs
NA data not available
In support of this hypothesis of drug insufficiency, Fletcher et al. reported in a recent prospective study that HIV+ patients on oral antiretroviral therapy exhibited lower drug concentrations in cells of numerous lymph nodes compared to those in blood [12]. Multiple drug concentrations in lymph nodes were as much as 99 % lower than those in blood for the same patient. An association was also noted between low drug concentrations and persistent viral replication [12]. Since the lymphatic system is a known reservoir of HIV, lack of drug penetration into these tissues could be the source of viral rebound [13]. Fletcher et al.’s data confirmed our initial 2003 finding on lymphatic indinavir insufficiency in treated patients and extended this hypothesis to multiple classes of HIV drugs.
Lymphatic drug insufficiency of oral HIV therapies is generally led by several biological barriers. First, limited drug absorption via the gut and biotransformation or elimination by the liver and kidney result in only a fraction of orally administered drug distributing to the blood (typically measured as plasma drug concentrations) [14]. Second, drugs in plasma exist in either protein-bound form or free soluble form, and generally, only drug in free soluble form can penetrate across the blood-lymphatic barriers [15]. Meanwhile, the fraction of soluble drug in lymph nodes can, in return, diffuse readily out of nodes with little resistance. Thus, strategies intended to increase lymphoid drug exposure require not only improved drug distribution but also enhanced retention in the lymphatic system. By understanding the target system and developing new drug formulations with a focus on these elements, we have developed a strategy to deliver and trap anti-HIV drug particles in lymph nodes in order to overcome the drug insufficiency observed in patients on oral HAART regimens.
Long-acting and enhanced intracellular exposure of anti-HIV therapy may address patient compliance and overcome lymphatic drug insufficiency
While a number of oral antiretroviral combination drug therapies are effective in producing undetectable viral blood levels and delaying disease progression, clinical experience points to patient compliance and adherence as a significant barrier to treatment success. Patient compliance and inability to adhere to an oral, frequent dosing schedule (once a day or more) has posed significant challenges, which is further exacerbated by the chronic nature of the treatment. Many scientists and clinicians now believe that non-adherence is a key reason that leads to the emergence of drug resistance and has weakened therapeutic effectiveness. Poor maintenance of drug levels may lead to persistent viral levels, increased risk of harboring drug-resistant virus, and potential treatment failure. Without patient compliance and adherence to HIV medication, the efficacy of viral suppression would never be achieved. Without a drug combination that provides sufficient cellular and plasma drug levels for an extended period and reduces the frequency of dosing, patient compliance issues (as in the case of oral fixed drug combination) will remain.
Recently, Hall and colleagues [16] reported that only about 25 % of 1.1 million HIV-infected patients exhibit sufficient viral suppression, while 63 % of the patients have not remained in care. This HIV therapeutic treatment gap, reported in 2013, is often referred to as a “leaky cascade of care.” This 63 % gap in treatment is a large proportion of the reported population that could have remained in care if an emphasis was placed on patient compliance. By developing a drug combination with less frequent dosing as well as fewer side effects, patient compliance would be expected to improve. A new formulation that incorporates multiple anti-viral therapies in a single subcutaneous administration may overcome barriers to patient compliance seen with high-frequency oral dosing and reduce patient pill burden. In addition to benefits in compliance, a focused delivery of anti-viral therapies to the lymphatic system can also attenuate the need for high systemic drug levels and reduce dose-dependent, peripheral toxicities. This therapeutic strategy, along with outreach efforts, would be able to bridge the gap and directly address the “leaky cascade of care.” Remedy of the “leaky cascade” is essential to achieve the final eradication of HIV.
In addition to the large fraction of patients who are out of care or poorly adherent to therapy, inconsistent drug penetration and insufficiency of drug exposure outside of highly perfused organs such as the kidney, liver, and lung pose a different set of challenges. Drug access to HIV host T cells within the lymph nodes after oral drug dosing is subject to limited absorption, first-pass and enzyme-mediated drug inactivation, P glycoprotein drug efflux, and elimination in the gut and liver. As shown in Fig. 3, the significantly lower drug fraction that appears in the blood is faced with blood-to-lymphatic barriers before reaching the lymph nodes. It is possible that poor drug penetration into lymph nodes is the key mechanism leading to low but detectable and persistent virus in the lymph nodes. In addition to the challenge in providing sufficient drug exposure in lymphoid and other tissues with limited blood perfusion, the frequent dosing requirement for oral combination HIV drug therapy poses an additional patient compliance challenge among several key demographics. As a chronic treatment, one or more daily dosing causes a stigma for adolescents and a practical/emotional challenge for single parents. Many HIV patients who are single parents face emotional and job demands, which can lead to missed doses. A chronic and frequent dosing schedule also poses a significant challenge for the drug-dependent and drug abuse population, who carry a high rate of HIV transmission. Many patients are also unable to take oral medications while admitted in care facilities for other treatments. With all these factors in mind, a long-acting and injectable dosage form that provides enhanced blood and tissue drug exposure is urgently needed to ameliorate patient compliance and practical needs. At the same time, if the long-acting dosage form could address the tissue drug insufficiency, a cure for AIDS would be possible.
Fig. 3.
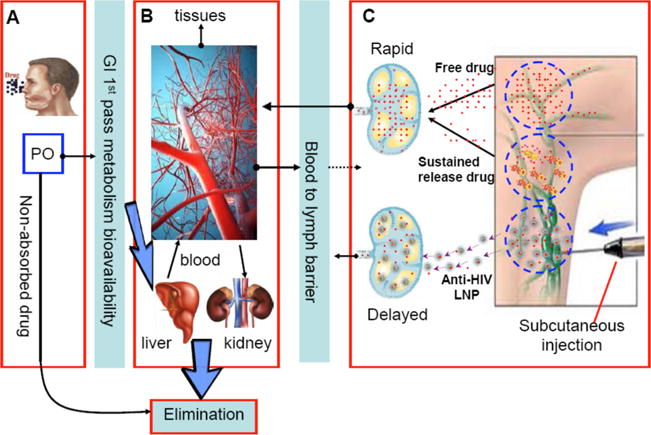
Distribution of drug based on oral (a) or subcutaneous (c) route of administration. A visual representation of the challenges faced in overcoming lymphatic drug insufficiency. Oral dosing (a) of anti-viral therapy has to overcome factors such as solubility of the drug, absorption of free drug through the GI, first-pass metabolism, and other elimination processes. After entering the systemic circulation (b), free drug can accumulate in peripheral tissues and only a small fraction of drug can enter the subcutaneous space (c). Free drug that does enter the lymphatic system can also be rapidly reabsorbed into the blood. Subcutaneous administration of drug can provide higher concentrations of drug in the lymph nodes; however, free drug rapidly transits to the blood with low residence time in the target tissues. Sustained release systems slow the release of free drug but do not alleviate rapid absorption into the blood. Anti-HIV lipid nanoparticles (LNP) accumulate in the lymph nodes and trap the drug at the target site allowing for increased concentration of drug as well as residence time in the lymph
Systems Approach to anti-HIV nanomedicine intended to enhance intracellular drug exposure with anti-HIV drug combination particles
There are a number of sustained drug release systems that are designed to provide long-lasting plasma drug levels. Many dosage forms are designed to slowly release the drug by encasing drug within polymeric matrices of nanoparticles, hydrogels, or transdermal patches. The long-acting drug delivery systems will allow the drug to slowly release from the matrices deposited at the site of injection or within the gastrointestinal (GI) tract and then appear in the blood at a defined rate, providing plasma drug concentrations within a therapeutically useful range for extended time (Fig. 3c).
Unlike these typical sustained release drug delivery systems, which are designed to provide sustained drug levels in blood, the Systems Approach is built on the knowledge of HIV infection pathophysiology and target tissues of viral replication as well as the understanding of drug distribution and clearance mechanisms. Under the guidance of the System Approach, a desirable drug dosage form could be designed to not only deliver a major fraction of HIV drugs to HIV host cells and tissues but also maintain drug levels in these tissues for prolonged periods, addressing both issues of drug insufficiency and patient non-adherence due to frequent dosing schedules. In other words, the goal is not just maintaining plasma drug levels over time as in a sustained release strategy but to transport and accumulate anti-HIV drug particles to drug-insufficient sites, such as lymph nodes, so that the overall drug exposure in lymphoid tissue and cells would be enhanced (Fig. 3c). For clinical translation, we favor the simplest tissue and cell localization strategy to provide cost-effective and scalable pharmaceutical preparation to accelerate product development. In the case of the HIV treatment context, by increasing drug exposure at HIV reservoir sites (i.e., the lymphatic system), we hope to reduce the incidence of viral rebound.
To address drug insufficiency, we have systematically developed and evaluated anti-HIV drug particles that are capable of gaining first-pass access to the lymph nodes and spreading throughout the lymphatic system with little or no blood redistribution. As a result, metabolic liabilities and toxicities due to oral dosing and resulting first-pass GI and liver drug exposure are greatly reduced or avoided. This concept, initially developed with a protease inhibitor indinavir in a proof-of-principle primate study, is based on the ability of lipid to stabilize water-insoluble indinavir in 50–90 nm drug-lipid particles at neutral pH (7–8). The drug releases from particles readily at pH 4–5 in solution. When indinavir particles were given subcutaneously to primates, they resulted in greatly enhanced drug concentrations in lymph nodes throughout the body when compared to free indinavir given via the traditional oral route (Table 2). These particles also distributed to lymph nodes throughout the lymphatic system, from inguinal and mesenteric to submandibular and other visceral and peripheral nodes, a finding that we have validated in subsequent studies [11, 17]. Additionally, HIV-infected primates given the indinavir-lipid particles showed a reduction in viral load in plasma and lower residual virus levels in lymph nodes, as well as reversal of CD4 T cell decline, indicating a disruption in the natural course of disease progression [17]. These findings demonstrate that lipid-bound drug not only targets the system of interest (namely the lymphatic system) but also retains pharmaceutical activity.
Table 2.
Indinavir concentration in plasma and lymph node tissuea in HIV-infected macaques treated with either oral soluble indinavirb or subcutaneous lipid-associated indinavirc at a normalized dose of 25 mg/kg. Lymph node drug concentration is expressed as lymph node-to-plasma ratio
| Animal ID | Plasma (ng/mL) | Lymph node:plasma ratio (indinavir)
|
|||
|---|---|---|---|---|---|
| Inguinal | Axillary | Mesenteric | Ileocecal | ||
| Free indinavir (PO) | |||||
| 94079 | 9.8 | 0.03 | 0.01 | – | – |
| 94094 | 5.2 | 0.01 | 0.02 | – | – |
| 94096 | 39.8 | 0.01 | <0.01 | – | – |
| Lipid-associated indinavir (SC) | |||||
| M98165 | 79.5 | 3.43 | 2.46 | 4.99 | 32.55 |
| J98328 | 52.0 | 6.27 | 2.48 | 16.26 | 7.01 |
For lymph node drug concentration, 1–10 mg of lymph node tissue was homogenized and drug was extracted as described previously [17]
Free drug was administered orally in solution at a dose of 25 mg/kg, and samples were collected 2–4 h after administration. Mesenteric and ileocecal lymph nodes were not sampled
Lipid-associated drug was administered subcutaneously at 10 mg/kg (data normalized to a dose of 25 mg/kg for comparison), and samples were collected 26–28 h after administration
These proof-of-principle studies with indinavir-lipid nanoparticles demonstrate the potential of the Systems Approach in developing a method to target HIV drug to sites of viral persistence, overcome drug insufficiency, and enhance viral suppression. However, due to the high potential for resistance in HIV, drug monotherapy is not indicated as a treatment paradigm. Rather, a combination of at least three drugs targeting at least two viral proteins is required to sufficiently combat HIV. Thus, to move our delivery system forward in a clinically relevant direction, we pursued a combination anti-HIV drug nanoparticle formulation to increase clinical efficacy and reduce the risk of harboring HIV drug-resistant virus.
Guided by our work with lipid-associated indinavir, we developed lipid particles containing three drugs and targeting two different viral proteins. Lopinavir and ritonavir are protease inhibitors that are highly stable and highly hydrophobic, resulting in strong natural lipid association [18]. Tenofovir (PMPA) is a hydrophilic nucleotide analogue reverse transcriptase inhibitor, a key class among first-line anti-HIV therapy, and is phosphorylated and retained intracellularly [19]. Tenofovir is commercially available as tenofovir disoproxil fumarate, a prodrug oral dosage form of active PMPA. In our formulation, we have chosen to incorporate the active PMPA instead of the prodrug (which is converted and presented in plasma as PMPA) form because of the preferential transit of our particles into the site of interest as demonstrated by the indinavir studies. When combined into lipid-drug nanoparticles, the lipid association of lopinavir and ritonavir was highly efficient and stable, with simultaneous association of a significant fraction of hydrophilic PMPA in a stable and reproducible manner [20]. Accumulated data suggests the importance of drug particles maintaining their size in vivo for optimal localization in the lymph nodes [21]. While lopinavir has been recently replaced by darunavir, a more potent protease inhibitor, in HIV treatment guidelines, lopinavir continues to be clinically useful in oral HAART regimens. Current guidelines published by the National Institute of Health (NIH) for first-line HIV combination regimens are shown in Table 3. Other drug combinations, including those recommended in the most current guidelines, are currently under investigation for use in combination lipid-drug particles.
Table 3.
Recommended first-line treatments of HIV-1-infected adults and adolescents
| Integrase inhibitor based |
| Dolutegravir/abacavir/lamivudinea |
| Dolutegravir plus tenofovirb/emtricitabine |
| Elvitegravir/cobocistat/tenofovir/emtricitabinec |
| Raltegravir plus tenofovir/emtricitabine |
| Protein inhibitor based |
| Darunavir/ritonavir plus tenofovir/emtricitabine |
Only for patients who are HLA-B*5701 negative
Tenofovir presented in this table is the tenofovir disoproxil fumarate form
Only for patients with pre-ART CrCl>70 mL/min
To validate the ability of this lipid nanoparticle platform to enhance delivery of drugs to lymphatic tissues and cells, the nanoparticle formulation composed of lopinavir, ritonavir, and PMPA was evaluated in primates [20]. As with lipid-associated indinavir, subcutaneous administration of these combination anti-HIV lipid particles resulted in enhanced intracellular drug concentrations in peripheral lymph nodes when compared to drug administered in free form (Table 4). In primates treated with a subcutaneous free drug formulation of all three compounds, drug levels were low or undetectable in plasma after 24 h, and only intracellular PMPA was detected in the lymph node mononuclear cells (LNMC). In contrast, primates treated with lipid-associated drug combination were found to have markedly higher drug concentrations both in plasma and intracellularly in lymph nodes at 24 h after administration with the exception of PMPA. More recent studies indicate that lipid-association can extend intracellular drug exposure in lymph nodes to over 7 days, providing much higher drug exposure than is achievable with subcutaneous injection of free drug (data not shown). These data verify that anti-HIV drug nanoparticles are superior to free drug in improving intracellular drug exposure in lymph nodes after subcutaneous administration.
Table 4.
Drug concentrations (ng/mL) in plasma and lymph node mononuclear cells (LNMCs) of primates 24 h after subcutaneous administration of a three-drug combination in either free or lipid-associated forma
| Lopinavir
|
Ritonavir
|
PMPA
|
||||
|---|---|---|---|---|---|---|
| Plasma | LNMC | Plasma | LNMC | Plasma | LNMC | |
| Free drug combination | ||||||
| M10066 | 8.12 | 0.0 | 0.00 | 0.0 | 0.00 | 224.2 |
| M10068 | 17.64 | 0.0 | 0.00 | 65.3 | 0.00 | 287.2 |
| Lipid-associated drug combination | ||||||
| R10142 | 740.07 | 1222.2 | 266.32 | 1660.8 | 117.26 | 167.1 |
| Z11084 | 696.49 | 1202.5 | 286.63 | 1622.8 | 89.99 | 212.7 |
Macaques received lopinavir (25 mg/kg), ritonavir (14.3 mg/kg), and PMPA (17.1 mg/kg) as a single subcutaneous bolus in either a free drug suspension or associated with lipid nanoparticles, as described previously [20]. Plasma and inguinal lymph nodes were collected 24 h after administration. LNMCs were isolated, and intracellular concentration was calculated assuming an average intracellular volume of 4×10−9 mL per cell [22]
In addition to improving intracellular drug exposures in the nodes, we also surveyed mononuclear cells in blood. These lipid-drug complexes also enhanced and prolonged intracellular drug concentrations of all three drugs in plasma and mononuclear cells in the blood, suggesting their potential as a long-acting antiretroviral agent (Table 5). Plasma drug concentrations in animals treated with free drug formulations exhibit no detectable drug level by 7 days. In contrast, significant and detectable plasma drug concentrations persist in primates treated with lipid-associated drug combination. We also noted persistence of drug in peripheral blood mononuclear blood cells treated with lipid-associated drug combination, which generally paralleled plasma concentrations. Overall, the average plasma drug exposure per dose, as measured by area under the curve (AUC) from 0 to 168 h, improved 18-, 14-, and 7-fold for lopinavir, ritonavir, and PMPA, respectively, by using lipid-associated drug combination in comparison to free drug. Therefore, the simultaneous and long-acting delivery of multiple anti-HIV drugs in a targeted manner to the lymphatic system could address not only issues of drug insufficiency but also challenges of patient compliance seen with daily oral therapy.
Table 5.
Drug concentrations (ng/mL) in plasma and peripheral blood mononuclear cells (PBMCs) over time in primates (n=4) after subcutaneous administration of a three-drug combination in either free or lipid-associated form
| Time (h) | Lopinavir
|
Ritonavir
|
PMPA
|
|||
|---|---|---|---|---|---|---|
| Plasma | PBMC | Plasma | PBMC | Plasma | PBMC | |
| Free drug combinationa | ||||||
| 1 | 88.2±89.5 | 482.5±295.7 | 45.8±25.9 | 730±655.1 | 16,138.8±3924.4 | 887.5±1052.6 |
| 24 | 24.5±26.3 | 40.0±69.4 | 5.9±9.3 | 0±0 | 0.9±1.1 | 2.5±3.4 |
| 168 | 0±0 | 0±0 | 0±0 | 0±0 | 0±0 | 0±0 |
| 0–168 (AUC)b | 3.83±4.04 | – | 1.39±1.18 | – | 56.6±17.04 | – |
| Lipid-associated drug combinationa | ||||||
| 1 | 358.9±76.9 | 462.5±278.1 | 142.9±47.2 | 642.5±260.1 | 10,415.5±1665.4 | 322.5±232.8 |
| 24 | 596.7±122.7 | 815.0±561.9 | 163.6±115.6 | 1022.5±724.5 | 3598.9±3508.5 | 175.0±35.3 |
| 168 | 7.1±4.0 | 20.0±26.0 | 7.8±3.9 | 330±284.7 | 814.9±805.7 | 62.5±21.0 |
| 0–168 (AUC)b | 69.6±10.7 | – | 19.4±12.2 | – | 395.0±344.5 | – |
Macaques received lopinavir (25 mg/kg), ritonavir (14.3 mg/kg), and PMPA (17.1 mg/kg) as a single subcutaneous bolus in either a free drug suspension or associated with lipid nanoparticles, as described previously [20]. Venous blood was collected at the indicated time points, and plasma and PBMCs were isolated. Intracellular concentration was calculated assuming an average intracellular volume of 4 × 10−9 mL per cell [22]
Area under the plasma time course curve (AUC) was calculated from 0 to 168 h using the trapezoidal rule
Summary
With the advent of new technologies that allow for the elucidation of physiological systems and drug targeting in disease states, an improvement in the approach to drug development should follow. The well-defined pathophysiology, cellular targets, recognized sites of viral persistence, and availability of effective and selective drugs in HIV lend themselves to the Systems Approach. A Systems Approach in drug targeting has been applied in developing a simple, yet effective means to overcome HIV drug insufficiency associated with oral HIV drug therapy and to improve intracellular drug exposure for an extended time. Due to the prolonged drug exposure and enhanced drug levels in the lymphatic system, our lipid-drug nanoparticles can be used to address the current clinical challenges of HIV therapy, which include noncompliance, peripheral toxicities, and drug insufficiency. While the doses used in the data presented are more in line with proof-of-concept, single-dose studies and would require optimization for clinical use, the concept and application of Systems Approach are still valid. By systematically identifying the lymphatic system and associated mononuclear cells as relevant targets, developing an in vivo delivery method, and demonstrating utility with a clinically relevant drug regimen, the Systems Approach has been shown to be a powerful tool in the development of novel therapeutics.
Acknowledgments
This work is supported in part by NIH grants AI-077390, AI077390-S1, AI-077390-S2, AI-077390-S3, UM1 AI-120176, and 1UL1-RR025014. RJY Ho is also supported by Milo Gibaldi endowment. This work is also supported in part by the National Natural Science Foundation of China (Nos. 81472767 and 81201709). JS is now holding an appointment at Fuzhou University.
Footnotes
Conflict of interest The authors declare that they have no competing interests.
References
- 1.Ho RJY, Gibaldi M. Biotechnology and biopharmaceuticals: transforming proteins and genes into drugs. 2nd. Hoboken: Wiley-Blackwell; 2013. [Google Scholar]
- 2.Qin C, et al. Therapeutic target database update 2014: a resource for targeted therapeutics. Nucleic Acids Res. 2014;42:D1118–1123. doi: 10.1093/nar/gkt1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 4.Butler J, et al. Improving cardiovascular clinical trials conduct in the United States: recommendation from clinicians, researchers, sponsors, and regulators. Am Heart J. 2015;169:305–14. doi: 10.1016/j.ahj.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Liu Z, et al. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16:83–92. doi: 10.1097/00042560-199710010-00003. [DOI] [PubMed] [Google Scholar]
- 6.Mitsuya H, Yarchoan R, Broder S. Molecular targets for AIDS therapy. Science. 1990;249:1533–44. doi: 10.1126/science.1699273. [DOI] [PubMed] [Google Scholar]
- 7.Castagna A, et al. Lamivudine monotherapy in HIV-1-infected patients harbouring a lamivudine-resistant virus: a randomized pilot study (E-184V study) AIDS. 2006;20:795–803. doi: 10.1097/01.aids.0000218542.08845.b2. [DOI] [PubMed] [Google Scholar]
- 8.Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–35. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 9.Montaner JS, et al. Association of highly active antiretroviral therapy coverage, population viral load, and yearly new HIV diagnoses in British Columbia Canada: a population-based study. Lancet. 2010;376:532–9. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrigan PR, Whaley M, Montaner JS. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS. 1999;13:F59–62. doi: 10.1097/00002030-199905280-00001. [DOI] [PubMed] [Google Scholar]
- 11.Kinman L, et al. Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J Acquir Immune Defic Syndr. 2006;42:155–61. doi: 10.1097/01.qai.0000214822.33905.87. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher CV, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A. 2014;111:2307–12. doi: 10.1073/pnas.1318249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schacker T, et al. Rapid accumulation of human immunodeficiency virus (HIV) in lymphatic tissue reservoirs during acute and early HIV infection: implications for timing of antiretroviral therapy. J Infect Dis. 2000;181:354–7. doi: 10.1086/315178. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Benet LZ. The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P-glycoprotein. Clin Pharmacokinet. 2001;40:159–68. doi: 10.2165/00003088-200140030-00002. [DOI] [PubMed] [Google Scholar]
- 15.Trainor GL. The importance of plasma protein binding in drug discovery. Expert Opin Drug Discovery. 2007;2:51–64. doi: 10.1517/17460441.2.1.51. [DOI] [PubMed] [Google Scholar]
- 16.Hall HI, et al. Differences in human immunodeficiency virus care and treatment among subpopulations in the United States. JAMA Intern Med. 2013;173:1337–44. doi: 10.1001/jamainternmed.2013.6841. [DOI] [PubMed] [Google Scholar]
- 17.Kinman L, et al. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34:387–97. doi: 10.1097/00126334-200312010-00005. [DOI] [PubMed] [Google Scholar]
- 18.Koehn J, Ho RJ. Novel liquid chromatography-tandem mass spectrometry method for simultaneous detection of anti-HIV drugs lopinavir, ritonavir, and tenofovir in plasma. Antimicrob Agents Chemother. 2014;58:2675–80. doi: 10.1128/AAC.02748-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piliero PJ. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr. 2004;37(Suppl 1):S2–S12. doi: 10.1097/01.qai.0000137001.40505.56. [DOI] [PubMed] [Google Scholar]
- 20.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res Hum Retrovir. 2014 doi: 10.1089/aid.2014.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oussoren C, Zuidema J, Crommelin DJ, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid composition and lipid dose. Biochim Biophys Acta. 1997;1328:261–72. doi: 10.1016/s0005-2736(97)00122-3. [DOI] [PubMed] [Google Scholar]
- 22.Alberts B. Molecular biology of the cell. 4th. Science; Garland: 2002. [Google Scholar]