

INTRODUCTION
Recombinant biological products have revolutionized modern medicine by providing both remarkably effective vaccines to prevent disease and therapeutic drugs to treat a wide variety of unmet medical needs. Since the early 1980s, dozens of new therapeutic protein drugs and macromolecular vaccines have been commercialized, which have benefitted millions of patients worldwide. The pharmaceutical development of these biological products presented many scientific and technical challenges, some of which continue today with newer candidates including recombinant protein-based vaccines with novel adjuvants, peptide and RNA-based drugs, and stem cellular therapies. Compared with small molecule drugs, the characterization, stabilization, formulation, and delivery of biomolecules share common hurdles as well as unique challenges. This area of drug development research has been referred to as “pharmaceutical biotechnology”, in recognition of the critical role that recombinant DNA technology plays in the design and production of most of these biological products. Current research focus areas in this field include (i) determination of structural integrity of the primary sequence, post-translational modifications, and higher-order three dimensional shapes, (ii) assessment of physicochemical degradation pathways and their effects on biological activity and potency, (iii) formulation design and development to optimize stability and delivery, (iv) evaluating and optimizing process development steps including lyophilization and fill-finish, (v) analytical method development and applications of new instruments and data visualization tools, (vi) design and development of drug delivery approaches, and (vii) studies of biological effects including pharmacokinetics, pharmacodynamics, and adverse immunogenicity.
During the early days of pharmaceutical biotechnology research, there were numerous scientific challenges because the analytical characterization approaches needed for development of recombinant biological molecules in “real world” pharmaceutical dosage forms were essentially unknown. Furthermore, understanding critical drug product manufacturing issues (e.g., stability of biological compounds during processing, storage, and shipping as well as reproducibility of fill-finish production technologies) and behavior during and after patient administration was often achieved by “on-the-job” training. Fortunately, the pioneers in the field regularly presented research at key conferences and started publishing early in pharmaceutical sciences journals such as Journal of Pharmaceutical Sciences. Recognizing this critically important new field, the then Editor of the journal, Professor Bill Higuchi, instituted a new “pharmaceutical biotechnology” category for research papers. This insightful move was coupled with an equally wise decision to recruit Dr. C. Russell Middaugh as the new Associate Editor for the new research category. As will be detailed below, under Dr. Middaugh’s diligent and expert guidance, pharmaceutical biotechnology papers have grown in number, scope, and impact over the past 20 years, and these days, the Journal of Pharmaceutical Sciences is viewed by scientific leaders in the field as the “go to” place for publication of the most important results and descriptions of innovations in pharmaceutical biotechnology.
TWENTY YEARS OF PHARMACEUTICAL BIOTECHNOLOGY IN JOURNAL OF PHARMACEUTICAL SCIENCES
The number of pharmaceutical biotechnology papers published in the Journal of Pharmaceutical Sciences from 1992 to 2013 is shown in Figure 1, both by year and cumulative number. These papers are categorized according to the pharmaceutical development of three different types of biotechnology-based product candidates: protein-based therapeutics, other biological molecules (including peptides, polysaccharides, DNA/RNA), and finally various macromolecular antigens (and adjuvants) being developed as vaccines. In 1994, Dr. C. Russell Middaugh joined the editorial board of Journal of Pharmaceutical Sciences as the first dedicated pharmaceutical biotechnology Editor. As can be seen in Figure 1, only a handful of biotechnology papers were published in 1993. From 1994 to the present, under Professor Middaugh’s ongoing editorial guidance, approximately 1000 pharmaceutical biotechnology papers have now appeared, with about half of the papers being published since 2007. For the first 6 months of 2014, 47 additional papers had been published (data not shown). This dramatic growth in pharmaceutical biotechnology papers in the Journal of Pharmaceutical Sciences parallels two major general trends in the biopharmaceutical industry over the past two decades: the emergence of therapeutic mAb drugs to address unmet medical needs for patients with a variety of disorders, especially cancer and autoimmune diseases, as well as the development of many new vaccines to protect both children and adults against a wide range of infectious diseases.
Figure 1.
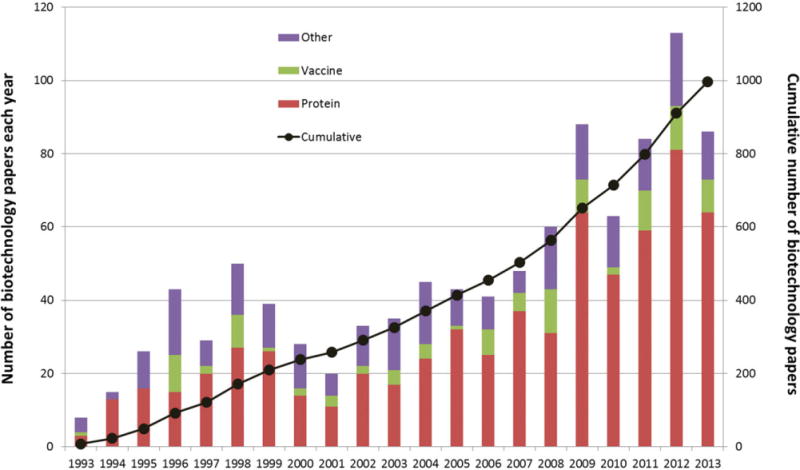
The number of pharmaceutical biotechnology papers published in the Journal of Pharmaceutical Sciences from 1993 to 2013. Data are shown by year (left axis, bar chart) and cumulative number (right axis, black circle). Dr. C. Russell Middaugh joined the editorial board as the dedicated editor for biotechnology papers in January 1994 and continues in that role to the present. Papers covering different aspects of pharmaceutical biotechnology including development of protein-based therapeutics, other biological molecules as drug candidates (peptides, polysaccharides, DNA/RNA), and vaccine candidates (macromolecular antigens and adjuvants) are indicated by color in the bar charts. Data were collected from review of table of contents from 252 issues of the journal from 1993 to 2013.
mAb DRUG APPROVALS OVER THE PAST TWENTY YEARS
To illustrate the tremendous growth in development of therapeutic mAb treatments over the past two decades, we focus on United States Food and Drug Administration (US FDA) approvals, although similar are trends would be observed with worldwide regulatory approvals. The first therapeutic mAb product approved for human use by the US FDA was Orthoclone OKT®3 in 1986; a mouse IgG2a antibody against the CD3 receptor on T-cells for treatment of acute rejection of organ transplants. For the following 8–10 years, it was unclear whether therapeutic mAbs would live up to their potential as “magic bullet” pharmaceutical treatments, and no additional full-length mAbs were approved. During this time period, however, great advances were achieved in the area of antibody engineering allowing for the humanization of mouse antibodies resulting in the ability to produce chimeric, humanized, and fully human mAbs (approximately 75%, 95%, and 100% human amino acid sequences, respectively).1 As shown in Figure 2, in 1994, the second mAb-based product was approved by the US FDA, a chimeric antibody fragment (anti-glycoproteinIIb/IIIa Fab) used as a platelet aggregation inhibitor (ReoPro®). Starting in 1997 to the present, approximately 30–35 new therapeutic mAbs have been approved for commercial use with one to four new mAb approvals per year (except for no US FDA approvals in 1999 and 2005).2–5 When examining the overall trend over the past 20 years, mAbs have grown and evolved into the major category of protein-based therapeutics, and this trend is expected to continue as newer technologies such as antibody–drug conjugates, bispecific antibodies, and various types of antibody-based fragments and fusion proteins become available as therapeutic molecules.
Figure 2.
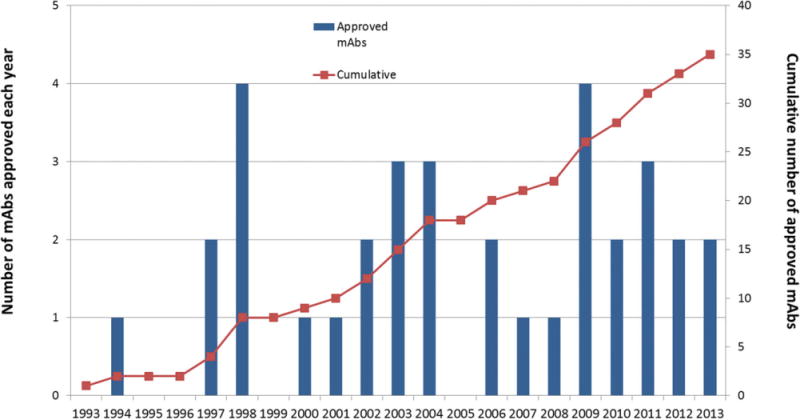
The number of therapeutic mAbs approved by the US FDA from 1993 to 2013. Data are shown by year (left axis, bar chart) and cumulative number (right axis, red squares) (2–5). In 1993, only one therapeutic mAb drug had been approved for commercial use versus 35 mAb therapeutics in 2013 (see text).
Approximately two-thirds of the therapeutic mAbs on the market are administered by intravenous (i.v.) injection, with most of the other mAb treatments injected subcutaneously, along with a few other administration routes including intramuscular and intravitreal (based on a review of online package inserts). In terms of pharmaceutical dosage forms, mAbs have been formulated as either liquid solutions (~2/3 of total) or freeze-dried powders (~1/3 of total) (based on a review of online package inserts). Liquid mAb formulations are filled and packaged into either glass vials or prefilled syringes (PFS), the latter can be used with auto-injectors as mAb drug-device combination products. Both pharmaceutical dosage forms and related administration procedures for therapeutic mAb treatments have become more sophisticated over the past 20 years. For example, in 1998 the first approved anti-tumor necrosis factor alpha (anti-TNF) mAb treatment, Remicade® produced by Centocor (now part of J&J), consists of a chimeric mAb which is lyophilized at 100 mg/vial, reconstituted with 10 mL sterile water for injection, and administered i.v. by medical professionals. In 2009, the same company introduced a newer anti-TNF mAb treatment (Simponi®) as a fully human mAb, formulated as a 100 mg/mL high concentration aqueous solution. A patient-convenient dosage form was utilized with the liquid formulation filled into a PFS, which in turn, is placed into an auto-injector allowing for self-administration at home/doctor’s office via subcutaneous (SC) injection.6
NEW VACCINE APPROVALS OVER THE PAST TWENTY YEARS
The past 20 years have also witnessed a surge in development and commercialization of new vaccines, both in terms of new vaccine antigens and new formulations and delivery technologies. As shown in Table 1, there has been an impressive and diverse array of macromolecules and microorganisms developed as new vaccines including polysaccharide-protein conjugates (e.g., pneumococcal and meningococcal vaccines), killed and live, attenuated viruses (e.g., hepatitis A, rotavirus and shingles vaccines) as well as recombinant protein technologies including virus-like particles (human papillomavirus or HPV vaccine) and a recombinant hemagglutinin flu vaccine.7 In addition, new formulations and delivery technologies have been introduced to improve and expand the utility of vaccines such as influenza (e.g., nasal and intradermal delivery, new adjuvants). Finally, new formulations containing mixtures of older, already approved vaccines have also been developed to decrease the complexity of the vaccination schedule and better ensure compliance (e.g., measles, mumps, rubella and varicella; and, although not listed in Table 1, diphtheria, tetanus toxoid, acellular pertussis, hepatitis B and inactivated poliovirus vaccines).
Table 1.
Examples of New Vaccines and New Vaccine Formulations Approved for Human Use in the Past 20 Years (1993–2013)
| Vaccine Antigen | Year | Protection Against | Vaccine Product
|
Vaccine Formulation
|
|||
|---|---|---|---|---|---|---|---|
| Name | Manufacturer | Dosage | Adjuvant | Admin Route | |||
| Polysaccharide-protein conjugates | 1993 | Haemophilus influenzae type b | ActHIB | Sanofi Pasteur | Lyo | None | IM |
| 2000/2010 | Pneumococcal | Prevnar/Prevnar 13 | Pfizer | Liquid | Aluminum | IM | |
| 2005/2010 | Meningococcal | Menactra/Menveo | Sanofi Pasteur/Novartis | Liquid/Lyo | None | IM | |
| Live, attenuated virus | 1995 | Varicella (Chicken Pox) | Varivax | Merck | Lyo | None | SC |
| 2005 | Measles, mumps, rubella and chickenpox | ProQuad | Merck | Lyo | None | SC | |
| 2006 | Varicella (Shingles) | Zostavax | Merck | Lyo | None | SC | |
| 2006/2008 | Rotavirus | Rotateq/Rotarix | Merck/GSK | Liquid/Lyo | None | Oral | |
| Inactivated virus | 1995/1996 | Hepatitis A | Havrix/Vaqta | GSK/Merck | Liquid | Aluminum | IM |
| 2009 | Japanese encephalitis | IXIARO | Novartis | Liquid | Aluminum | IM | |
| Recombinant protein virus-like-particle | 2006/2009 | Human papillomavirus (HPV) | Gardasil/Cervarix | Merck/GSK | Liquid | Aluminum/AS04 | IM |
| New flu vaccines | |||||||
| (live vaccine) | 2003 | Influenza | FluMist | Medimmune | Liquid | None | Nasal |
| (intradermal delivery) | 2011 | Fluzone Intradermal |
Sanofi Pasteur | Liquid | None | ID | |
| (cell culture) | 2012 | Fluarix | Novartis | Liquid | None | IM | |
| (recombinant protein) | 2013 | Flublok | Protein Science | Liquid | None | IM | |
| (biodefense stockpile only) | 2013 | Influenza A (H5N1) | Influenza A (H5N1) virus monovalent vaccine, adjuvanted | GSK | Liquid | AS03 | IM |
This table is not a comprehensive list of approved vaccines by regulatory agencies, but rather provides illustrative examples from US FDA approvals of vaccine development trends in terms of types of new vaccine antigens, formulations, and delivery routes (7).
From a pharmaceutical dosage form perspective, live attenuated viral vaccines tend to be lyophilized and do not require adjuvants. This can be attributed to the inherent complexity and instability of microorganisms along with their ability to replicate upon administration and thus better mimic a natural infection. One exception in the 1993–2013 time period is the successful development of a liquid formulation of the orally administered rotavirus vaccine, which required identification of stabilizers to ensure stability of a pentavalent virus mixture when stored at 2–8°C in a plastic squeeze tube, and at the same time, provide sufficient acid neutralizing capacity to protect the viruses from gastric acid degradation during oral administration.8
Another trend to note in Table 1 is that inactivated viral and subunit vaccines (e.g., polysaccharide-protein conjugates, recombinant protein virus-like particle vaccines) tend to be formulated as liquid solutions containing adjuvants to enhance their immunogenicity. Aluminum salts have been used as adjuvants for decades, and most new vaccines still contain this conventional adjuvant. It is interesting to note that despite decades of research to identify and develop new adjuvants, US FDA approvals have occurred only relatively recently including GSK produced HPV vaccine with AS04 (aluminum adjuvant and monophosphoryl lipid A) in 2009, and a biodefense flu vaccine in the US stockpile with AS03 (emulsion containing squalene, polysorbate 80 and DL-alpha-tocopherol) in 2013. Additional vaccine formulations containing new adjuvants have been approved by European regulatory agencies including oil-in-water emulsions as adjuvants for flu vaccines (not shown). One interesting pharmaceutical biotechnology-related case study for new protein-based vaccines is the stabilization and formulation of the human papillomavirus virus-like particles (HPV VLPs). When the recombinant viral surface protein was recombinantly expressed in yeast and then assembled into virus-like particles in vivo, the purified VLPs were observed to form a mixture of fully and partially assembled particles. An in vitro large scale disassembly and reassembly procedure was developed and implemented to ensure the formation of correctly assembled virus-like particles, resulting in enhanced potency and improved stability (accelerated and long-term storage) for a quadrivalent, aluminum adsorbed HPV VLP vaccine formulation.9
DEVELOPMENT TRENDS OVER THE PAST 20 YEARS IN PHARMACEUTICAL BIOTECHNOLOGY
Physicochemical Stability of Protein Drugs
After three decades of diligent research, pharmaceutical scientists now know that the safety and efficacy of therapeutic protein drug products can be compromised not only via post-translational modifications in the cell, but also by well-defined physical and chemical degradation pathways. It has also become apparent that sometimes even trace amounts of modified or degraded protein can result in suboptimal to adverse effects in patients (see immunogenicity section below). We have learned that proteins can be extremely susceptible to such degradation and that such damage can occur at all stages of a protein product’s life history, from fermentation, purification, formulation/storage to patient administration (and perhaps in vivo within the patient). Furthermore, the starting bulk drug substance can contain a wide range of molecular variants, e.g., subpopulations with different glycosylation patterns or charged isoforms, which can have different pharmaceutical properties such as receptor binding, pharmacokinetic profiles and propensity to aggregate.
Physical Stability
Currently, the study of protein physical stability is understood to encompass characterization of degradation products such as “soluble” aggregates (e.g., oligomers), “particles” (submicron, subvisible and visible) and larger precipitates. In addition, a particular concern for low dose drug products is the loss of potency due to adsorption of protein molecules to the container/closure (e.g., wall of glass vials or rubber stopper) and/or the delivery system (e.g., bags for i.v. administration). In addition, physical stability can refer to the key properties of the native protein under various solution conditions; e.g., the effects of pH or ionic strength on a protein’s conformational and colloidal stability. In turn, it is now better understood that these physical properties can govern the rates of degradation of a given protein (e.g., aggregation rate). Furthermore, pharmaceutically unacceptable physical properties can include opalescent appearing solutions, liquid-liquid phase separation and high solution viscosity. These properties are particularly problematic with the development of high concentration (e.g., 100–200 mg/ml) formulations of mAbs.
Moreover, it has been well established that there are exposures to numerous stresses during a protein product’s life history that readily induce aggregation, particle formation and/or loss of protein molecules from solution due to adsorption at interfaces. These include freeze-thawing, exposure to extremes of pH, filtration steps, pumping during fill-finish and fluid transfers, exposure to various surfaces and interfaces in primary containers and delivery systems, as well as agitation and other stresses during shipping. Many of the stresses result in exposure of protein molecules to interfaces to which they can adsorb, resulting in assembled networks of native and/or structurally altered protein molecules on the interface. Disruption of the assembled films or gels formed at the interfaces (e.g., during agitation) can result in protein aggregates or particles in the bulk solution. In addition, extrinsic, foreign particles can be shed from essentially any material to which protein solutions are exposed, (e.g., silicone oil-in-water, stainless steel particles-in-water, glass delamination) and protein molecules can readily adsorb to the foreign particle-liquid interfaces resulting in heterogeneous particle formation. These may, in turn, stimulate further protein aggregation and particle formation, especially upon exposure to pharmaceutically relevant stresses (e.g., freeze-thawing or agitation).
In concert with the efforts to delineate the causes of protein physical degradation, tremendous progress has been made in understanding mechanisms by which proteins can be stabilized against such damage and in developing effective means to minimize degradation. This work includes both theoretical and experimental advances, which have led to much more rationale design of protein stabilizers, high-throughput formulation development approaches to optimize protein formulation composition, and to more powerful data processing/data visualization methods. Also, approaches to reduce the detrimental impact of processing conditions have been studied at lab scale, and resulting mitigation strategies have been scaled-up and implemented in commercial manufacturing settings.
Thirty years ago the field was not aware of many of the numerous problems that can arise during the scale-up, manufacturing and shipping/handling of therapeutic proteins, because the development efforts for these products were in their infancy. Through rigorous and insightful research over the intervening years, and the publication of important results and theoretical insights, pharmaceutical biotechnology has made remarkable progress. Many of the key papers have been (and continue to be) published in Journal of Pharmaceutical Sciences. These publications include impactful Commentaries and Reviews, as well as numerous seminal research papers. For example, over the years, higher resolution, more sensitive and increasing reliable characterization and quantitation assays for monitoring protein aggregation and particle formation have been developed. This progress in developing improved analytical tools has greatly increased our insights into how protein physical degradation can readily occur and our understanding that if even minute fractions of a protein product become degraded, there may be detrimental impacts on subsequent protein stability and product quality, and on product safety and efficacy in patients (as discussed in more detail below).
Chemical Stability
Since the inception of development of therapeutic proteins, in parallel with research efforts to understand and control protein physical stability, intensive research also has been focused on the chemical stability of proteins, which is also critically important for safety and efficacy of therapeutic protein products. Decades of advancements in the field were required to develop the theoretical and practical understanding – and the requisite advances in analytical capabilities – that we now have about chemical degradation of therapeutic proteins. There are many different pathways for chemical degradation that have been elucidated in extensive mechanistic detail including oxidation, deamidation, and hydrolysis of certain amino acid residues. This in turn has led to a better understanding of chemical hotspots in protein molecules (e.g., Asn deamidation, Asp isomerization, Met oxidation, and Trp photo-degradation). In addition to chemical degradation, chemical heterogeneity in therapeutic protein products is also well-established, for example, disulfide isoforms, charged variants, C-terminal lysine and N-terminal pyroglutamate variants, and even proteolytic clippings.
Early efforts often focused on investigations of causes and control of known chemical degradation pathways and discovery of new pathways. It was quickly realized that conditions encountered during processing, storage, shipping, and delivery could lead to rapid chemical degradation of specific amino acid residues and/or the polypeptide backbone. These conditions included exposures to: metals from processing equipment, primary containers and/or excipients; peroxides from surfactants; extremes of pH; and light. Current control strategies now include screening of excipient lots for metals and peroxides, and the replacement of stainless steel processing equipment with single-use plastic systems. But the plastic systems have not been without problems, for example, substances leaching from the plastics have been found in some cases to cause chemical degradation of therapeutic proteins. For some products, despite these efforts, chemical degradation is so extensive in solution that freeze-drying is required to ensure chemical stability and a multi-year shelf life.
Light exposure during manufacturing is minimized with approaches such as running a side stream from a chromatography system through the UV detector while the main flow is not exposed to light. In addition, using secondary packaging helps to reduce light exposure in the final product container. But there are still unintended and poorly controlled situations in which light exposure and potential damage can occur to the protein product. For example, when patients are warming a PFS prior to administration at home, they may place it on the kitchen counter in direct sunlight. Similarly, during preparation and administration of i.v. products, there is exposure to room lights and potentially to sunlight. Such exposures can cause substantial photochemical (and physical) degradation of proteins.
There also has been much effort on developing formulation approaches to minimize chemical damage to proteins in the drug substance and in the final formulated drug product. Some successful approaches that have been implemented include the selection of the optimal solution pH and the inclusion of free radical scavengers (e.g., methionine) and/or metal ion chelators in the formulation. It has also been discovered that other additives, such as the pharmaceutical anti-oxidant ascorbate, may actually accelerate protein chemical degradation by catalyzing the generation of free radicals in solution under certain conditions. In other cases, inclusion of appropriate excipients has been documented to inhibit light-induced degradation in some proteins, whereas in other studies they have been found to be ineffective for another protein. The difference might be due to the locations of the damage-sensitive amino acid residues within the protein molecule. For example, residues located on the protein surface may be protected to some degree by stabilizing excipients in the solution, whereas residues that are degraded because of photon absorption in the interior of the protein may not be protected by components in the formulation.
Chemical damage and physical stability are often linked. For example, oxidation of amino acid residues within a protein may also lead to protein aggregation, perhaps including covalent crosslinks. Another example is creation of a protein species with reduced solubility via fragmentation or proteolytic clipping of the polypeptide backbone, leading to protein precipitation. Conversely, perturbation of the tertiary structure of a protein molecule may result in more rapid degradation of amino acid residues that were previously buried in the most compact species in the native state ensemble. The exact nature of such linkages between chemical and physical stability cannot be predicted – nor can the consequences of the damage. Consequently, Arrhenius kinetics cannot be relied on to give accurate predictions of degradation rates or shelf life, adding complexity to formulation development for biologics compared to small molecules. Careful studies are needed to characterize the degradation profile for each given protein and to develop formulations to minimize both physical and chemical degradation pathways. Often times, a compromise is required to identify conditions that lead to optimal overall protein stability, conditions which in turn, may not be optimal for every individual physical or chemical degradation pathway.
As has been the case for publications on physical stability of therapeutic proteins, many of the key research papers, Reviews and Commentaries have been, and continue to be, published on the topic of chemical stability of protein drug candidates in Journal of Pharmaceutical Sciences. These papers are further evidence of the important roles that the journal has played in the advancement of the field for the past two decades.
Immunogenicity of Biotech Drugs
For millions of patients, therapeutic protein products are miracle drugs that save and improve lives. However, for many patient populations these miracle drugs, which are initially highly effective, eventually fail in a fraction of the initial responders. Depending on the product and patient group, the fraction of these so-called “secondary non-responders” may reach 50% or higher, with many patients developing treatment failure in less than a year or two. In some cases, (e.g., rheumatoid arthritis patients treated with anti-TNF therapies) patients can be switched to another product in the same class with a restoration of therapeutic effectiveness. But even these patients may experience subsequent treatment failures. For other patients there is no alternative therapy or they have already used all of the approved biologics. In these cases, they suffer from loss of treatment with the protein miracle drugs, resulting in morbidity or even death.
It is now widely documented that treatment failure is usually due to adverse immunogenicity caused by the protein drug product. Since the 1960s, many clinical investigations and animal studies have shown that a major contributing factor to immunogenicity is the presence of protein aggregates and particles. Even trace amounts of these degradation products can stimulate an immune response leading to generation of antibodies that neutralize the drug’s activity and/or promote its rapid clearance from the body. Furthermore, immune response to aggregates and particles can be greatly enhanced if the protein molecules are absorbed onto pharmaceutically relevant foreign particles (e.g., glass from vials or stainless steel from filling pumps) and/or are chemically degraded (e.g., oxidized). Therefore, major research efforts are now devoted to understanding and controlling the levels of such protein aggregates, particles and chemically degraded species. Concurrently regulatory expectations in this area are becoming increasingly more stringent. As with the other categories considered in this Commentary, many of the most innovative and influential research papers, Reviews and Commentaries in the area of immunogenicity of protein drugs, its causes, mechanisms and related regulatory expectations have been published in the Journal of Pharmaceutical Sciences.
Pharmaceutical Dosage Forms
In the 1980s and early 1990s, some of the new therapeutic protein products were being developed in traditional “small molecule” pharmaceutical companies. Often there was a philosophical, as well as a physical separation of biotechnology R&D/production from small molecule pharmaceutical R&D/production. These organizational silos not only led to differences in the education and training of scientists and managers, but to differences in overall management approaches. At the same time, some of the new protein therapeutics were being developed in stand-alone biotechnology companies. Often the protein biochemists and molecular biologists had completely different trainings and experiences than the drug development experts who were brought into the new biotech companies from traditional pharmaceutical companies. For example, biotechnology researchers often worked in systems that operated relatively freely and had an attitude toward research and development more akin to that which they experienced in academic universities. In contrast, the traditional pharmaceutical scientists and process development experts were focused on implementation of current good manufacturing practices (cGMP), reducing time to market, and commercial scale-up and distribution.
Overall, new thinking and interfacing was needed in both the start-up biotechnology and traditional pharmaceutical companies to successfully develop protein drugs. With therapeutic proteins, the starting point for drug product formulation was not a dry, crystalline powder but a “bulk solution”. The term “preformulation of proteins” is sometimes used to refer to the activities related to developing a bulk protein solution into a well characterized pharmaceutical dosage form with a “drug product history” including frozen and thawed, heated for viral deactivation, pH adjusted, dialyzed, diluted and reformulated, as well as sterile filtered, filled into final containers, and in some cases lyophilized.
Approaches to protein drug product development have changed dramatically over the last 25–30 years. Many companies have been successful in managing the interface between biotechnology R&D and more traditional pharmaceutical product development approaches, although some companies still struggle with this challenge. Also, in many cases, the gaps between drug substance and final drug product formulation have vanished, and bulk protein solutions are developed such that they are provided ready to fill. In addition, formulation work has been shifted towards earlier phases of R&D, including the selection of the final protein molecule candidate, via an early evaluation of physicochemical properties and product stability (i.e., “developability” or “drugability” assessments). The early paradigm of drug selection followed by formulation studies that must somehow find a way to stabilize the molecule has largely been abandoned and replaced by a more integrated approach, at least in the hands of more experienced biologics therapeutics developers. This approach is widely used, for example, with mAb products for which several high-affinity candidate molecules may be developed for a given target, followed by a selection process to identify the candidate with the best pharmaceutical properties of stability, solubility and manufacturability. In some cases, compatibility of the candidate molecules with the final anticipated primary containers/closures (e.g., PFSs) may be assessed early as part of the candidate selection screening process.
The vast majority of protein drugs require parenteral delivery and therefore sterile packaging. The rise of the therapeutic protein products is correlated with the decline of the ampule, the classical primary container often used in parenteral delivery. When the first protein products came out in the late 1980s and into the 1990s, certain standard pharmaceutical glass vials and stoppers were the dominant, or in some cases, the only choice. From a practical standpoint they could be used for both aqueous solution and lyophilized formulations. Little innovation in glassware occurred in the first years, but there were important improvements in the rubber stoppers. For example, stoppers coated with polytetrafluoroethylene and similar materials came to the market to minimize leachable compounds that could affect protein stability. Although these new stoppers were slow to be adopted by the industry primarily because of the relatively high price, today these coated stoppers are widely used.
Another trend in the choice of primary packaging, which represented new thinking in drug delivery and self-administration of therapeutic proteins, was the increased use of “ready to use” prefilled syringes (PFSs). In the 1990s, the change to a PFS container was typically only implemented as a life-cycle management strategy and only for the most commercially successful products. Today, PFSs are considered standard primary packaging for many therapeutic protein product candidates. PFSs are now produced in the hundreds of millions units annually. The widely available “nested” pre-sterilized syringes in closed trays now make it possible to operate exactly the same configuration for filling and packaging from the first 100-piece developmental batch up to large-scale commercial manufacturing. With these products in hand, millions of patients today self-inject their life changing medications at home. On the basis of these improvements in PFS technology, autoinjectors have been brought to the customer too, making self-injection even more convenient.
A major development challenge in the switch of a therapeutic protein product from a glass vial to a PFS (or even when a PFS is the first container/closure option investigated) is that a formulation that may have worked well in terms of maintaining protein stability in a glass vial may not provide adequate stability in a PFS. This effect is usually because of the destabilization of protein molecules that can occur upon adsorption to the silicone oil that is used for plunger lubrication. Furthermore, the combination of protein adsorption to the oil–water interface and exposure to the air–water interface that can occur in PFS has been shown to promote protein aggregation and particle formation. However, with a combination of mechanistic and practical studies, rational formulation strategies have been devised for developing stable aqueous solution protein formulations in PFSs. Descriptions of many of the key studies in this area have also been published as Research Papers and Reviews in Journal of Pharmaceutical Sciences.
Although there is much interest in needle-free injection (NFI), NFI is still not readily available for parenteral administration of protein drugs. Barriers to NFI adoption include relatively higher costs, lack of bioequivalency compared with regular subcutaneous injection via a needle, and finally, experience demonstrating that “needle-free” is not necessarily “pain-free”. Nevertheless, because needle stick injuries are a global problem and reuse of syringe and needles is a serious issue in some countries, evaluation of NFI technologies will undoubtedly continue for both currently available therapeutic protein and vaccine products and for new clinical candidates.
Development of stable dosage forms to ensure long term storage has always been a challenge and is critically important for commercialization of biotechnology-based drugs and vaccines. In an effort to meet this challenge, lyophilization has become a successful alternative to provide the required stability in commercial formulations. There have been several interesting trends in this field over the last 20 years. Firstly, for the fundamental scientific studies in the 1980s and 1990s began to elucidate the mechanisms by which certain excipients stabilize a protein during freezing, drying, storage in the solid state, and finally, upon reconstitution. At the same time, empirical studies in industry screened for effective excipients that were practically useful; that is, compounds that stabilized proteins which were also used in approved parenteral products and could be lyophilized relatively economically on a commercial scale. With the groundbreaking work of academic and industry researchers around the globe, there is now a solid understanding of how to rationally select excipients and process parameters for successful lyophilization of a protein drug. Furthermore, the essential analytical methods to monitor protein stability—such as solid-state differential scanning calorimetry (DSC) and IR spectroscopy—and the associated physical properties that govern structural integrity of a lyophilized cake, have now been well established. Also, advances in process control and freeze-drying cycle development approaches have resulted in more economical, robust, and consistent commercial scale manufacturing of lyophilized protein formulations.
Over the past 20 years and continuing to today, many critical Research Papers and Reviews on mechanisms for protein stabilization during lyophilization and subsequent storage in the freeze-dried state in various formulations have been published in Journal of Pharmaceutical Sciences. In parallel, several key papers on new analytical methods and process development/control strategies have appeared in the journal, and it is still a leading venue for publication in this field. There are many new challenges in development of lyophilized formulations, including those for ultra-high concentration mAb products and dual chambered syringes, and there will be many more groundbreaking studies in the field. The Journal of Pharmaceutical Sciences will continue to be an important repository for the Research Papers describing this new work and for Reviews summarizing the key advances.
Novel Drug Delivery Approaches
It is interesting to note that during the first ~10 years of Pharmaceutical Biotechnology papers being published in the Journal of Pharmaceutical Sciences (1994–2004), only about 4 biomolecules reached over $1 billion annual sales.10 For the second ~10 years, the increased rate in pharmaceutical biotechnology manuscripts being published in Journal of Pharmaceutical Sciences (Fig. 1) coincided with the rapid growth in commercially available mAb therapeutics (Fig. 2). This growth in therapeutic biomolecule approvals, combined with investments by small, medium and large pharmaceutical sponsors, has been reflected in the annual sales of biotherapeutic products. For example, many of the anti-TNF antibody products have reached annual sales exceeding $6 billion, and have become top income producer of major pharmaceutical companies.11 Although mAbs are administered to patients by injections, there is a growing interest in how protein pharmaceutics could potentially be made more effective, safer and convenient for patients through novel drug delivery technologies. As patient compliance and therapeutic adherence are issues central to the overall outcomes clinical care, scientists across disciplines are taking integrated drug delivery approaches to address these issues.
The overarching goal of drug delivery is to improve the therapeutic index by enhancing safety and/or efficacy, or to improve convenience—and preferably both. The field encompasses a very broad range of technologies, from device innovations such as portability and biofeedback regulation (e.g., insulin delivery devices linked to glucose sensors that monitor insulin effects in patient) to platforms such as colloid, micro, and nano drug carriers or particles which require knowledge of physiologic mechanisms in biopharmaceutics as well as the pharmacokinetic characteristics of the drug (which can include proteins and peptide as well as most of the DNA/RNA or nucleic acid-based biologicals). By one estimate, the annual revenue of drug delivery products, including biotechnology-based and small molecule drugs, is over $57 billion and is projected to have an annual growth rate of 5%–10%.11 Advances in protein, peptide and DNA/RNA research, combined with clinical experience using these biopharmaceuticals as therapeutics, have provided key information on distinctive disposition characteristics, the role of protein sequence variations, details of the barrier dynamics to protein absorption, and to some degree, drug target distribution in the body as it relates to drug effects. The growing knowledge of these properties, collectively referred to as biopharmaceutical characteristics and pharmacokinetic profiles as they are linked to therapeutic responses, has allowed for further development of novel drug delivery strategies. Many of these discoveries, development and drug delivery technologies are documented in Journal of Pharmaceutical Sciences, enabling scientists across disciplines and around the world to gain access to the vast knowledge in drug delivery research in academic and pharmaceutical industry.
These drug delivery strategies, placed in the context of human physiological interactions—referred to as a “systems approach” to drug delivery—can be categorized as control release, permeation enhancement, modulation of drug clearance, targeting to the site of action, as well as molecular optimization. However, these delivery systems remain very challenging to develop for biologics. For example a polymeric sustained release formulations of recombinant human growth hormone (rhGH) (Nutropin Depot) was approved by the US FDA and marketed for several years. However, a variety of manufacturing and delivery challenges were encountered with this dosage form leading to a decision by the sponsor to discontinue production of this product.12 Despite the challenges, development of sustained release dosage forms for biologics continues and has led more recently to the 2012 approval and commercialization of Bydureon®, a sustained release formulation of a 39 amino acid peptide, glucagon-like peptide-1 (GLP-1) agonist.
The clinical impact of drug delivery systems may be viewed in the context of how a particular approach improves the therapeutic index, for example by dose reduction or through minimizing untoward effects after extended dosing. The best results are often achieved with drug localization strategies that enhance exposure to target tissues and cells but reduce drug exposure in organs linked to toxicity. The biodistribution of all drug targets in cells and tissues within the body is yet to be fully understood. However, virus- or bacteria-laden cell distribution, and by extension their protein target localization in cells and tissues, are now mapped, elucidated, and validated for defined organs within the body. Some of the recent efforts in developing and validating pharmacokinetic relationship with therapeutic outcomes or pharmacodynamics as well as pharmacometrics definitions would add to these knowledge base to guide dose selection and the type of drug delivery strategies to maximize therapeutic effects. Such knowledge has led to successful development of targeted drug delivery systems to improve the therapeutic index. Research to discover sites of drug action through proteomic modeling and high-throughput screening has yielded targets that have been used to construct fusion proteins with added effector function to enhance pharmacologic activity. The efficiency of identifying drug targets has improved and cloning technology has matured. Through a systems approach, pharmaceutical formulation and effective delivery to the target cells and tissues central to potentiating therapeutic effects have become one the key rate-limiting steps for bringing new molecular entities to market.
Although the development of gene- and cell-based medicine has been slow, it continues to mature. Although gene therapy intended for cancer treatment and enzyme replacement may be a distance away, the use of DNA-based vaccines, and more recently RNA-based vaccines, is on the horizon. Nucleic acids, which are polar and poor cell penetrants, often face challenges in reaching the intracellular targets after injection treatment in animal models and in human clinical trials. A number of nucleic acid delivery systems, including lipid- or protein-based technologies as well as viral or vector-based technologies, also pose significant challenges in terms of off-target cellular and tissue distribution (e.g., liver) and/or a low degree of functional impact within the target cells. Thus, even with the exciting work with siRNA and RNAi for a number of target therapeutic genes in vitro and in animal models, the inability to deliver a sufficient fraction of these agents into target cells has prompted a scale back in clinical development programs within major pharmaceutical companies, although efforts continue in dedicated start-ups and other settings. Although low-volume cell-based therapeutics, including stem cell and immune cell therapeutics currently in clinical trials for skin and other cancers, are promising, they are still in the clinical research stage. These highly publicized stem-cell therapeutics—in vitro or in situ—will likely mature with time.
There is now substantial and cumulative clinical experience using different classes of protein molecules—mAb, enzymes, interferons, cytokines, hematopoietic growth and coagulation factors, hormone and peptides, vaccines, and nucleic acid (including aptamers)—that can be applied to development of biopharmaceuticals. This knowledge has given insight to the strengths and weaknesses in localization, distribution, and pharmacokinetics, as well as disposition and elimination pathways for specific biomolecule classes. Leveraging this knowledge, drug delivery systems are being developed to improve safety while enhancing effectiveness through better localization and exposure to target cells and tissues.
Advances in Analytical Methods
The primary structure or amino acid sequence of a therapeutic protein drug, along with any post-translational modifications produced by a recombinant expression system, has to be confirmed and monitored. Around two decades ago, this could be a very difficult task experimentally. However new tools, especially mass spectrometry, have emerged as key analytical techniques for the determination of the primary structure of a therapeutic protein. In particular, mass spectrometry combined with HPLC (LC–MS) has dramatically changed the reliability, sensitivity, and speed of sequence analysis.
Before mass spectrometry, confirmation of primary structure could be achieved by the combination of results from different analytical methods. The full amino acid composition of the protein was determined by amino acid analysis. The amino acid sequence of the protein and/or of peptides fragmented by single or multiple enzymatic treatments of the protein were assessed mainly by N-terminal sequence analysis using the Edman degradation method. Advancements of mass spectrometers and related methods over the past two decades, especially highly improved resolution of mass determination, has enabled us to more directly analyze proteins and peptide fragments. Molecular mass of the protein without enzymatic digestion can be estimated by mass spectrometry with high accuracy. In most cases, electron spray ionization or matrix assisted laser desorption ionization is used for the ionization, followed by mass measurement via quadrupole, ion-trap, time-of-flight, or Fourier transform mass spectrometer, and/or a combination of these approaches. Typically, more than 95% of the total sequence is covered by mass spectrometry of enzymatically digested peptides. Patterns of disulfide bonding are also determined by mass spectrometry of digested protein under non-reducing conditions. Today, even trace amounts of degraded protein can be correctly analyzed by mass spectrometry, as can the inherent post-translational heterogeneity of a therapeutic protein drug such as the glycosylation pattern.
Capillary electrophoresis also is now a standard method, often replacing traditional polyacrylamide gels, to estimate molecular weight of a protein and to quantify levels of disulfide cross-linked aggregates. Moreover, two decades ago, net charge variations of proteins were characterized by ion exchange chromatography and/or isoelectric focusing electrophoresis using immobilized pH gradient gels. Today, charge heterogeneity properties of protein drugs are routinely analyzed by capillary electrophoresis approaches including capillary IEF (cIEF). This approach provides higher resolution results in a relatively short time.
Several methods have been used in protein biochemistry to study the higher order structures of proteins. X-ray crystal structure analysis has long history in the determination of protein three-dimensional structure at high resolution. In addition, for more than 20 years nuclear magnetic resonance analysis has been applied to the high resolution structure analysis of proteins in solution. However, these methods require elaborate work up, and remain relatively complicated techniques requiring specialized and costly equipment, highly skilled researchers, and considerable expenditure of time. Thus, these methods, although useful for research applications, are not used on a routine basis, especially in pharmaceutical dosage forms containing a variety of excipients.
Instead, for therapeutic proteins, lower resolution methods have been used for higher-order structural characterization and comparisons. For example, circular dichroism (CD) measurement has been used for the secondary structure (far-UV range) and for tertiary structure (near-UV range) of the proteins. Non-linear regression analysis of the far-UV CD spectrum provides contents of secondary structure. Intrinsic fluorescence of a tryptophan residue reflects the local environment of the residue, and thus fluorescence measurements have been used to monitor changes in the tertiary structure of proteins. These methods can also be interfaced with automated temperature controlled cuvette holders, allowing for determination of the thermal stability of the secondary and tertiary structure of proteins, often a useful surrogate for examining higher-order structural integrity of proteins. To this end, DSC can provide thermodynamic parameters of protein stability directly and has used for this purpose in basic protein research for the past two decades. Improved sensitivity, reproducibility and throughput of calorimeters now enable its use for the assessment of relative thermal stability of therapeutic proteins, an approach that is valuable in product characterization, comparability studies and formulation screening. Hydrogen deuterium exchange mass spectrometry (HDX-MS), the most recently emerging technique for higher order structure analysis, has been used in a growing number of studies of therapeutic proteins in recent years. HDX-MS can provide site specific higher-order structural information at several amino acid residues resolution, which is difficult to acquire by other analytical methods, especially in the presence of the excipients that are often required to stabilize therapeutic protein drugs.
In addition to monitoring the structural integrity and conformational stability of therapeutic proteins, analysis of hydrodynamic properties such as overall size and shape of the molecule is important, especially as related to monitoring protein aggregation. Several analytical approaches are commonly used including size-exclusion chromatography (SEC) as well as dynamic and static light scattering. Analytical ultracentrifugation has been also been widely used for quaternary structure studies and native-state self-assembly, as well as for characterization and quantitation aggregates due to physical degradation. As discussed below, these aggregates can form larger assemblies, often referred to as particles, which can raise regulatory concerns.
As in the case of other research areas discussed in this Commentary, many seminal Research Papers, Reviews, and Commentaries on protein structural analysis have been and continue to be published in Journal of Pharmaceutical Sciences. The strength of the journal in this important area of pharmaceutical biotechnology is evidenced not only by the many high quality papers from this field, but also the numerous members of the Editorial Advisory and Scientific Advisory Boards of Journal of Pharmaceutical Sciences who are leaders in areas of protein biophysical chemistry and its application to therapeutic protein characterization.
Regulatory Expectations
Along with the many scientific advances in pharmaceutical biotechnology made over the past two decades, regulatory expectations for product quality and appropriate analytical methods have increased substantially. Often, there has been an iterative process by which a group of researchers makes critical advances in an area and publish their work, which then triggers the regulatory authorities to begin asking for these new advances to be employed by sponsors. The Journal of Pharmaceutical Sciences has played a key role in not only publishing the latest scientific findings and reviews in pharmaceutical biotechnology, but also as a forum for highlighting and debating these emerging topics “in real time” via the journal’s commentary section.
For example, in the early days of the field until today, SEC has been the method of choice for characterization and quantitation of protein aggregates. A group of researchers with academic training in protein native state self-assembly began using analytical ultracentrifugation as a complementary method to study aggregation in therapeutic protein products. They quickly realized that results with SEC often did not accurately reflect the actual aggregate levels and types in a protein product. They then created research approaches that used AUC during SEC method development to assure that the latter method was accurate and provided proper results. Because much of this work was published, including commentaries in Journal of Pharmaceutical Sciences,13 regulators at agencies such as the US FDA soon came to expect that this approach to monitor protein aggregation be used for during development of most (if not all) therapeutic protein products.
More recently, it has been recognized that there was an “analytical gap” in studies of protein physical degradation because subvisible particles smaller than 10 micron were not being routinely studied and characterized for therapeutic protein products. This became a topic of several commentaries in Journal of Pharmaceutical Sciences.14,15 Based on instrument advances and calls from regulators to “fill this gap,” the field is now routinely quantifying and characterizing micron and submicron sized particles in therapeutic protein products. Today control strategies for protein degradation in manufacturing, storage, shipping, and delivery now include a focus on subvisible particles. These examples highlight the important role that research papers, reviews and commentaries published in Journal of Pharmaceutical Sciences can play in elucidating the nature, and debating the current status, of critical product quality issues affecting protein drugs. The journal continues to be the choice for industry, regulatory agencies, and academia alike to publish new insights and calls for improvements in assessment of product quality affecting clinical safety and efficacy.
CONCLUDING REMARKS
The purpose of this commentary is not only to reflect on the scientific and technical advances that have occurred in pharmaceutical biotechnology over the past 20 years, but to illustrate the key role played by Journal of Pharmaceutical Sciences in presenting and debating these achievements. As shown in Figure 1, approximately 1000 pharmaceutical biotechnology papers have appeared in the past 20 years in the Journal of Pharmaceutical Sciences under the leadership of the Biotechnology Editor, Professor C. Russell Middaugh. As highlighted by the guest editors, many specific pharmaceutical biotechnology development issues have been researched, and in many cases now successfully addressed, for protein, peptide and nucleic acid-based drug and vaccine candidates under development: elucidation of physicochemical mechanisms of instability, development and scale-up of pharmaceutical dosage forms, challenges in novel drug delivery challenges, and the key role of analytical characterization and developing new analytical approaches.
What does the next 20 years have in store for pharmaceutical biotechnology? As highlighted in Figure 2 and Table 1, essentially an entire new class of protein drugs (therapeutic mAbs) and many new and important vaccines were developed and approved for human use over the past 20 years. What new class of biotechnology products will account for the new therapies and vaccines in the next 20 years? What pharmaceutical development challenges will emerge to develop, manufacture and commercialize them to successfully address today’s unmet medical needs? Although we can only guess at possible answers to such questions, we do know with certainty we can look forward to the next 1000 pharmaceutical biotechnology papers in Journal of Pharmaceutical Sciences to find out!
References
- 1.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 2.Mullard A. 2011 FDA drug approvals. Nat Rev Drug Discov. 2012;11:91–94. doi: 10.1038/nrd3657. [DOI] [PubMed] [Google Scholar]
- 3.Mullard A. 2012 FDA drug approvals. Nat Rev Drug Discov. 2013;12:87–90. doi: 10.1038/nrd3946. [DOI] [PubMed] [Google Scholar]
- 4.Mullard A. 2013 FDA drug approvals. Nat Rev Drug Discov. 2014;13:85–89. doi: 10.1038/nrd4239. [DOI] [PubMed] [Google Scholar]
- 5.Reichert JM. Marketed therapeutic antibodies compendium. mAbs. 2012;4:413–415. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shealy DJ, Cai A, Staquet K, Baker A, Lacy ER, Johns L, Vafa O, Gunn G, 3rd, Tam S, Sague S, Wang D, Brigham-Burke M, Dalmonte P, Emmell E, Pikounis B, Bugelski PJ, Zhou H, Scallon BJ, Giles-Komar J. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs. 2010;2(4):428–39. doi: 10.4161/mabs.2.4.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.US Food and Drug Administration Website. Biological Approvals by Year. Accessed July, 2014 at: http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/BiologicalApprovalsbyYear/
- 8.Clark HF, Burke CJ, Volkin DB, Offit P, Ward RL, Bresee JS, Dennehy P, Gooch WM, Malacaman E, Matson D, Walter E, Watson B, Krah DL, Dallas MJ, Schodel F, Kaplan KM, Heaton P. Safety, immunogenicity and efficacy in healthy infants of G1 and G2 human reassortant rotavirus vaccine in a new stabilizer/buffer liquid formulation. Pediat Infect Dis J. 2003;22:914–20. doi: 10.1097/01.inf.0000091887.48999.77. [DOI] [PubMed] [Google Scholar]
- 9.Mach H, Volkin DB, Troutman RT, Wang B, Luo Z, Jansen KU, Shi L. Disassembly and reassembly of yeast-derived recombinant human papillomavirus virus-like particles (HPV VLPs) J Pharm Sci. 2006;95:2195–2206. doi: 10.1002/jps.20696. [DOI] [PubMed] [Google Scholar]
- 10.Ho RJY, Gibaldi M. Introduction to biopharmaceuticals. In: Ho RJY, Gibaldi M, editors. Biotechnology and biopharmaceuticals: Transforming proteins and genes into drugs. New York: John Wiley and Sons; 2003. pp. 3–7. [Google Scholar]
- 11.Ho RJY, Chien JY. Drug delivery trends in clinical trials and translational medicine: Growth in biologic molecule development and impact on rheumatoid arthritis, Crohn’s disease, and colitis. J Pharm Sci. 2012;101(8):2668–74. doi: 10.1002/jps.23154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho RJY. Advanced drug delivery. In: Ho RJY, editor. Biotechnology and biopharmaceuticals: Transforming proteins and genes into drugs. 2nd. New York: John Wiley and Sons; 2013. pp. 427–465. [Google Scholar]
- 13.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G. Potential inaccurate quantitation and sizing of protein aggregates by size exclusion chromatography: Essential need to use orthogonal methods to assure the quality of therapeutic protein products. J Pharm Sci. 2010;99:2200–2208. doi: 10.1002/jps.21989. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G, Fan YX, Kirshner S, Verthelyi D, Kozlowski S, Clouse KA, Swann PG, Rosenberg A, Cherney B. Overlooking subvisible particles in therapeutic protein products: Gaps that may compromise product quality. J Pharm Sci. 2009;98:1201–1205. doi: 10.1002/jps.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh SK, Afonina N, Awwad M, Bechtold-Peters K, Blue JT, Chou D, Cromwell M, Krause HJ, Mahler HC, Meyer BK, Narhi L, Nesta DP, Spitznagel T. An industry perspective on the monitoring of subvisible particles as a quality attribute for protein therapeutics. J Pharm Sci. 2010;99:3302–3321. doi: 10.1002/jps.22097. [DOI] [PubMed] [Google Scholar]