

Abstract
Corynebacterium pseudotuberculosis strain VD57 (Cp_VD57), a highly virulent, nonmotile, non-sporulating, and a mesophilic bacterium, was isolated from a goat’s granulomatous lesion in the municipality of Juazeiro, Bahia State, Brazil. Here, we describe a set of features of the strain, together with the details of its complete genome sequence and annotation. The genome comprises of a 2.5 Mbp long, single circular genome with 2,101 protein-coding genes, 12 rRNA, 49 tRNA and 47 pseudogenes and a G + C content of 52.85 %. Genetic variation was detected in Cp_VD57 using C. pseudotuberculosis strain 1002 as reference, wherein small genomic insertions and deletions were identified. The comparative analysis of the genome sequence provides means to better understand the host pathogen interactions of this strain and can also help us to understand the molecular and genetic basis of virulence of this bacterium.
Keywords: Biovar ovis, Gram-positive pathogen, Caseous lymphadenitis, Corynebacterium pseudotuberculosis, Goat, Genome sequencing, Ion Torrent PGM
Introduction
Corynebacterium pseudotuberculosis is the etiologic agent of caseous lymphadenitis in sheep and goats, the organism has also been associated with mastitis [1–3] and can cause ulcerative lymphangitis in horses and cattle [4]. CL is a chronic disease that is characterized by the formation of granulomas in lymph nodes and internal organs, as a response of the host’s immune system against this bacterium that resists to the bactericidal action of phagocytic cells [3].
CL is considered as one of the economically important diseases of small ruminants with losses attributed to reduced wool and hide yields, carcass condemnation, morbidity and rarely mortality [5, 6]. The prevalence of CL has been observed worldwide, including South Africa, Brazil, the USA, Canada, Australia, New Zealand, United Kingdom and Egypt [7].
The pangenome analysis of 15 strains of the pathogen was completed recently [8]. However, as C. pseudotuberculosis is a relatively clonal organism [9–13], the identification of the virulence mechanisms or nucleotide modifications responsible for making a strain more virulent than another, have not yet been identified.
Sequencing of new genomes coupled with a deeper comparative analysis between the genomes and associating such analyses with the host pathogen interactions can help us understand and identify the differences between genomes and virulence factors. In this context, the present study reports the sequence the genome of the highly virulent strain VD 57 and to understand its virulence factors.
Organism information
Classification and features
C. pseudotuberculosis is a Gram-positive bacteria and belong to a CMNR (Corynebacterium,Mycobacterium,Nocardia and Rhodococcus) group that shares characteristics including an outer lipid layer, mycolic acids in the cell wall along with its derivatives including phospholipids and lipomannans [7]. C. pseudotuberculosis is a facultative intracellular pathogen showing pleomorphic forms like coccoids and filamentous rods, non-motile, non-sporulating and possessing fimbriae, with sizes ranging between 0.5–0.6 μm and 1.0–3.0 μm [7].
The C. pseudotuberculosis strain VD57 (Cp_VD57) was isolated from a goat’s granulomatous lesion in the municipality of Juazeiro, Bahia State, Brazil. The bacterial identification was made through Gram’s staining, colonies’ morphology analysis, synergic hemolysis with Rhodococcus equi in Brain Heart Infusion, Blood Agar Medium, and biochemical assays using the API Coryne system (BioMérieux). The strain is maintained in BHI broth at the Microbiology Laboratory of the Federal University of Bahia [14, 15].
C. pseudotuberculosis strain VD57 has been shown to be highly pathogenic to goats and mice [14]. This Cp_VD57 strain was able to induce IFN-gamma production in goats on day 5 after infection. Additionally, it induced a positive antibody titer between 6 and 11 days after infection [16]. Using a murine experimental model, it was observed that, the strain was able to induce a high mortality, when compared to the T1 attenuated strain, confirming its virulent profile [15]. Moura-Costa et al. used Cp_VD57 strain to challenge goats that were immunized with the attenuated T1 strain, obtaining a protection of 33.3 % and a strong humoral response, but the immunization was not able to prevent the spread of this virulent bacteria in the majority of the vaccinated animals [14].
One of the most important fields in the C. pseudotuberculosis study is the definition of genes that are differentially expressed in bacterial cultures and inside the granulomatous lesions. In this regard, VD57 strain was used in a study with the objective to determine reference genes to be used in quantitative real time PCR. It was found that eight of these genes (atpA, dnaG, efp, fusA, gyrA, gyrB, rpoB, and rpoC), mostly participating in DNA replication and transcription, can be useful as candidate reference genes, while DNA gyrase subunit A (gyrA) and elongation factor P (fusA) presented the most suitable profiles to be used in qPCR studies [17]. Figure 1 shows a phylogenetic tree of Corynebacterium pseudotuberculosis strain VD57 based on rpoB gene (β subunit of RNA polymerase). All the classification and general features of C. pseudotuberculosis strain VD57 are summarized in Table 1.
Fig. 1.
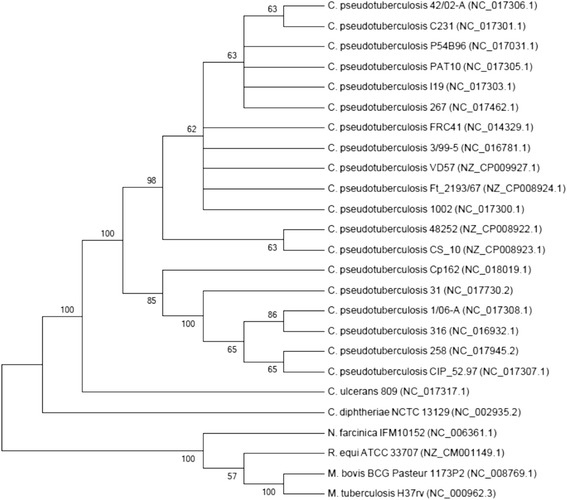
Phylogenetic tree of C. pseudotuberculosis strain VD57 representing its position relative to type strains in Corynebacteriaceae along with some other type strains of CMNR group. The tree was inferred from 3,537 aligned characters of the rpoB gene sequence using maximum likelihood method and then checked for its agreement with the current classification in Table 1. The branch lengths represent the expected number of substitutions per site. Numbers adjacent to the branches are support values from 1,000 bootstrap replicates, indicated when larger than 60 %. Calculations to determine the phylogenetic distances were done by the software MEGA v6 [40]. The GenBank accession numbers are shown in parentheses
Table 1.
Classification and general features of Corynebacterium pseudotuberculosis strain VD57 according to the MIGS recommendations [19]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [30] | |
| Phylum Actinobacteria | TAS [31] | ||
| Class Actinobacteria | TAS [32] | ||
| Order Actinomycetales Suborder Corynebacterineae | TAS [32, 33] | ||
| Family Corynebacteriaceae | TAS [32–35] | ||
| Genus Corynebacterium | TAS [36–38] | ||
| Species Corynebacterium pseudotuberculosis | TAS [37, 39] | ||
| Gram stain | Positive | TAS [14] | |
| Cell shape | Bacilli | TAS [14] | |
| Motility | Non-motile | TAS [14] | |
| Sporulation | Non-sporulating | TAS [14] | |
| Temperature range | Mesophilic | NAS | |
| Optimum temperature | 37 °C | TAS [14, 18] | |
| pH range; Optimum | 7.0–7.2 | TAS [7] | |
| Carbon source | Glucose | TAS [14] | |
| MIGS-6 | Habitat | Host | TAS [32] |
| MIGS-6.3 | Salinity | Not reported | |
| MIGS-22 | Oxygen requirement | Aerobic and Obligate Aerobic | TAS [14, 18] |
| MIGS-15 | Biotic relationship | Intracellular facultative pathogen | TAS [7, 14, 15] |
| MIGS-14 | Pathogenicity | Goat | TAS [14] |
| MIGS-4 | Geographic location | Bahia State, Brazil | TAS [14] |
| MIGS-5 | Sample collection time | 2005 | [NAS] |
| MIGS-4.1 | Latitude | 9°24’S | [IDA] |
| MIGS-4.2 | Longitude | 40°30’W | [IDA] |
aEvidence codes - IDA Inferred from Direct Assay, TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence)
De Souza et al. employed VD57 strain to verify the intracellular signaling cascade activation during the infection of splenocytes with the bacterium, and the importance of signaling pathways in the production of different cytokines. The results showed that VD57 strain was able to induce the production of TNF-alpha through the MAPK p38, and IL-10 induction via ERK-1 and −2 pathways. The complete genome sequencing and analysis will help in identifying the genetic background and the genes that may be involved in the infections [18].
Genome sequencing information
Genome project history
In the present study, we determined the nucleotide sequence of the C. pseudotuberculosis strain VD57 (Cp_VD57) genome, isolated from a goat granulomatous lesion. Sequencing, assembly, and annotation were performed at Laboratory of Cellular and Molecular Genetics (LGCM), Federal University of Minas Gerais, Belo Horizonte, Minas Gerais, Brazil and Aquacen - National Reference Laboratory for Aquatic Animal Diseases, Federal University of Minas Gerais, Brazil. The Cp_VD57 complete genome sequence and annotation data were deposited in the GenBank under the accession number CP009927. Table 2 presents the project information in accordance with the Minimum Information about a Genome Sequence (MIGS) [19].
Table 2.
Genome sequencing project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Fragments |
| MIGS 29 | Sequencing platforms | Semiconductor Ion Torrent PGM |
| MIGS 31.2 | Fold coverage | 78.22-fold |
| MIGS 30 | Assemblers | MIRA .4.0CLC Genome Workbench 4.7.2 |
| MIGS 32 | Gene calling method | Glimmer v3.02 |
| Locus Tag | CpVD57 | |
| Genbank ID | CP009927 (chromosome) | |
| GenBank Date of Release | January 06, 2015 | |
| BIOPROJECT | PRJNA267107 | |
| MIGS 13 | Source Material Identifier | BHI broth, VD57 |
| Project relevance | Animal Pathogen, Medical |
Growth conditions and genomic DNA preparation
Cp_VD57 strain was grown in brain-heart-infusion media (BHI-HiMedia Laboratories Pvt. Ltd, India) under rotation at room temperature (37 °C). Extraction of chromosomal DNA was performed using 30 mL of 48–72 h culture of bacteria, centrifuged at 4 °C and 4000 rpm for 15 min. Re-suspension of cell pellets was done in 600 μL Tris/EDTA/NaCl [10 mM Tris/HCl (pH7.0), 10 mM EDTA (pH 8.0), and 300 mM NaCl], and transferred to tubes with beads for cell lysis using Precellys®24-Dual (2 cycles of 15 s at 6500 rpm with 30 s between them). Thereafter, purification of DNA with phenol/chloroform/isoamyl alcohol (25:24:1) was followed by precipitation with ethanol/NaCl/glycogen (2.5v, 10 % NaCl and 1 % glycogen). The DNA was re-suspended in 30 μL MilliQ®. The concentration was determined by spectrophotometer, and the DNA was visualized in ethidium bromide-stained 0.7 % agarose gel.
Genome sequencing and assembly
The Ion Personal Genome Machine® System (Life Technologies) platform was used for sequencing, using fragment library. The reads with good quality was assembled using de novo strategy through Mira 4.0 software [20]. The assembly produced a total of 15 contigs, coverage of 78.22x with a N50 contig length of 405.436. Additionally, a scaffold was created using the CONTIGuator 2 software [21], taking the genome sequence of C. pseudotuberculosis strain 1002 (NC_017300.1) as reference. The gaps were closed manually using CLC Genomics Workbench 7 software [22].
Genome annotation
The annotation of genes was transferred by our in-house scripts using C. pseudotuberculosis strains 1002, 258 (NC_017945.2) and FRC41 (NC_014329.1) as reference. Manual annotation was performed using Artemis software [23]. Other elements such as rRNA, tRNA, and repetitive regions were predicted using RNAmmer [24], tRNAscan-SE [25], and Tandem Repeat Finder [26], respectively. Enzyme Commission Numbers (EC number) prediction were performed using RAST tool [27].
Genome properties
The genome is 2,337,177 bp long and comprises one main circular chromosome with a 52.19 % GC content. A total of 2,148 genes were predicted, among which 2,101 were protein coding genes, and 61 RNAs. Forty seven pseudogenes were also identified. The properties and statistics of the Cp_VD57 strain genome are listed in Table 3. The distributions of genes according to the COGs functional categories is presented in Table 4, followed by a cellular overview diagram in Fig. 2 and a summary of metabolic network statistics shown in Table 5.
Table 3.
Genome Statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 2,337,177 | 100.0 |
| DNA coding (bp) | 1,998,286. | 85.5 |
| DNA G + C (bp) | 1,235,198 | 52.9 |
| DNA scaffolds | 1 | |
| Total genesa | 2,148 | 100.0 |
| Protein coding genesa | 2,101 | 97.8 |
| RNA genes | 61 | 2.83 |
| Pseudo genes | 47 | 2.2 |
| Genes in internal clusters | NA | NA |
| Genes with function prediction | 1,578 | 73.5 |
| Genes assigned to COGs | 1,629 | 75.8 |
| Genes with Pfam domains | 1,682 | 80,1 |
| Genes with signal peptides | 158 | 7.36 |
| Genes with transmembrane helices | 605 | 28.8 |
| CRISPR repeats | NA | NA |
aThe total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome
Table 4.
Number of genes associated with the general COG functional categories
| Code | Value | % agea | Description |
|---|---|---|---|
| J | 148 | 7.04 | Translation, ribosomal structure and biogenesis |
| A | 2 | 0.09 | RNA processing and modification |
| K | 113 | 5.37 | Transcription |
| L | 104 | 4.95 | Replication, recombination and repair |
| B | 0 | 0.00 | Chromatin structure and dynamics |
| D | 20 | 0.95 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.00 | Nuclear structure |
| V | 31 | 1.47 | Defense mechanisms |
| T | 51 | 2.42 | Signal transduction mechanisms |
| M | 93 | 4.42 | Cell wall/membrane biogenesis |
| N | 5 | 0.23 | Cell motility |
| Z | 1 | 0.04 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 33 | 1.57 | Intracellular trafficking and secretion |
| O | 82 | 3.90 | Posttranslational modification, protein turnover, chaperones |
| C | 100 | 4.75 | Energy production and conversion |
| G | 115 | 5.47 | Carbohydrate transport and metabolism |
| E | 191 | 9.09 | Amino acid transport and metabolism |
| F | 69 | 3.28 | Nucleotide transport and metabolism |
| H | 103 | 4.90 | Coenzyme transport and metabolism |
| I | 62 | 2.95 | Lipid transport and metabolism |
| P | 128 | 6.09 | Inorganic ion transport and metabolism |
| Q | 31 | 1.47 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 193 | 9.18 | General function prediction only |
| S | 141 | 6.71 | Function unknown |
| - | 472 | 22.46 | Not in COGs |
| Totalb | 2288 | 104.42 |
aThe percentage is based on the total number of protein coding genes in the annotated genome
bThe total does not correspond to 1,537 CDSs, because some genes are associated with more than one COG functional categories
Fig. 2.
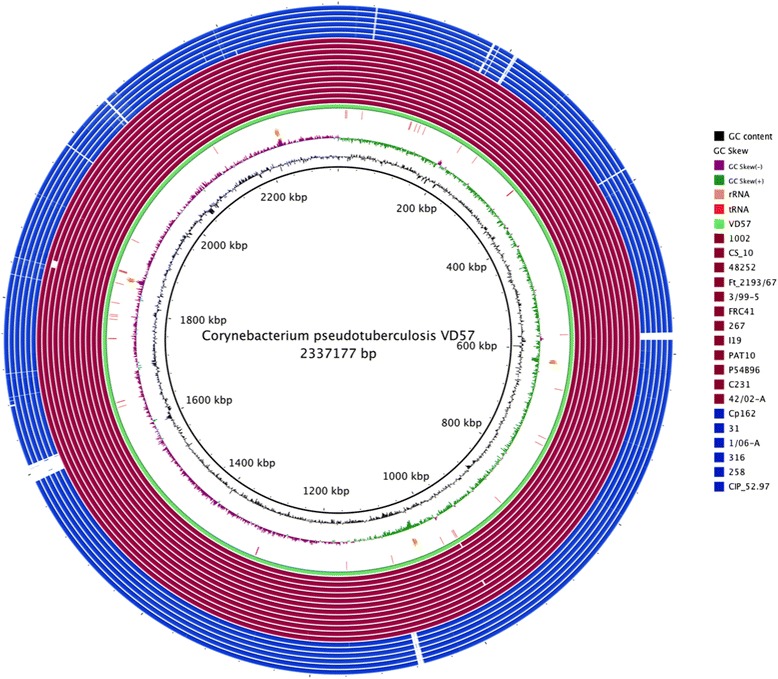
Graphical circular map of the genome [41]. From center to the outside: In wine Ovis strains, in blue Equi strains, RNA genes (tRNAs green, rRNAs orange, tRNAs red), GC content in black, GC skew
Table 5.
Metabolic Network Statistics
| Attribute | Value |
|---|---|
| Total genes | 2145 |
| Enzymes | 599 |
| Enzymatic reactions | 1197 |
| Metabolic pathways | 232 |
| Compounds | 912 |
Insights from the genome sequence
Genetic variation seems to be limited in C. pseudotuberculosis, which has been shown previously as genetically homogenous [9–13]. The MLST findings of the 64 biovar ovis strains show seven STs and all were clonally derived by eBURST analysis when a complex was deemed to share 7/8 loci; the strain Cp_VD57 was included in this analysis [28]. Although it is evident that there is very little genetic variation, we analyzed the fully sequenced Cp_VD57 genome to detect the presence of SNPs. The detected SNPs are listed in Table 6.
Table 6.
Total number of SNP’s in C. pseudotuberculosis VD57 in comparison to other strains
| Reference | Total SNPs | SNP coding regions | SNP intergenic regions |
|---|---|---|---|
| C. pseudotuberculosis 31 Equi | 25,609 | 19,811 | 5,798 |
| C. pseudotuberculosis 258 Equi | 25,706 | 21,303 | 4,403 |
| C. pseudotuberculosis 106A Equi | 24,352 | 18,085 | 6,267 |
| C. pseudotuberculosis 5297 Equi | 25,866 | 20,017 | 5,849 |
| C. pseudotuberculosis 162 Equi | 24,274 | 18,501 | 5,773 |
| C. pseudotuberculosis 316 Equi | 25,905 | 20,911 | 4,994 |
| C. pseudotuberculosis 1002 Ovis | 35 | 28 | 7 |
| C. pseudotuberculosis C231 Ovis | 952 | 741 | 211 |
| C. pseudotuberculosis P54B56 Ovis | 999 | 754 | 245 |
| C. pseudotuberculosis I19 Ovis | 968 | 762 | 206 |
| C. pseudotuberculosis FRC41 Ovis | 471 | 374 | 97 |
| C. pseudotuberculosis 267 Ovis | 2,404 | 1,869 | 535 |
| C. pseudotuberculosis PAT10 Ovis | 1,060 | 804 | 256 |
| C. pseudotuberculosis 4202 Ovis | 956 | 735 | 221 |
| C. pseudotuberculosis 3/99-5 Ovis | 502 | 411 | 91 |
| C. pseudotuberculosis 48252 | 521 | 394 | 127 |
| C. pseudotuberculosis CS_10 | 516 | 392 | 124 |
| C. pseudotuberculosis Ft_2193 | 492 | 380 | 112 |
To run SNP detection programs with MUMmer [29], default parameters were assigned. The results for SNP are in agreement with the literature, despite the fact that these strains were isolated from several hosts in different countries thereby verifying that C. pseudotuberculosis strains show limited genetic differences between worldwide strains.
Small genomic insertions and deletions were identified using the reference strain 1002, which is closer to Cp_VD57. MUMmer [29] identified 425 indels in Cp_VD57, 18 of which were in coding regions. However, three major regions of indel were identified comparing 1002 and VD57 strains: two insertion regions and one deletion. The first insertion region is located at coordinates 966430 to 968875 and comprises 2445 pb; this region has 4 genes and is present in biovar Equi strains. The second insertion region is located at coordinates 1182765 to 1182855 (90 pb), and is located within a hypothetical protein. Finally, the deletion region is located at 1002 strain (1575360–1576000) and comprises 640pb aceF pseudogenes.
Conclusions
Isolates from the C. pseudotuberculosis are genetically homogenous. Multi-locus sequence typing and comparative genomic analysis show that the isolates ovis seem to fall into the same clades. Despite the general similarity between the strains from C. pseudotuberculosis, some are more virulent, as C. pseudotuberculosis strain VD57 presented in this paper. Comparative studies with genome sequences of different C. pseudotuberculosis strains and Cp_VD57 can be performed and these analyses may be useful in identification of genome variations.
Acknowledgements
We would like to acknowledge the help of all the team members & the financing agencies. Scholarship from the CNPq under the “TWAS-CNPq Postgraduate Fellowship Programme” for doctoral studies. This work was partially executed by Rede Paraense de Genômica e Proteômica supported by FAPESPA (Fundação de Amparo à Pesquisa do Estado do Pará), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brasil), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brasil), FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais, Brasil) and FAPESB (Fundação de Amparo à Pesquisa do Estado da Bahia)
Abbreviations
- CL
caseous lymphadenitis
- Cp_VD57
Corynebacterium pseudotuberculosis strain VD57
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SA: wrote the manuscript, performed manual curation, and analyzed the data. ST: drafted the manuscript and analyzed the data. DM: genome assembly and analysis of raw data. FS and FAD performed laboratory experiments. SBJ, RTR and NC: annotated the genome. LFM, RP and RM: performed the microbiology and molecular biology studies. FLP: development of scripts for analysis of raw data. SCS, CAGL and AFC: analysis of raw data. VA: wrote the manuscript. VA, AS, DB, PG and HF: Contributed reagents/materials/analysis tools. All authors read and approved the final manuscript.
Contributor Information
Sintia Almeida, Email: sintiaalmeida@ufmg.br.
Vasco Azevedo, Email: vasco@icb.ufmg.br.
References
- 1.Hommez J, Devriese LA, Vaneechoutte M, Riegel P, Butaye P, Haesebrouck F. Identification of nonlipophilic corynebacteria isolated from dairy cows with mastitis. J. Clin. Microbiol. 1999;37:954–957. doi: 10.1128/jcm.37.4.954-957.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeruham I, Elad D, Friedman S, Perl S. Corynebacterium pseudotuberculosis infection in Israeli dairy cattle. Epidemiol. Infect. 2003;131:947–955. doi: 10.1017/S095026880300894X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braga WU, Chavera AE, González AE. Clinical, humoral, and pathologic findings in adult alpacas with experimentally induced Corynebacterium pseudotuberculosis infection. Am. J. Vet. Res. 2006;67:1570–1574. doi: 10.2460/ajvr.67.9.1570. [DOI] [PubMed] [Google Scholar]
- 4.Steinman A, Elad D, Shpigel NY. Ulcerative lymphangitis and coronet lesions in an Israeli dairy herd infected with Corynebacterium pseudotuberculosis. Vet. Rec. 1999;145:604–606. doi: 10.1136/vr.145.21.604. [DOI] [Google Scholar]
- 5.Paton MW, Mercy AR, Wilkinson FC, Gardner JJ, Sutherland SS, Ellis TM. The effects of caseous lymphadenitis on wool production and bodyweight in young sheep. Aust. Vet. J. 1988;65:117–119. doi: 10.1111/j.1751-0813.1988.tb14429.x. [DOI] [PubMed] [Google Scholar]
- 6.Rizvi S, Green LE, Glover MJ. Caseous lymphadenitis: an increasing cause for concern. Vet. Rec. 1997;140:586–587. [PubMed] [Google Scholar]
- 7.Dorella FA, Pacheco LGC, Oliveira SC, Miyoshi A, Azevedo V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet. Res. 2006;37:201–218. doi: 10.1051/vetres:2005056. [DOI] [PubMed] [Google Scholar]
- 8.Soares SC, Silva A, Trost E, et al. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PloS One. 2013;8:e53818. doi: 10.1371/journal.pone.0053818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Songer JG, Beckenbach K, Marshall MM, Olson GB, Kelley L. Biochemical and genetic characterization of Corynebacterium pseudotuberculosis. Am. J. Vet. Res. 1988;49:223–226. [PubMed] [Google Scholar]
- 10.Sutherland SS, Hart RA, Buller NB. Genetic differences between nitrate-negative and nitrate-positive C. pseudotuberculosis strains using restriction fragment length polymorphisms. Vet Microbiol. 1996;49:1–9. doi: 10.1016/0378-1135(95)00146-8. [DOI] [PubMed] [Google Scholar]
- 11.Literák I, Horváthová A, Jahnová M, Rychlík I, Skalka B. Phenotype and genotype characteristics of the Slovak and Czech Corynebacterium pseudotuberculosis strains isolated from sheep and goats. Small Rumin. Res. 1999;32:107–111. doi: 10.1016/S0921-4488(98)00174-6. [DOI] [Google Scholar]
- 12.Connor KM, Quirie MM, Baird G, Donachie W. Characterization of United Kingdom isolates of Corynebacterium pseudotuberculosis using pulsed-field gel electrophoresis. J. Clin. Microbiol. 2000;38:2633–2637. doi: 10.1128/jcm.38.7.2633-2637.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connor KM, Fontaine MC, Rudge K, Baird GJ, Donachie W. Molecular genotyping of multinational ovine and caprine Corynebacterium pseudotuberculosis isolates using pulsed-field gel electrophoresis. Vet. Res. 2007;38:613–623. doi: 10.1051/vetres:2007013. [DOI] [PubMed] [Google Scholar]
- 14.Moura-Costa LF, Bahia RC, Carminati R, et al. Evaluation of the humoral and cellular immune response to different antigens of Corynebacterium pseudotuberculosis in Canindé goats and their potential protection against caseous lymphadenitis. Vet. Immunol. Immunopathol. 2008;126:131–141. doi: 10.1016/j.vetimm.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 15.Vale VLC. Avaliação de aspectos da resposta imune de camundongos balb/c contra linhagem T1 de Corynebacterium pseudotuberculosis [Evaluation of Balb/c mice imune response against the Corynebacterium pseudotuberculosis T1 strain] 2005. [Google Scholar]
- 16.Paule BJA, Azevedo V, Regis LF, et al. Experimental Corynebacterium pseudotuberculosis primary infection in goats: kinetics of IgG and interferon-gamma production, IgG avidity and antigen recognition by Western blotting. Vet. Immunol. Immunopathol. 2003;96:129–139. doi: 10.1016/S0165-2427(03)00146-6. [DOI] [PubMed] [Google Scholar]
- 17.Carvalho DM, de Sá PH, Castro TLP, et al. Reference genes for RT-qPCR studies in Corynebacterium pseudotuberculosis identified through analysis of RNA-seq data. Antonie Van Leeuwenhoek. 2014;106:605–614. doi: 10.1007/s10482-014-0231-3. [DOI] [PubMed] [Google Scholar]
- 18.De Souza A, Vale V, Silva M, et al. MAPK involvement in cytokine production in response to Corynebacterium pseudotuberculosis infection. BMC Microbiol. 2014;14:230. doi: 10.1186/s12866-014-0230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Field D, Garrity G, Gray T, et al. The minimum information about a genome sequence (MIGS) specification. Nat. Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chevreux B, Wetter T, Suhai S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. In: ISB, ed. Hannover, Germany. 1999:45–46. Available at: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.23.7465.
- 21.Galardini M, Biondi EG, Bazzicalupo M, Mengoni A. CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol. Med. 2011;6:11. doi: 10.1186/1751-0473-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CLCBio. CLC Genomics Workbench. Accessed 2014. Available at: http://www.clcbio.com/products/clc-genomics-workbench.
- 23.Rutherford K, Parkhill J, Crook J, et al. Artemis: sequence visualization and annotation. Bioinforma. Oxf. Engl. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 24.Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aziz RK, Bartels D, Best AA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolt F. The population structure of the Corynebacterium diphtheriae group. http://wrap.warwick.ac.uk/1759/ (2009). Accessed 25 Out 2009.
- 29.Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U. S. A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2001. pp. 119–169. [Google Scholar]
- 32.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a New Hierarchic Classification System, Actinobacteria classis nov. Int. J. Syst. Bacteriol. 1997;47:479–491. doi: 10.1099/00207713-47-2-479. [DOI] [Google Scholar]
- 33.Zhi X-Y, Li W-J, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int. J. Syst. Evol. Microbiol. 2009;59:589–608. doi: 10.1099/ijs.0.65780-0. [DOI] [PubMed] [Google Scholar]
- 34.Skerman VBD, McGowan VF, Sneath PHA, et al. Approved lists of bacterial names. Washington, D.C.: American Society for Microbiology; 1989. [PubMed] [Google Scholar]
- 35.Lehmann K, Neumann R. Atlas und Grundriss der Bakteriologie und Lehrbuch der speziellen bakteriologischen Diagnostik. München: J.F. Lehmann; 1907. [Google Scholar]
- 36.Lehmann KBNR. Atlas und Grundriss der Bakteriologie und Lehrbuch der speziellen bakteriologischen Diagnostik. 1. München: J.F. Lehmann; 1896. pp. 1–448. [Google Scholar]
- 37.Skerman VBDMV, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [Google Scholar]
- 38.Bernard KA, Wiebe D, Burdz T, et al. Assignment of Brevibacterium stationis (ZoBell and Upham 1944) Breed 1953 to the genus Corynebacterium, as Corynebacterium stationis comb. nov., and emended description of the genus Corynebacterium to include isolates that can alkalinize citrate. Int. J. Syst. Evol. Microbiol. 2010;60:874–879. doi: 10.1099/ijs.0.012641-0. [DOI] [PubMed] [Google Scholar]
- 39.Eberson F. A Bacteriologic Study of the Diphtheroid Organisms with Special Reference to Hodgkin’s Disease [with Discussion] J. Infect. Dis. 1918;23:1–42. doi: 10.1086/infdis/23.1.1. [DOI] [Google Scholar]
- 40.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]