

Abstract
The accelerated metabolic demands of the working muscle cannot be met without a robust response from the liver. If not for the hepatic response, sustained exercise would be impossible. The liver stores, releases, and recycles potential energy. Exercise would result in hypoglycemia if it were not for the accelerated release of energy as glucose. The energetic demands on the liver are largely met by increased oxidation of fatty acids mobilized from adipose tissue. Adaptations immediately following exercise facilitate the replenishment of glycogen stores. Pancreatic glucagon and insulin responses orchestrate the hepatic response during and immediately following exercise. Like skeletal muscle and other physiological systems, liver adapts to repeated demands of exercise by increasing its capacity to produce energy by oxidizing fat. The ability of regular physical activity to increase fat oxidation is protective and can reverse fatty liver disease. Engaging in regular physical exercise has broad ranging positive health implications including those that improve the metabolic health of the liver.
The liver is a battery, a rechargeable battery at that. It releases stored energy at times of high metabolic demand and replenishes energy stores during the nutrient excess associated with a meal. The liver is a recycler converting metabolites into macronutrients, amino acids into proteins, and transforming potential energy into chemical energy. The liver is a detoxifier removing nitrogenous molecules, hemoglobin, hormones, foreign substances, immunoglobulin, and other compounds from the circulation. The muscle contracts, the adipose tissue stores fat, and the heart pumps blood. The functions of the liver are far too vast to describe with a single dominant process, but all make broad contributions to arterial homeostasis and thereby homeostasis of numerous cell types. Physical exercise poses a unique challenge to the liver as metabolic demands of working muscles require the liver to mobilize energy stores, recycle metabolites, and convert compounds that are toxic in excess to innocuous forms. The focus of this review will be on how the liver adapts to the metabolic demands of physical exercise.
1. LIVER RESPONSE TO ACUTE EXERCISE
1.1 Mobilization of Liver Energy Stores Maintains Glucose Homeostasis
The energy requirements of exercise necessitate a marked increase in glucose uptake by muscle, as well as increased utilization of lipids and muscle glycogen. Energy for working muscle may also be derived from branched chain amino acids. The mechanism for the increase in muscle fuel utilization is discussed in detail previously1 and elsewhere in this volume. The requirement of glucose uptake for the working muscle is transferred in part to the liver, which must release glucose at a rate that matches the accelerated rate of glucose uptake to maintain glucose homeostasis.2 There may be deviations from glucose homeostasis during high intensity and/or prolonged exercise. During high-intensity exercise, the stimulus to release glucose from the liver exceeds glucose utilization causing a rise in arterial glucose.3 In contrast, during prolonged exercise, hypoglycemia may result as liver glycogen nears depletion.4 Hypoglycemia can cause a more rapid onset of fatigue, perhaps due to neuroglycopenia. In cases of hypoglycemia, fatigue can be delayed by ingestion of glucose or glucose polymers.5
Accelerated muscle glucose uptake during exercise is matched by the sum of increased mobilization of hepatic glycogen and gluconeogenesis. Potential energy in the form of glycogen is at its highest concentration in the liver. With the onset of exercise, this energy store is hydrolyzed by activation of glycogen phosphorylase,6 contributing to the release of glucose from the liver. Moreover, exercise causes the liver to discharge chemical energy with the hydrolysis of adenosine triphosphate (ATP). Adenosine monophosphate (AMP) and adenosine diphosphate (ADP) accumulate,7,8 resulting in a precipitous fall in liver energy charge (Fig. 1). This discharge of chemical energy with exercise is necessary for gluconeogenesis and reactions that support this pathway (e.g., fatty acid activation, ureagenesis). Studies using 5′-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) have aided in establishing a role for hepatic adenine nucleotides in the regulation of liver glucose metabolism. AICAR is converted by phosphorylation to an AMP analog (ZMP) inside cells.9 Infusing AICAR at rates to create liver ZMP concentrations that match liver AMP concentrations evident during exercise creates a potent breakdown of liver glycogen,9,10 due presumably to increased allosteric activation of glycogen phosphorylase. Thus, the AMP produced with chemical energy discharge may combine with or mediate endocrine stimuli to enhance the release of potential energy by mobilization of hepatic glycogen.
Figure 1.
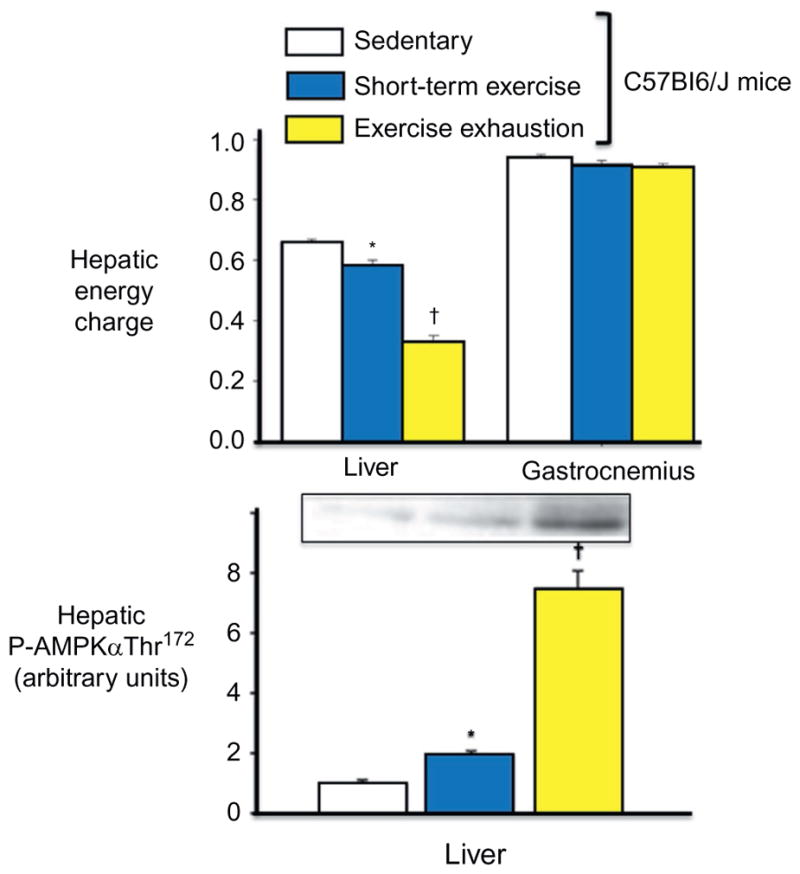
Hepatic energy discharge in response to short term and exhaustive exercise in C57Bl/6J mice. Hepatic energy charge decreases with exercise and becomes critically low with exhaustive exercise (top). Skeletal muscle energy charge is not affected by exercise. The decrease in energy charge is associated with an increase in hepatic AMPK activation (bottom). Energy charge is calculated by the equation (ATP+ 0.5ADP)/(ATP + ADP + AMP). Data are mean ± SE. *Significantly different from SED (P<0.05). †Significantly different from SED and ST (P<0.05). Modified from Camacho et al.8
AMP-activated protein kinase (AMPK) serves as a transducer, sensing energy state and activating metabolic pathways in accordance with metabolic needs.11 Free fatty acids are mobilized from adipose tissue and delivered to the liver as the body transitions into a gluconeogenic mode during prolonged exercise.12 AMPK activation increases the oxidation of fatty acids delivered to the liver resulting in the production of chemical energy from extrahepatic sources.13 This process is reflected by increased hepatic oxygen uptake and ketone body output. Elevated fatty acid oxidation also increases the liver mitochondrial redox state, which favors gluconeogenic flux.12
Accelerated gluconeogenic flux during exercise requires more than activation of reactions within the liver. The increased flux through gluconeogenesis requires increased gluconeogenic precursor delivery from extrahepatic sites to the liver and transport across the membrane by the liver (Fig. 2).
Figure 2.

Gluconeogenesis is regulated by gluconeogenic precursor supply to the liver, extraction by the liver, and conversion to glucose within the liver. All these processes are accelerated by physical exercise.
1.2 Pancreatic Hormones Stimulate Hepatic Glucose Output
Exercise is characterized by complex neural and endocrine responses. If exercise is sustained (>~20 min), a decrease in insulin secretion and increases in glucagon, catecholamines, and cortisol secretion, among other hormones, are observed.2 The signal for these hormonal and autonomic changes has been difficult to elucidate. An increase in afferent nerve activity originating at the working limb, a deficit in fuel availability, and a neural feed-forward mechanism have all been postulated as possible stimuli.2 Afferent sensors in the carotid sinus area are required for the full increases in glucagon and norepinephrine during exercise.14 Surprisingly, denervation of the pancreas does not impair the glucagon and insulin responses to exercise,15 suggesting that an endocrine or a paracrine factor may be involved. The myokine interleukin 6 (IL-6) is released in response to physical exercise16 and has been shown to stimulate glucagon release from the pancreatic alpha-cell under stressful conditions.17 The pancreas is highly sensitive to small changes in blood glucose during exercise.18–21 In light of this, it is surprising that preventing the exercise-induced increase in muscle glucose uptake by deleting the exercise-sensitive glucose transporter, Glut4, prevents neither the pancreatic hormone response nor the increase in hepatic glucose output during exercise.22 This suggests that the endocrine and hepatic responses to exercise are not due to a feedback signal from accelerated blood glucose removal as once proposed.
Understanding regulation of the liver has proven difficult, particularly during exercise, since it is inaccessible in conscious humans. Moreover, movement makes magnetic resonance spectroscopy of the liver during exercise untenable. The dog has proven to be a useful experimental model for gaining insights into liver function because the portal vein, which perfuses the liver, and hepatic vein, which drains it, can be accessed using implanted catheters. The small size of rodent models is not conducive to these deep abdominal catheterizations or the blood sample volume associated with multiple sampling ports. Despite evidence showing the exquisite sensitivity of the liver to glucagon in humans,23 a vital role of glucagon for the exercise-induced increase in hepatic glucose output has been slow to gain acceptance. This is because the increment in arterial glucagon with exercise is delayed and dampened with respect to the increase in glucose released from the liver.2 In fact, depending on the duration and intensity of exercise, arterial glucagon may not increase at all. Glucagon released from the pancreas first perfuses the liver delaying its entry into the peripheral circulation. Since the liver extracts glucagon, the increase in arterial glucagon is attenuated.24,25 Portal vein glucagon is increased during exercise to a much greater extent than in arterial and hepatic vein blood. The portal vein to arterial glucagon gradient is increased by approximately 10-fold in response to exercise.24,25 The placement of the liver between the pancreas and general circulation is efficient since it allows for increased glucagon in the blood perfusing the liver without the high glucagon secretion rates needed to rapidly fill the general circulation.
Studies conducted in exercising dogs and humans defined the specific roles of insulin and glucagon in control of hepatic glucose output.26–33 Studies have shown that the rise in glucagon32,33 and the fall in insulin19,33,34 are major determinants of glucose production during moderate exercise. The rise in glucagon is required for the full increment in hepatic glycogenolysis and gluconeogenesis,32,33 while the fall in insulin33,34 is necessary for hepatic glycogenolysis. Although changes in glucagon and insulin are individually very important, the interaction of these hormones is an essential component of the stimulus.2 An increase in glucagon in the physiological context of exercise is considerably more potent than the effects of an experimental increase in glucagon of the same magnitude. A twofold increase in glucagon causes a peak increase in hepatic glucose output of ~1 mg kg−1 min−1 in the sedentary dog,35 while the same increase during exercise causes a peak increase of ~5 mg kg−1 min−1.32 Glucagon action is fully manifested during exercise for three reasons (Fig. 3). First, the increased glucose utilization of working muscle prevents the hyperglycemia that accompanies an experimental increase in glucagon. Second, as mentioned earlier, prolonged exercise creates a physiological environment that supports gluconeogenesis.12 This includes mobilization of gluconeogenic substrates from muscle, adipose, and intestine. Finally, exercise causes a fall in insulin that sensitizes the liver to glucagon.2
Figure 3.

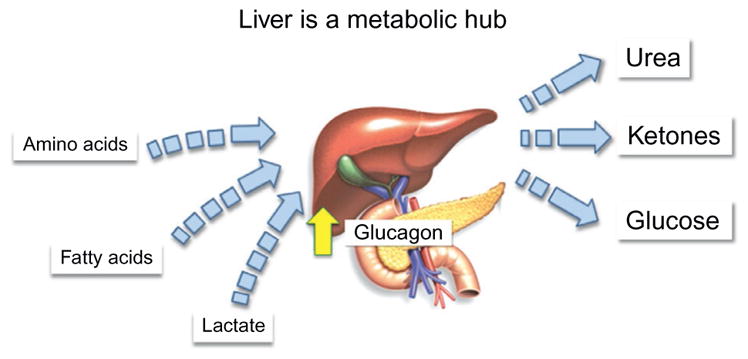
The liver is a metabolic hub where pathways for amino acid, fat, and glucose metabolism are integrated. The integration of these pathways serves to provide energy in the form of glucose, recycle carbon-based metabolites, and prevent nitrogen toxicity. The demand on these pathways is accentuated during exercise and driven by glucagon secreted from pancreatic alpha-cells.
1.3 Evidence Lacking for Adrenergic Stimulation of Hepatic Glucose Output
The robust increase in arterial catecholamine concentrations that occurs in response to exercise led to a long-standing assumption that norepinephrine and epinephrine stimulate the observed increase in hepatic glucose output.36 It is now clear that changes in arterial catecholamine concentrations do not translate to changes in concentrations at the hepatocyte. Hepatic norepinephrine spillover (reflecting sympathetic drive) is not increased during moderate exercise, and the portal vein epinephrine concentration at the liver is markedly attenuated as the gastrointestinal tract extracts 50% of it.37 In line with this, a broad range of experimental approaches have failed to show an appreciable effect of either hepatic sympathetic nerves or circulating epinephrine in stimulation of hepatic glucose output during moderate exercise. There are instances such as high-intensity exercise or exercise in specific populations (e.g., poorly controlled diabetics) where the adrenergic response is unusually high, while the pancreatic hormone response is not.3 For this reason, the regulation of hepatic glucose output under conditions such as these has been postulated to be different.38 It has been hypothesized that during high-intensity exercise, control of glucose production shifts from the pancreatic hormones to the catecholamines.38,39 This is based on two observations. First, circulating blood norepinephrine and epinephrine can increase by 10- to 20-fold,39 whereas the increase in the glucagon to insulin ratio in peripheral blood is considerably less and in some cases undetectable.31 Second, when high-intensity exercise is performed during a pancreatic clamp (portal insulin and glucagon fixed at basal), hepatic glucose output increases normally even though the increase in arterial glucagon is blunted and the fall in insulin is absent.40
Despite the evidence cited above, the role of the catecholamines in control of hepatic glucose output with high-intensity exercise (>80% maximum O2 uptake) remains unclear. Studies that have assessed the role for catecholamines during high-intensity exercise using whole-body pharmacological adrenergic receptor blockade have uniformly been without an effect on hepatic glucose production.31,39,41 Such studies are difficult to interpret due to the lack of specificity of these pharmacological agents. Intraportal propranolol and phentolamine infusion has been used in a dog model to create selective hepatic adrenergic blockade.42,43 With this technique, hepatic adrenergic blockade can be achieved without extrahepatic effects. Hepatic adrenergic blockade did not impair the increase in hepatic glucose output or affect blood glucose homeostasis during high-intensity exercise, resulting in a threefold increase in hepatic norepinephrine spillover.42 Similar results are seen in dogs treated with the β-cell toxin, alloxan.43 Alloxan-diabetic dogs in poor metabolic control have sevenfold higher rates of hepatic norepinephrine spillover than nondiabetic dogs during moderate exercise. Even with the greater hepatic norepinephrine spillover in diabetic dogs, selective hepatic adrenergic receptor blockade did not affect the exercise-induced increase in hepatic glucose output.43 Thus, hepatic glucose output in the dog model is not reliant on hepatic adrenergic receptor stimulation even during heavy exercise or in diabetes, which is associated with excessive sympathetic drive.
There are factors involved in the stimulation of glucose production during exercise that are as yet undefined. One could postulate that myokines such as IL-6,44 retinol-binding protein 4,45 apelin,45 and myonectin46 play a role in regulation of hepatic glucose production. More studies are needed to determine the role of these proteins on liver metabolism.
1.4 Recharging Liver Glycogen Stores after Exercise
Despite the important role of the liver in fuel homeostasis and the added metabolic demands placed on it by exercise, very little is known about how this organ is regulated during the recovery period following exercise. Prior exercise increases the capacity of the liver to consume glucose in response to a simulated meal.47 Studies following 150 min of exercise in the chronically catheterized dog exhibited a twofold increase in hepatic glucose uptake in response to a twofold increase in glucose load compared to sedentary dogs. These data show that the well-established increase in whole-body glucose tolerance in the postexercise state is due, in part, to an increased ability of liver to take up glucose. Indirect assessments in the anesthetized rabbit support findings in the dog, showing that liver deposition of a fluorescent glucose analog via either an oral bolus or a continuous 120 min intraportal infusion was greater after hind limb contraction.48 These data were also supported by studies using magnetic resonance spectroscopy that showed that ingestion of 13C-glucose immediately after completion of prolonged moderate exercise in human subjects increased liver glycogen resynthesis by ~0.7 mg kg−1 min−1 over a period of 4 h of postexercise recovery.49
The liver is more insulin sensitive after exercise and this could explain the improved ability of the liver to take up glucose. During a hyperinsulinemic euglycemic clamp, net hepatic glucose output was suppressed to a greater extent following prolonged exercise, compared to their sedentary controls.50 These results provide a basis for the hypothesis that hepatic insulin sensitivity could be the cause of increased net hepatic glucose uptake during a hepatic glucose load after exercise. This hypothesis was tested in studies in which net hepatic glucose uptake was measured in sedentary and exercised dogs during an increased portal venous glucose supply (simulated meal) with either basal or elevated insulin.51 The increase in net hepatic glucose uptake (Fig. 4) and fractional glucose extraction with hyperinsulinemia was approximately 50% greater in exercised compared to sedentary dogs. These findings were consistent with subsequent findings in mice that show that acute exercise induced a rapid and pronounced transcriptional effect in the liver and regulated hepatic insulin receptor substrate (IRS) proteins leading to improved cellular insulin signaling.52 Results from these studies provide a further physiological basis for recommending exercise for patients with insulin resistance.
Figure 4.

Net hepatic glucose uptake in dogs following 150 min of exercise or an equivalent sedentary period. Glucose was infused into a portal vein to increase the liver glucose supply by twofold, and arterial glucose was clamped at 180 mg dl−1. Insulin was infused at basal rates (0.2 mU kg−1 min−1) or at rates designed to simulate a meal (1.2 mU kg−1 min−1). *p ≤0.05 versus basal insulin. †p ≤0.05 versus sedentary high insulin. Data are mean±SE. Modified from Pencek et al.51
As discussed earlier, the exercise-induced increase in glucagon and/or decrease in insulin are the major stimuli for the accelerated mobilization of glucose from the liver. The hypothesis that exercise-induced changes in pancreatic hormones could increase the ability of the liver to consume glucose following exercise by depleting hepatic glycogen stores was tested. Adaptation of the liver following exercise was tested in dogs in which somatostatin was used to suppress glucagon and insulin. Glucagon and Insulin were replaced at either basal rates or rates that simulated the exercise response.53 Preventing the response of glucagon and insulin to exercise prevents hepatic glucose output and glycogen breakdown. Simulation of the glucagon and insulin responses to exercise resulted in a threefold increase in hepatic glucose output and marked glycogen breakdown. Despite the differences in glycogen mobilization in the two protocols, hepatic glucose uptake was increased equally in response to a glucose load and hyper-insulinemia, exceeding rates in sedentary dogs. However, when pancreatic hormone responses were simulated and hepatic glucose output was accelerated during exercise, a greater fraction of the glucose consumed by the liver was directed to glycogen.53 Thus, the increase in insulin-stimulated hepatic glucose uptake observed after exercise is attributable to a factor other than those adaptations resulting from the exercise-induced pancreatic hormone responses. The pancreatic hormone response is a critical determinant of the fate of glucose consumed by the liver, with a greater fraction directed to liver glycogen. This unique finding is consistent with studies that show a greater fraction of the glucose taken up by the liver after prolonged exercise in dogs is metabolized primarily nonoxidatively.54 A similar result is seen after a prolonged, glycogen-depleting fast.55
Exercise also leads to a number of other endocrine changes. It is possible, for example, that the exercise-stimulated glucocorticoid response may prime the liver to take up more glucose since high doses of this hormone can stimulate hepatic glycogen deposition.56 The growing list of myokines and adipokines, such as adiponectin and irisin, may also have implications for the effects of prior exercise on liver insulin sensitivity and glycogen repletion. Adiponectin released from adipocytes57 has insulin-sensitizing effects at the liver.58 Irisin released from muscle has also been associated with improved liver insulin action.59 Exercise has persistent effects on processes and enzymes involved in liver glucose metabolism that are sustained well after the cessation of exercise.60 It is possible that some facet of the exercise response activates enzymes involved in hepatic glucose uptake and metabolism.
Clearly, there are effects of prior exercise directly at the liver that facilitate hepatic glucose uptake and glycogen storage. It is important to consider the integrated response of the body when considering the physiological response to ingested glucose. In this regard, replenishment of hepatic glycogen stores is facilitated by intestinal adaptations that cause an increase in absorption of glucose into the portal circulation. Hepatic glucose uptake after exercise is facilitated by increased absorption of ingested glucose.54,61,62 Through the use of the isotopic glucose analogs 3-O-[3H]methylglucose (absorbed via transporter-mediated and passive processes) and L-[14C]glucose (absorbed passively), it was determined that the increase in gut glucose absorption seen following exercise was primarily due to an increase in passive absorption across the intestinal cell wall.62
2. THE LIVER RECYCLES CARBONS AND DISPOSES OF EXCESS METABOLITES
The liver plays a vital role in recycling potential energy and thereby conserving it with maximal efficiency. The liver conserves energy by recycling carbon-based molecules and turning them into potential energy by the gluconeogenic pathway. Consistent with the role of glucagon in stimulating hepatic gluconeogenesis, glucagon plays a vital role in stimulating the hepatic extraction of metabolites released during exercise.63–65 Glucagon stimulates both the N66 and A67 amino acid transport systems causing accelerated transport of gluconeogenic amino acids, glutamine and alanine into the liver. Lactate and pyruvate produced by glycolytically and glycerol produced by lipolysis are also recycled into potential energy in the liver gluconeogenic pathway. It is fascinating that gluconeogenic precursors are not only channeled from working muscle68 and adipose tissue but are also released from nonworking muscle as lactate and the gastrointestinal tract as amino acids.69,70 Recycling energy from metabolites and amino acids into glucose has an energetic cost. Fatty acid mobilization leads to increased fat oxidation, which provides energy for gluconeogenesis and associated pathways. This fatty acid oxidation, like other vital pathways in the liver during exercise, is driven by the fall in insulin71 and the increase in glucagon.65
3. THE LIVER DETOXIFIES BY CONVERTING EXCESS NITROGEN TO UREA
During exercise, amino acid and AMP deamination lead to an increased formation of NH3 by skeletal muscle.72 Moreover, increased protein breakdown from the skeletal muscle73,74 or the gastrointestinal tract69,70 results in the release of amino acids into the circulation. The amino acids are only a minor direct fuel source for the muscle. The carbon skeleton of a number of amino acids including alanine, glutamine, glutamate, serine, threonine, and valine is delivered to the liver where the carbons are recycled into glucose. A corollary to the scavenging of amino acid carbons is that the associated nitrogen must be converted to a nontoxic form and excreted. Free or amino acid-associated NH3 formed during exercise, in large part, enters the urea cycle. Urea is secreted from the liver during exercise and is filtered in the kidney, prior to excretion.
As with mobilization of energy and the recycling of carbons, the incorporation of nitrogen is largely governed by glucagon. Increased glucagon during exercise stimulates the liver uptake of amino acids during exercise.32,63 In the absence of the rise in glucagon, amino acid concentrations increase. It has been shown in the exercising dog model that the increase in glucagon is required for the transfer of isotopically labeled glutamine nitrogen into urea nitrogen.63 Thus, there is compelling evidence that glucagon plays an essential role in preventing toxic nitrogen accumulation during times of amino acid formation and delivery to the liver. The role of the exercise-induced increase in glucagon in the integration of metabolic pathways involved in carbohydrate, fat, and amino acid metabolism is illustrated schematically in Fig. 5.
Figure 5.
The liver is exquisitely sensitive to the actions of glucagon, exceeding glucagon sensitivity in the sedentary state (inset). Glucagon action is fully manifested during exercise because (i) the increased glucose utilization of working muscle prevents the hyperglycemia that accompanies an experimental increase in glucagon; (ii) prolonged exercise creates a physiological environment that supports gluconeogenesis as gluconeogenic substrates are mobilized from muscle, adipose, and intestine; and (iii) exercise causes a fall in insulin that sensitizes the liver to glucagon.
4. HEPATIC ADAPTATIONS TO REGULAR PHYSICAL ACTIVITY
It has become increasingly evident that just as muscle adapts to habitual physical activity, so does the liver. These adaptations that influence macro-nutrient metabolism are summarized in Fig. 6. The hepatic adaptations may be important in increasing exercise capacity. More significantly, the hepatic adaptations reduce fatty liver, which is a major risk factor for the constellation of conditions that comprise metabolic syndrome. Given the role of liver as an energy balance maintenance system, it is no surprise that it undergoes a dynamic remodeling in response to a metabolically demanding process such as exercise. Physical activity is currently maintained as a first line treatment for obesity and any number of the comorbidities that accompany this disease state. While many studies have examined exercise training, relatively few of these examine specific effects of exercise on liver-related endpoints.
Figure 6.

Exercise training elicits complex adaptations in liver metabolic processes.79–83,85,86,90,93–97 Decreased delivery of substrates (a), constant rates of VLDL-TG synthesis (b), increased mitochondrial oxidation of lipid (c), and decreased lipid anabolic processes (d) may contribute to a net decrease in hepatic lipid stores (e). Increased mitochondrial function (f) and/or content (g) may serve to elevate lipid oxidation rates. An increase in PEPCK levels (h), but not G6Pase (i), may indicate elevated gluconeogenic potential elicited through training. An improved ability of the liver to respond to hormonal stimuli, such as insulin (j), may underlie some of the metabolic adaptations seen. Nuclear transcription and protein translation alterations may underlie adaptive changes of this organ.
Among the literature focused on liver outcomes in relation to exercise, a significant portion consists of epidemiological associations of exercise or physical activity with general health outcomes or superficial markers of liver function.75 Typically, these studies conclude that exercise correlates with decreased hepatic fat content76 and decreased prevalence of nonalcoholic fatty liver disease (NAFLD).77,78 These studies are valuable for assessment of large medical populations with broad diagnostic criteria but are inherently limited in mechanistic insights.
Animal models have served an invaluable role regarding this general lack of mechanisms consequent to exercise in liver. The Otsuka Long-Evans Tokushima Fatty (OLETF) rat, a rat model of hyperphagic induced obesity and type 2 diabetes, has been carefully characterized in relation to the response to exercise training. In this model, exercise has been used as an intervention and prevention technique for parameters related to NAFLD. Outside of improvements in general measures of glucose tolerance and calculated insulin sensitivity, both study designs demonstrate that exercise seems to modify the liver cellular machinery related to lipid metabolism and gluconeogenesis. One of the underlying causes of decreased liver lipid content and improvements in steatotic phenotypes may be decreased lipid anabolism coupled with increased lipid catabolism.79–82 This is demonstrated by decreases in the lipogenic proteins fatty acid synthase (Fas) and acetyl-CoA carboxylase (Acc) with relative increases in deactivation of Acc by phosphorylation as well. This decrease in Fas protein persists in a model of swim training in C57Bl6/J mice fed a standard chow or a high-fat diet.83 An increase in complete oxidation of palmitate to CO2 in the OLETF model has been interpreted as improved coupling of mitochondrial oxidation to the TCA cycle. Interestingly, rats bred for an intrinsically higher aerobic capacity also show increases in fatty acid oxidation metrics within the liver in absence of training.84 Whether the intrinsic aerobic capacity is afferent or efferent to hepatic lipid oxidative phenotypes is currently unknown. Other markers such as Cpt-1 activity, β-Had activity, citrate synthase activity, Tr4 protein, and cytochrome c oxidase component levels are increased suggesting an increase in mitochondrial function and/or content,79,80,85–87 which may be dependent on presence of Pgc-1α.86 Alterations in mitochondrial FA oxidation have typically been accompanied by a decrease in extramitochondrial palmitate oxidation in the OLETF rat model.79,81,87 This decrease in extramitochondrial palmitate oxidation is hypothesized to represent a decrease in peroxisomal contributions to oxidation. This concomitant increase in mitochondrial oxidation and decreased peroxisomal oxidation may be representative of a more “metabolically healthy” lipid oxidation phenotype resulting in decreased oxidative stress,88 a hypothesized contributor to liver injury in NAFLD,89 being a potential result.
Among the caveats of evaluating the molecular underpinnings of exercise in liver function is an inability to dissociate effects of a lifestyle intervention from the pleiotropic benefits of weight loss. This proves difficult to resolve in animals without implementation of complicated pair feeding strategies. Despite this challenge, several human studies have shown that exercise, independent of weight loss effects, decreases hepatic lipid content.90–93 This seems to be independent of lipid delivery to the liver as concentrations of TG and FFA only decrease when weight loss is evident.79–83,90,91,94–96 Recently, it was confirmed that altered hepatic lipid content is independent of any effects on VLDL-TG or VLDL-ApoB100 secretion rates.93 Increased utilization of lipid substrates at rest or during exercise may be the underlying cause of these decreases.90,97 One study specifically delineated the effects of exercise with and without weight loss on liver insulin sensitivity using a modified clamp technique in human subjects.98 This study concludes that exercise, in absence of weight loss, improves the ability of insulin to suppress glucose production of liver; however, combining exercise with ~6% weight loss in this study led to even further improvements in this metric. This improved liver metabolic profile may be due to an altered localization of adipose tissue from visceral to subcutaneous depots resulting in a lower constitutive provision of FFAs to the portal circulation.91,92 This hypothesis is far from resolved as metabolic improvements have been reported in the absence of decreased visceral adipose tissue area as well.90
As described previously, energy demand from the muscle is met with increased release of glucose from the muscle in a glucagon-dependent manner. Training increases the liver’s sensitivity to glucagon.99–102 This effect is associated with upregulation of glucagon receptors in the liver.103 The processes contributing to glucagon signaling and gluconeogenesis consume ATP resulting in a reduced AMP/ATP ratio in the liver.7,104 Decreased energy charge of the liver activates AMPK resulting in a host of molecular cascades leading to a net decrease in lipogenic processes with a complementary increase in lipid oxidation.105,106 Chronic exercise stimulus does not seem to rely on transient energy charge effects, but rather on a bulk shift in lipid metabolic machinery and oxidative processes. This is seen in the consistent increase in several parameters relating to mitochondria previously noted.
5. SUMMARY
The accelerated metabolic demands of the working muscle cannot be met without a robust response from the liver. If not for the hepatic response, sustained exercise would be impossible. If not for the increased hepatic glucose output with exercise, hypoglycemia would result due to inadequate breakdown of glycogen stores and recycling of metabolites through the gluconeogenic pathway. The energetic demands on the liver are largely met by increased oxidation of fatty acids mobilized from adipose tissue. Adaptations immediately following exercise facilitate the replenishment of glycogen stores. Pancreatic glucagon and insulin responses orchestrate the hepatic response during and immediately following exercise. Like skeletal muscle, liver adapts to repeated demands of exercise by increasing its capacity to produce energy by oxidizing fat. The ability of regular physical activity to increase fat oxidation is protective and can reverse fatty liver disease. Engaging in regular physical exercise has broad ranging positive health implications including those that improve the metabolic health of the liver.
Acknowledgments
Grants from the NIH are gratefully acknowledged (R01DK054902 and R37 DK050277). A.S.W. received support from the Molecular Endocrinology Training Program (T32 DK07563). The Vanderbilt Diabetes Research and Training Center (P30 DK20593) and Vanderbilt Mouse Metabolic Phenotypiing Centers (U24 DK059637) are acknowledged.
References
- 1.Wasserman DH, Kang L, Ayala JE, Fueger PT, Lee-Young RS. The physiological regulation of glucose flux into muscle in vivo. J Exp Biol. 2011;214(pt 2):254–262. doi: 10.1242/jeb.048041. http://dx.doi.org/10.1242/jeb.048041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab. 2009;296(1):E11–E21. doi: 10.1152/ajpendo.90563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sigal RJ, Kenny GP, Wasserman DH, Castaneda-Sceppa C. Physical activity/exercise and type 2 diabetes. Diabetes Care. 2004;27(10):2518–2539. doi: 10.2337/diacare.27.10.2518. [DOI] [PubMed] [Google Scholar]
- 4.Felig P, Cherif A, Minageref A, Wahren J. Hypoglycemia during prolonged exercise in normal men. N Engl J Med. 1982;306:895–900. doi: 10.1056/NEJM198204153061503. [DOI] [PubMed] [Google Scholar]
- 5.Wasserman DH, Zinman B. American diabetes association technical review on exercise in individuals with insulin-dependent diabetes mellitus. Diabetes Care. 1994;17:924–937. doi: 10.2337/diacare.17.8.924. [DOI] [PubMed] [Google Scholar]
- 6.Wasserman DH. Regulation of glucose fluxes during exercise in the postabsorptive state. Annu Rev Physiol. 1995;57:191–218. doi: 10.1146/annurev.ph.57.030195.001203. [DOI] [PubMed] [Google Scholar]
- 7.Berglund ED, Lee-Young RS, Lustig DG, et al. Hepatic energy state is regulated by glucagon receptor signaling in mice. J Clin Invest. 2009;119(8):2412–2422. doi: 10.1172/JCI38650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camacho RC, Donahue EP, James FD, Berglund ED, Wasserman DH. Energy state of the liver during short-term and exhaustive exercise in C57BL/6J mice. Am J Physiol Endocrinol Metab. 2006;290(3):E405–E408. doi: 10.1152/ajpendo.00385.2005. [DOI] [PubMed] [Google Scholar]
- 9.Camacho RC, Pencek RR, Lacy DB, James FD, Donahue EP, Wasserman DH. Portal venous 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion overcomes hyperinsulinemic suppression of endogenous glucose output. Diabetes. 2005;54(2):373–382. doi: 10.2337/diabetes.54.2.373. [DOI] [PubMed] [Google Scholar]
- 10.Pencek RR, Shearer J, Camacho RC, et al. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside causes acute hepatic insulin resistance in vivo. Diabetes. 2005;54:355–360. doi: 10.2337/diabetes.54.2.355. [DOI] [PubMed] [Google Scholar]
- 11.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23(12):1112–1119. doi: 10.1002/bies.10009. http://dx.doi.org/10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 12.Wasserman DH, Lacy DB, Bracy D, Williams PE. Metabolic regulation in peripheral tissues and transition to increased gluconeogenic mode during prolonged exercise. Am J Physiol. 1992;263:E345–E354. doi: 10.1152/ajpendo.1992.263.2.E345. [DOI] [PubMed] [Google Scholar]
- 13.Foretz M, Viollet B. Regulation of hepatic metabolism by AMPK. J Hepatol. 2011;54(4):827–829. doi: 10.1016/j.jhep.2010.09.014. http://dx.doi.org/10.1016/j.jhep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Koyama Y, Coker RH, Denny JC, et al. Role of carotid bodies in the neuroendocrine response to exercise. Am J Physiol. 2001;281:E742–E748. doi: 10.1152/ajpendo.2001.281.4.E742. [DOI] [PubMed] [Google Scholar]
- 15.Coker RH, Koyama Y, Lacy DB, Williams PE, Rheaume N, Wasserman DH. Pancreatic innervation is not essential for exercise-induced changes in glucagon and insulin or glucose kinetics. Am J Physiol. 1999;277:E1122–E1129. doi: 10.1152/ajpendo.1999.277.6.E1122. [DOI] [PubMed] [Google Scholar]
- 16.Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8(8):457–465. doi: 10.1038/nrendo.2012.49. http://dx.doi.org/10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- 17.Barnes TM, Otero YF, Elliott AD, et al. Interleukin-6 amplifies glucagon secretion: coordinated control via the brain and pancreas. Am J Physiol Endocrinol Metab. 2014;307(10):E896–E905. doi: 10.1152/ajpendo.00343.2014. http://dx.doi.org/10.1152/ajpendo.00343.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wasserman DH, Lickley HLA, Vranic M. Interactions between glucagon and other counterregulatory hormones during normoglycemic and hypoglycemic exercise in dogs. J Clin Invest. 1984;74:1404–1413. doi: 10.1172/JCI111551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wasserman DH, Lacy DB, Colburn CA, Bracy DP, Cherrington AD. Efficiency of compensation for the absence of the fall in insulin during exercise. Am J Physiol. 1991;261:E587–E597. doi: 10.1152/ajpendo.1991.261.5.E587. [DOI] [PubMed] [Google Scholar]
- 20.Berger CM, Sharis PJ, Bracy DP, Lacy DB, Wasserman DH. Sensitivity of exercise-induced increase in hepatic glucose production to glucose supply and demand. Am J Physiol. 1994;267:E411–E421. doi: 10.1152/ajpendo.1994.267.3.E411. [DOI] [PubMed] [Google Scholar]
- 21.Jenkins AB, Chisholm DJ, Ho KY, Kraegen EW. Exercise induced hepatic glucose output is precisely sensitive to the rate of systemic glucose supply. Metabolism. 1985;34:431–434. doi: 10.1016/0026-0495(85)90208-2. [DOI] [PubMed] [Google Scholar]
- 22.Fueger PT, Li CY, Ayala JE, et al. Glucose kinetics and exercise tolerance in mice lacking the GLUT4 glucose transporter. J Physiol. 2007;582(pt 2):801–812. doi: 10.1113/jphysiol.2007.132902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lins PE, Wajngot A, Adamson U, Vranic M, Efendic S. Minimal increases in glucagon levels enhance glucose production in man with partial hypoinsulinemia. Diabetes. 1983;32(7):633–636. doi: 10.2337/diab.32.7.633. [DOI] [PubMed] [Google Scholar]
- 24.Coker RH, Lacy DB, Krishna MG, Wasserman DH. Splanchnic glucagon kinetics in exercising alloxan-diabetic dogs. J Appl Physiol. 1999;86(5):1626–1631. doi: 10.1152/jappl.1999.86.5.1626. [DOI] [PubMed] [Google Scholar]
- 25.Wasserman DH, Lacy DB, Bracy DP. Relationship between arterial and portal vein immunoreactive glucagon during exercise. J Appl Physiol. 1993;75:724–729. doi: 10.1152/jappl.1993.75.2.724. [DOI] [PubMed] [Google Scholar]
- 26.Tuttle KR, Marker JC, Dalsky GP, et al. Glucagon, not insulin, may play a secondary role in defense against hypoglycemia. Am J Physiol. 1988;254:E713–E719. doi: 10.1152/ajpendo.1988.254.6.E713. [DOI] [PubMed] [Google Scholar]
- 27.Wolfe RR, Nadel ER, Shaw JH, Stephenson LA, Wolfe MH. Role of changes in insulin and glucagon in glucose homeostasis in exercise. J Clin Invest. 1986;77(3):900–907. doi: 10.1172/JCI112388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wasserman DH, Lickley HL, Vranic M. Interactions between glucagon and other counterregulatory hormones during normoglycemic and hypoglycemic exercise in dogs. J Clin Invest. 1984;74(4):1404–1413. doi: 10.1172/JCI111551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wasserman DH, Lickley HL, Vranic M. Important role of glucagon during exercise in diabetic dogs. J Appl Physiol. 1985;59(4):1272–1281. doi: 10.1152/jappl.1985.59.4.1272. [DOI] [PubMed] [Google Scholar]
- 30.Hirsch IB, Marker JC, Smith LJ, et al. Insulin and glucagon in the prevention of hypoglycemia during exercise in humans. Am J Physiol. 1991;260:E695–E704. doi: 10.1152/ajpendo.1991.260.5.E695. [DOI] [PubMed] [Google Scholar]
- 31.Kjaer M, Engfred K, Fernandez A, Secher N, Galbo H. Regulation of hepatic glucose production during exercise in humans: role of sympathoadrenergic activity. Am J Physiol. 1993;265:E275–E283. doi: 10.1152/ajpendo.1993.265.2.E275. [DOI] [PubMed] [Google Scholar]
- 32.Wasserman DH, Spalding JS, Lacy DB, Colburn CA, Goldstein RE, Cherrington AD. Glucagon is a primary controller of the increments in hepatic glycogenolysis and gluconeogenesis during exercise. Am J Physiol. 1989;257:E108–E117. doi: 10.1152/ajpendo.1989.257.1.E108. [DOI] [PubMed] [Google Scholar]
- 33.Lavoie C, Ducros F, Bourque J, Langelier H, Chiasson JL. Glucose metabolism during exercise in man: the role of insulin and glucagon in the regulation of hepatic glucose production and gluconeogenesis. Can J Physiol Pharmacol. 1997;75(1):26–35. doi: 10.1139/cjpp-75-1-26. [DOI] [PubMed] [Google Scholar]
- 34.Wasserman DH, Williams PE, Lacy DB, Goldstein RE, Cherrington AD. Exercise-induced fall in insulin and hepatic carbohydrate metabolism during muscular work. Am J Physiol. 1989;256(4 pt 1):E500–E509. doi: 10.1152/ajpendo.1989.256.4.E500. [DOI] [PubMed] [Google Scholar]
- 35.Stevenson RW, Steiner KE, Davis MA, et al. Similar dose responsiveness of hepatic glycogenolysis and gluconeogenesis to glucagon in vivo. Diabetes. 1987;36:382–389. doi: 10.2337/diab.36.3.382. [DOI] [PubMed] [Google Scholar]
- 36.Christensen NJ, Galbo H. Sympathetic nerve activity during exercise. Annu Rev Physiol. 1983;45:139–153. doi: 10.1146/annurev.ph.45.030183.001035. [DOI] [PubMed] [Google Scholar]
- 37.Coker RH, Krishna MG, Lacy DB, Zinker B, Wasserman DH. Sympathetic drive to liver and nonhepatic splanchnic tissue during prolonged exercise is increased in diabetes. Metabolism. 1997;46:1327–1332. doi: 10.1016/s0026-0495(97)90239-0. [DOI] [PubMed] [Google Scholar]
- 38.Marliss EB, Simantirakis E, Purdon C, et al. Glucoregulatory and hormonal responses to repeated bouts of intense exercise in normal male subjects. J Appl Physiol. 1991;71:924–933. doi: 10.1152/jappl.1991.71.3.924. [DOI] [PubMed] [Google Scholar]
- 39.Marliss EB, Purdon C, Halter JB, Sigal RJ, Vranic M. Glucoregulation during and after intense exercise in control and diabetic subjects. In: Devlin J, Horton E, Vranic M, editors. Diabetes Mellitus and Exercise. London: Smith-Gordon and Company Limited; 1992. pp. 173–188. [Google Scholar]
- 40.Sigal RJ, Fisher SF, Halter JB, Vranic M, Marliss EB. The roles of catecholamines in glucoregulation in intense exercise as defined by the islet cell clamp technique. Diabetes. 1995;45:148–156. doi: 10.2337/diab.45.2.148. [DOI] [PubMed] [Google Scholar]
- 41.Sigal RJ, Fisher S, Marliss EB. Catecholamines are the primary regulators of glucose production (Ra) during intense exercise: definition using the islet-cell clamp (IC) Diabetes. 1994;43(suppl 1):159A. [Google Scholar]
- 42.Coker RH, Krishna MG, Lacy DB, Bracy DP, Wasserman DH. Role of hepatic alpha-and beta-adrenergic receptor stimulation on hepatic glucose production during heavy exercise. Am J Physiol. 1997;273(5 pt 1):E831–E838. doi: 10.1152/ajpendo.1997.273.5.E831. [DOI] [PubMed] [Google Scholar]
- 43.Coker RH, Lacy DB, Williams PE, Wasserman DH. Hepatic alpha- and beta-adrenergic receptors are not essential for the increase in Ra during exercise in diabetes. Am J Physiol. 2000;278:E444–E451. doi: 10.1152/ajpendo.2000.278.3.E444. [DOI] [PubMed] [Google Scholar]
- 44.Febbraio MA, Hiscock N, Sacchetti M, Fischer CP, Pedersen BK. Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes. 2004;53:1643–1648. doi: 10.2337/diabetes.53.7.1643. [DOI] [PubMed] [Google Scholar]
- 45.Besse-Patin A, Montastier E, Vinel C, et al. Effect of endurance training on skeletal muscle myokine expression in obese men: identification of apelin as a novel myokine. Int J Obes. 2014;38(5):707–713. doi: 10.1038/ijo.2013.158. http://dx.doi.org/10.1038/ijo.2013.158. [DOI] [PubMed] [Google Scholar]
- 46.Seldin MM, Lei X, Tan SY, Stanson KP, Wei Z, Wong GW. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J Biol Chem. 2013;288(50):36073–36082. doi: 10.1074/jbc.M113.500736. http://dx.doi.org/10.1074/jbc.M113.500736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galassetti P, Coker RH, Lacy DB, Cherrington AD, Wasserman DH. Prior exercise increases net hepatic glucose uptake during a glucose load. Am J Physiol. 1999;276:E1022–E1029. doi: 10.1152/ajpendo.1999.276.6.E1022. [DOI] [PubMed] [Google Scholar]
- 48.Matsuhisha M, Nishizawa I, Ikeda M, et al. Prior muscular contraction enhances disposal of a glucose analog in the liver and muscle. Metabolism. 1998;47:44–49. doi: 10.1016/s0026-0495(98)90191-3. [DOI] [PubMed] [Google Scholar]
- 49.Casey A, Mann R, Banister K, et al. Effect of carbohydrate ingestion on glycogen resynthesis in human liver and skeletal muscle, measured by 13C MRS. Am J Physiol. 2000;278:E65–E75. doi: 10.1152/ajpendo.2000.278.1.E65. [DOI] [PubMed] [Google Scholar]
- 50.Koyama Y, Galassetti P, Coker RH, et al. Prior exercise and the response to insulin-induced hypoglycemia in the dog. Am J Physiol. 2002;282:E1128–E1138. doi: 10.1152/ajpendo.00370.2001. [DOI] [PubMed] [Google Scholar]
- 51.Pencek RR, James FD, Lacy DB, et al. Interaction of insulin and prior exercise in control of hepatic metabolism of a glucose load. Diabetes. 2003;52:1897–1903. doi: 10.2337/diabetes.52.8.1897. [DOI] [PubMed] [Google Scholar]
- 52.Hoene M, Lehmann R, Hennige AM, et al. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. J Physiol. 2009;587(pt 1):241–252. doi: 10.1113/jphysiol.2008.160275. http://dx.doi.org/10.1113/jphysiol.2008.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pencek RR, James FD, Lacy DB, Camacho RC, Fueger PT, Wasserman DH. Exercise-induced changes in insulin and glucagon are not required for enhanced glucose uptake by the liver following exercise but influence the fate of glucose. Diabetes. 2004;53:3041–3047. doi: 10.2337/diabetes.53.12.3041. [DOI] [PubMed] [Google Scholar]
- 54.Hamilton KS, Gibbons FK, Bracy DP, Lacy DB, Cherrington AD, Wasserman DH. Effect of prior exercise on the partitioning of an intestinal glucose load between splanchnic bed and skeletal muscle. J Clin Invest. 1996;98:125–135. doi: 10.1172/JCI118756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Galassetti P, Reed E, Messina A, Lacy DB, Wasserman DH. Effect of prior fast duration on the appearance and disposal of an intraduodenal glucose load. Am J Physiol. 1999;276:E543–E552. doi: 10.1152/ajpendo.1999.276.3.E543. [DOI] [PubMed] [Google Scholar]
- 56.Long CNH, Katzen B, Fry EG. Adrenal cortex and carbohydrate metabolism. Endocrinology. 1940;26:309–351. [Google Scholar]
- 57.Bouassida A, Chamari K, Zaouali M, Feki Y, Zbidi A, Tabka Z. Review on leptin and adiponectin responses and adaptations to acute and chronic exercise. Br J Sports Med. 2010;44(9):620–630. doi: 10.1136/bjsm.2008.046151. http://dx.doi.org/10.1136/bjsm.2008.046151. [DOI] [PubMed] [Google Scholar]
- 58.Tao C, Sifuentes A, Holland WL. Regulation of glucose and lipid homeostasis by adiponectin: effects on hepatocytes, pancreatic beta cells and adipocytes. Best Pract Res Clin Endocrinol Metab. 2014;28(1):43–58. doi: 10.1016/j.beem.2013.11.003. http://dx.doi.org/10.1016/j.beem.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arias-Loste MT, Ranchal I, Romero-Gomez M, Crespo J. Irisin, a link among fatty liver disease, physical inactivity and insulin resistance. Int J Mol Sci. 2014;15(12):23163–23178. doi: 10.3390/ijms151223163. http://dx.doi.org/10.3390/ijms151223163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dohm GL, Kasperek GJ, Barakat HA. Time course of changes in gluconeogenic enzyme activities during exercise and recovery. Am J Physiol. 1985;249:E6–E11. doi: 10.1152/ajpendo.1985.249.1.E6. [DOI] [PubMed] [Google Scholar]
- 61.Maehlum S, Felig P, Wahren J. Splanchnic glucose and muscle glycogen metabolism after glucose feeding during post-exercise recovery. Am J Physiol. 1978;235:E255–E260. doi: 10.1152/ajpendo.1978.235.3.E255. [DOI] [PubMed] [Google Scholar]
- 62.Pencek RR, Koyama Y, Lacy DB, et al. Prior exercise enhances passive absorption of intraduodenal glucose. J Appl Physiol. 2003;95:1132–1138. doi: 10.1152/japplphysiol.01172.2002. [DOI] [PubMed] [Google Scholar]
- 63.Krishna MG, Coker RH, Lacy DB, Zinker BA, Halseth AE, Wasserman DH. Glucagon response to exercise is critical for accelerated hepatic glutamine metabolism and nitrogen disposal. Am J Physiol. 2000;279:E638–E645. doi: 10.1152/ajpendo.2000.279.3.E638. [DOI] [PubMed] [Google Scholar]
- 64.Wasserman DH, Williams PE, Lacy DB, Green DR, Cherrington AD. Importance of intrahepatic mechanisms to gluconeogenesis from alanine during prolonged exercise and recovery. Am J Physiol. 1988;254:E518–E525. doi: 10.1152/ajpendo.1988.254.4.E518. [DOI] [PubMed] [Google Scholar]
- 65.Wasserman DH, Spalding JS, Bracy DP, Lacy DB, Cherrington AD. Exercise-induced rise in glucagon and the increase in ketogenesis during prolonged muscular work. Diabetes. 1989;38:799–807. doi: 10.2337/diab.38.6.799. [DOI] [PubMed] [Google Scholar]
- 66.Nissim I, Brosnan M, Yudkoff M, Nissim I, Brosnan JT. Studies of hepatic glutamine metabolism in the perfused rat liver with 15N-labeled glutamine. J Biol Chem. 1999;274:28958–28965. doi: 10.1074/jbc.274.41.28958. [DOI] [PubMed] [Google Scholar]
- 67.Cariappa R, Kilberg MS. Plasma membrane domain localization and transcytosis of the glucagon-induced hepatic system A carrier. Am J Physiol. 1992;263(6 pt 1):E1021–E1028. doi: 10.1152/ajpendo.2006.263.6.E1021. [DOI] [PubMed] [Google Scholar]
- 68.Ahlborg G, Felig P. Lactate and glucose exchange across the forearm, legs, and splanchnic bed during after prolonged leg exercise. J Clin Invest. 1982;69:45–54. doi: 10.1172/JCI110440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams BD, Wolfe RR, Bracy DP, Wasserman DH. Gut contributes essential amino acids during exercise. Am J Physiol. 1996;270:E85–E90. doi: 10.1152/ajpendo.1996.270.1.E85. [DOI] [PubMed] [Google Scholar]
- 70.Wasserman DH, Geer RJ, Williams PE, Lacy DB, Abumrad NN. Interaction of gut and liver in nitrogen metabolism during exercise. Metabolism. 1991;40:307–314. doi: 10.1016/0026-0495(91)90115-d. [DOI] [PubMed] [Google Scholar]
- 71.Wasserman DH, Lacy DB, Goldstein RE, Williams PE, Cherrington AD. Exercise-induced fall in insulin and the increase in fat metabolism during prolonged exercise. Diabetes. 1989;38:484–490. doi: 10.2337/diab.38.4.484. [DOI] [PubMed] [Google Scholar]
- 72.Eriksson LS, Broberg S, Bjorkman O, Wahren J. Ammonia metabolism during exercise in man. Clin Physiol. 1985;5:325–336. doi: 10.1111/j.1475-097x.1985.tb00753.x. [DOI] [PubMed] [Google Scholar]
- 73.Felig P, Wahren J. Amino acid metabolism in exercising man. J Clin Invest. 1971;50:2703–2711. doi: 10.1172/JCI106771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Loon LJ. Is there a need for protein ingestion during exercise? Sports Med. 2014;44(suppl 1):S105–S111. doi: 10.1007/s40279-014-0156-z. http://dx.doi.org/10.1007/s40279-014-0156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caldwell S, Lazo M. Is exercise an effective treatment for NASH? Knowns and unknowns. Ann Hepatol. 2009;8(suppl 1):S60–S66. [PubMed] [Google Scholar]
- 76.Perseghin G, Lattuada G, De Cobelli F, et al. Habitual physical activity is associated with intrahepatic fat content in humans. Diabetes Care. 2007;30(3):683–688. doi: 10.2337/dc06-2032. http://dx.doi.org/10.2337/dc06-2032. [DOI] [PubMed] [Google Scholar]
- 77.Church TS, Kuk JL, Ross R, Priest EL, Biltoft E, Blair SN. Association of cardiorespiratory fitness, body mass index, and waist circumference to nonalcoholic fatty liver disease. Gastroenterology. 2006;130(7):2023–2030. doi: 10.1053/j.gastro.2006.03.019. http://dx.doi.org/10.1053/j.gastro.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 78.Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, et al. Role of leisure-time physical activity in nonalcoholic fatty liver disease: a population-based study. Hepatology. 2008;48(6):1791–1798. doi: 10.1002/hep.22525. http://dx.doi.org/10.1002/hep.22525. [DOI] [PubMed] [Google Scholar]
- 79.Borengasser SJ, Rector RS, Uptergrove GM, et al. Exercise and omega-3 polyunsaturated fatty acid supplementation for the treatment of hepatic steatosis in hyperphagic OLETF rats. J Nutr Metab. 2012;2012:268680. doi: 10.1155/2012/268680. http://dx.doi.org/10.1155/2012/268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rector RS, Thyfault JP, Morris RT, et al. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. 2008;294(3):G619–G626. doi: 10.1152/ajpgi.00428.2007. http://dx.doi.org/10.1152/ajpgi.00428.2007. [DOI] [PubMed] [Google Scholar]
- 81.Rector RS, Uptergrove GM, Morris EM, et al. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G874–G883. doi: 10.1152/ajpgi.00510.2010. http://dx.doi.org/10.1152/ajpgi.00510.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Linden MA, Fletcher JA, Morris EM, et al. Combining metformin and aerobic exercise training in the treatment of type 2 diabetes and NAFLD in OLETF rats. Am J Physiol Endocrinol Metab. 2014;306(3):E300–E310. doi: 10.1152/ajpendo.00427.2013. http://dx.doi.org/10.1152/ajpendo.00427.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schultz A, Mendonca LS, Aguila MB, Mandarim-de-Lacerda CA. Swimming training beneficial effects in a mice model of nonalcoholic fatty liver disease. Exp Toxicol Pathol. 2012;64(4):273–282. doi: 10.1016/j.etp.2010.08.019. http://dx.doi.org/10.1016/j.etp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 84.Morris EM, Jackman MR, Johnson GC, et al. Intrinsic aerobic capacity impacts susceptibility to acute high-fat diet-induced hepatic steatosis. Am J Physiol Endocrinol Metab. 2014;307(4):E355–E364. doi: 10.1152/ajpendo.00093.2014. http://dx.doi.org/10.1152/ajpendo.00093.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lezi E, Lu J, Burns JM, Swerdlow RH. Effect of exercise on mouse liver and brain bioenergetic infrastructures. Exp Physiol. 2013;98(1):207–219. doi: 10.1113/expphysiol.2012.066688. http://dx.doi.org/10.1113/expphysiol.2012.066688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Haase TN, Ringholm S, Leick L, et al. Role of PGC-1alpha in exercise and fasting-induced adaptations in mouse liver. Am J Physiol Regul Integr Comp Physiol. 2011;301(5):R1501–R1509. doi: 10.1152/ajpregu.00775.2010. http://dx.doi.org/10.1152/ajpregu.00775.2010. [DOI] [PubMed] [Google Scholar]
- 87.Linden MA, Fletcher JA, Morris EM, et al. Treating NAFLD in OLETF rats with vigorous-intensity interval exercise training. Med Sci Sports Exerc. 2014 doi: 10.1249/MSS.0000000000000430. http://dx.doi.org/10.1249/MSS.0000000000000430. [DOI] [PMC free article] [PubMed]
- 88.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. http://dx.doi.org/10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 89.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 90.Hallsworth K, Fattakhova G, Hollingsworth KG, et al. Resistance exercise reduces liver fat and its mediators in non-alcoholic fatty liver disease independent of weight loss. Gut. 2011;60(9):1278–1283. doi: 10.1136/gut.2011.242073. http://dx.doi.org/10.1136/gut.2011.242073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnson NA, Sachinwalla T, Walton DW, et al. Aerobic exercise training reduces hepatic and visceral lipids in obese individuals without weight loss. Hepatology. 2009;50(4):1105–1112. doi: 10.1002/hep.23129. http://dx.doi.org/10.1002/hep.23129. [DOI] [PubMed] [Google Scholar]
- 92.van der Heijden GJ, Wang ZJ, Chu ZD, et al. A 12-week aerobic exercise program reduces hepatic fat accumulation and insulin resistance in obese, Hispanic adolescents. Obesity. 2010;18(2):384–390. doi: 10.1038/oby.2009.274. http://dx.doi.org/10.1038/oby.2009.274. [DOI] [PubMed] [Google Scholar]
- 93.Sullivan S, Kirk EP, Mittendorfer B, Patterson BW, Klein S. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology. 2012;55(6):1738–1745. doi: 10.1002/hep.25548. http://dx.doi.org/10.1002/hep.25548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jenkins NT, Padilla J, Arce-Esquivel AA, et al. Effects of endurance exercise training, metformin, and their combination on adipose tissue leptin and IL-10 secretion in OLETF rats. J Appl Physiol. 2012;113(12):1873–1883. doi: 10.1152/japplphysiol.00936.2012. http://dx.doi.org/10.1152/japplphysiol.00936.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marques CM, Motta VF, Torres TS, Aguila MB, Mandarim-de-Lacerda CA. Beneficial effects of exercise training (treadmill) on insulin resistance and nonalcoholic fatty liver disease in high-fat fed C57BL/6 mice. Braz J Med Biol Res. 2010;43(5):467–475. doi: 10.1590/s0100-879x2010007500030. [DOI] [PubMed] [Google Scholar]
- 96.Tamura Y, Tanaka Y, Sato F, et al. Effects of diet and exercise on muscle and liver intracellular lipid contents and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2005;90(6):3191–3196. doi: 10.1210/jc.2004-1959. http://dx.doi.org/10.1210/jc.2004-1959. [DOI] [PubMed] [Google Scholar]
- 97.Fealy CE, Haus JM, Solomon TP, et al. Short-term exercise reduces markers of hepatocyte apoptosis in nonalcoholic fatty liver disease. J Appl Physiol. 2012;113(1):1–6. doi: 10.1152/japplphysiol.00127.2012. http://dx.doi.org/10.1152/japplphysiol.00127.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coker RH, Williams RH, Yeo SE, et al. The impact of exercise training compared to caloric restriction on hepatic and peripheral insulin resistance in obesity. J Clin Endocrinol Metab. 2009;94(11):4258–4266. doi: 10.1210/jc.2008-2033. http://dx.doi.org/10.1210/jc.2008-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Drouin R, Lavoie C, Bourque J, Ducros F, Poisson D, Chiasson JL. Increased hepatic glucose production response to glucagon in trained subjects. Am J Physiol. 1998;274(1 pt 1):E23–E28. doi: 10.1152/ajpendo.1998.274.1.E23. [DOI] [PubMed] [Google Scholar]
- 100.Drouin R, Robert G, Milot M, Massicotte D, Peronnet F, Lavoie C. Swim training increases glucose output from liver perfused in situ with glucagon in fed and fasted rats. Metabolism. 2004;53(8):1027–1031. doi: 10.1016/j.metabol.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 101.Podolin DA, Wills BK, Wood IO, Lopez M, Mazzeo RS, Roth DA. Attenuation of age-related declines in glucagon-mediated signal transduction in rat liver by exercise training. Am J Physiol Endocrinol Metab. 2001;281(3):E516–E523. doi: 10.1152/ajpendo.2001.281.3.E516. [DOI] [PubMed] [Google Scholar]
- 102.Bonjorn VM, Latour MG, Belanger P, Lavoie JM. Influence of prior exercise and liver glycogen content on the sensitivity of the liver to glucagon. J Appl Physiol. 2002;92(1):188–194. doi: 10.1152/jappl.2002.92.1.188. [DOI] [PubMed] [Google Scholar]
- 103.Legare A, Drouin R, Milot M, et al. Increased density of glucagon receptors in liver from endurance-trained rats. Am J Physiol Endocrinol Metab. 2001;280(1):E193–E196. doi: 10.1152/ajpendo.2001.280.1.E193. [DOI] [PubMed] [Google Scholar]
- 104.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi: 10.1038/nature11808. http://dx.doi.org/10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ruderman NB, Park H, Kaushik VK, et al. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol Scand. 2003;178(4):435–442. doi: 10.1046/j.1365-201X.2003.01164.x. http://dx.doi.org/10.1046/j.1365-201X.2003.01164.x. [DOI] [PubMed] [Google Scholar]
- 106.Park H, Kaushik VK, Constant S, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277(36):32571–32577. doi: 10.1074/jbc.M201692200. http://dx.doi.org/10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]