

Abstract
Bile acids are well known for their effects on cholesterol homeostasis and lipid digestion. Since the discovery of bile acid receptors, of which there are farnesoid X receptor (FXR), a nuclear receptor, and the plasma membrane G-protein receptor, as well as Takeda G-protein coupled receptor clone 5, further roles have been elucidated for bile acids including glucose and lipid metabolism as well as inflammation. Additionally, treatment with bile acid receptor agonists has shown a decrease in the amount of atherosclerosis plaque formation and decreased portal vascular resistance and portal hypotension in animal models. Furthermore, rodent models have demonstrated antifibrotic activity using bile acid receptor agonists. Early human data using a FXR agonist, obeticholic acid, have shown promising results with improvement of histological activity and even a reduction of fibrosis. Human studies are ongoing and will provide further information on bile acid receptor agonist therapies. Thus, bile acids and their derivatives have the potential for management of liver diseases and potentially other disease states including diabetes and the metabolic syndrome.
Keywords: Bile acids, FXR, TGR5, Liver disease
Introduction
Bile acids, the principal constituent of bile, are synthesized from cholesterol in the hepatocytes and secreted into the bile canaliculi to be stored in the gall bladder [1]. In response to a meal, the gall bladder contracts and bile flows into the small intestine. Being amphipathic molecules, bile acids aid in the emulsification of lipids and facilitate the absorption of lipid nutrients and lipid soluble vitamins [2]. Nearly 95 % of the bile acids that are secreted into the small intestine are transported back to the liver from the distal part of the ileum and colon via the enterohepatic circulation. Bile acid transport is achieved by both passive diffusion and active transport [3]. Although bile acids are efficiently cleared from the portal circulation by the liver, a fraction enters the systemic circulation. Fasting circulating bile acid levels are generally less than 5 μM and increase up to 15 μM postprandially [4]. The remaining 5 % of bile acids that escape absorption from the intestine is lost in the feces, a major route for the elimination of excess cholesterol from the body [1, 5]. In addition to their established roles in cholesterol homeostasis and lipid digestion, bile acids also act as signaling molecules. With the identification of the bile acid receptors, both the endocrine and paracrine functions of bile acids have become evident [6]. There has been an increasing development in understanding bile acid signaling, and bile acid receptors have become an attractive therapeutic target for the treatment of diseases such as metabolic syndrome, diabetes and nonalcoholic fatty liver disease (NAFLD).
Bile acid synthesis and regulation
Bile acid synthesis is a complex multistep process. There are two major bile acid biosynthetic pathways: the classical or neutral pathway and the acidic pathway. Bile acid synthesis from the classical pathway (75 %) is initiated by CYP7A1 (cholesterol 7 alpha-hydroxylase or Cytochrome P450 7A1), which is expressed only in the hepatocytes, whereas the acidic pathway (25 %), initiated by mitochondrial CYP27A1, is also expressed in macrophages and many other tissues [7]. It is worth noting that all the essential enzymes for the conversion of cholesterol into bile acids reside only in the liver, which is the main site of synthesis [8]. The rate-limiting steps in the classical pathway and acidic pathway are 7α-hydroxylation of cholesterol by CYP7A1 and the transport of free cholesterol to the inner mitochondrial membrane by StarD1, respectively [7, 9]. In humans, the primary bile acids produced in the liver are cholic acid (CA) and chenodeoxycholic acid (CDCA), whereas cholic acid and muricholic acid form the primary bile acid pool in mice. Primary bile acids are either glycine or taurine conjugated to render them more water-soluble. Upon reaching the large intestine, they undergo deconjugation and dehydroxylation by the bacteria present in the gut forming secondary bile acids, which consist of lithocholic acid (LCA) and deoxycholic acid (DCA) in humans [10, 11]. The secondary bile acids (unconjugated) are passively absorbed by the epithelial cells of the distal ileum, whereas the conjugated bile acids are actively transported via bile acid transporters [12]. Bile acids consist of a steroid core and a side chain with carboxyl group. The number and position of hydroxyl groups on the steroid core determine the hydrophilic or hydrophobic nature of the bile acid. The primary bile acids are more hydrophilic than the secondary bile acids, and taurine conjugates are more hydrophilic than the glycine conjugates [13].
Bile acid synthesis is highly regulated. The basal rate of synthesis of bile acids in the liver is ~0.6 g per day in healthy humans, the amount sufficient to replace the lost bile acids in the feces. The total bile acid pool in the gastrointestinal tract is ~3 g, and this pool recirculates ~4 to 12 times per day. If the reabsorption of bile acids is defective, de novo synthesis of bile acids in the liver increases up to 4–6 g per day. As high concentrations of bile acids are cytotoxic, bile acids regulate their own metabolism by exerting a negative feedback on their synthesis [5, 7, 14, 15]. The bile acid feedback repression mechanism is governed by the interplay of several nuclear receptors to inhibit the transcription of the CYP7A1 gene, the rate-limiting enzyme in bile acid synthesis [16]. In addition, factors such as diet, nutrients, cytokines and hormones also regulate the expression of CYP7A1 [17].
Bile acid transport
Bile acid homeostasis is maintained by the balance of bile acid synthesis, secretion and reabsorption [7, 9, 17]. Bile acids are absorbed by both passive and active mechanisms. Passive absorption of bile acids occurs along the entire small intestine and colon. At the normal pH of the intestine (5.5–6.5), the majority of the unconjugated bile acids exist in the protonated or neutral form, and a very small amount of glycine-conjugated or taurine-conjugated bile acids is protonated [11]. Thus, the unconjugated bile acids are absorbed by passive diffusion; however, it is less intense than the active absorption [3, 12]. The majority of bile acids in the ileum are conjugated and exist in anionic form. Hence, the entry and exit of conjugated bile acids in the intestine and hepatocytes require an active transport system as they cannot cross the plasma membrane [5, 7]. Bile acid transporters play an important role in modulating bile acid signaling by controlling the flux of bile acids [9, 18].
The conjugated bile acids are actively absorbed in the terminal ileum via apical sodium-dependent bile acid transporter (ASBT) [19]. The ileal bile acid-binding protein (IBABP) facilitates the intracellular transport of bile acids across the enterocyte, and the bile acids are effluxed to the portal blood via organic solute transporter α and β (OSTα/OSTβ) [20]. Other transporters such as MRP2, MRP4 and MRP3 are known to be involved in the transport of bile acids at the apical membrane and basolateral membrane of the enterocytes, respectively [21].
The circulating bile acids in the portal blood are taken up by the hepatocytes through the basolateral membrane by the Na+-dependent cotransporter system, NTCP [19, 22]. It is estimated that 90 % of the bile acid uptake is via Na+-taurocholate cotransporting polypeptide (NTCP) and to a lesser extent by organic anion transporting polypeptides (OATP1 and OATP4). At the canalicular membrane of the hepatocytes, the BSEP (bile salt export pump) plays a major role in the secretion of bile acids into the bile canaliculi, whereas MRP2 (multidrug resistance associated protein-2) mediates secretion of bile acids along with other organic solutes [21, 22]. The heterodimeric organic solute transporters (OSTα/OSTβ), MRP3 and MRP4 mediate secretion of bile acids into the circulation at the basolateral membrane of the hepatocytes [19, 20, 23].
Bile acid receptors
The importance of bile acids as signaling molecules in the regulation of physiological function has gained attention with the discovery of receptors for bile acids. The most prominent and best-studied bile acid receptors are the farnesoid X receptor (FXR) and Takeda G-protein-coupled receptor clone 5 (TGR5) (Fig. 1).
Fig. 1.
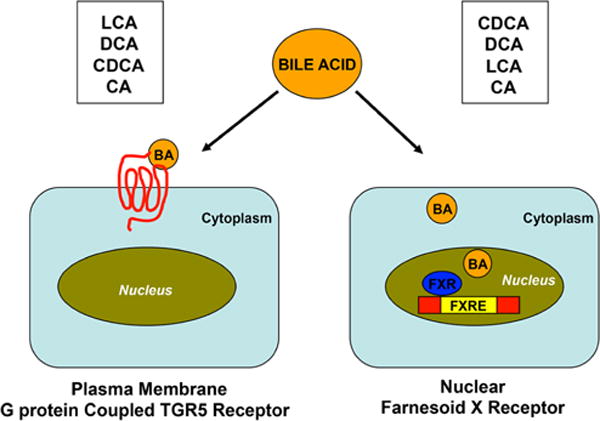
Schematic representation of the activation of TGR5 and FXR by bile acids. TGR5 is a plasma membrane G protein coupled receptor and FXR is a nuclear receptor that is activated by bile acids. The receptors are activated by both primary (CA and CDCA) and secondary bile acids (LCA and DCA). LCA is a potent activator of TGR5 while CDCA for FXR
FXR
In 1999 three independent research groups reported that bile acids act as ligands for the FXR [24–26]. The discovery of FXR was later followed by human steroid and xenobiotic receptor (SXR) and its rodent homolog pregnane X receptor (PXR), vitamin D receptor (VDR) and constitutive androstane receptor (CAR), all of which are bile acid-activated nuclear receptors, which are known to be functionally related [27–29].
Farnesoid X receptor (FXR) or NR1H4, a member of the nuclear receptor superfamily, was identified during the search for a new heterodimer that binds to the retinoid X receptor (RXR). FXR is highly expressed in liver, intestine, kidneys and adrenal glands and is selectively activated by CDCA (Table 1) [24–26]. FXR was identified as a master regulator playing a pivotal role in the feedback regulation of bile acid synthesis by repressing CYP7A1, the rate-limiting enzyme of the synthetic pathway [7, 9, 17]. FXR activates the expression of the bile acid export transporters (MRP2 and BSEP) and simultaneously represses bile acid import to regulate bile acid transport [30, 31].
Table 1.
Bile acid receptors and their respective expression and function
| Bile acid receptor | Cell/tissue expression | Function/effects |
|---|---|---|
| FXR | Liver | Downregulates triglyceride, fatty acid and cholesterol biosynthesis |
| Increases fatty acid oxidation | ||
| Increases lipolysis | ||
| Decreases gluconeogenesis | ||
| Promotes glycogen synthesis | ||
| Exerts antiinflammatory and antifibrotic effects | ||
| Promotes liver regeneration | ||
| Pancreas | Stimulates insulin secretion | |
| Intestine | Stimulates bile acid reabsorption | |
| Stimulates FGF19 production | ||
| Exerts antiinflammatory and antifibrotic effects | ||
| Skeletal muscle and adipocytes | Increases glucose uptake and insulin sensitivity | |
| Increases adipogenesis and lipid storage | ||
| Kidney | Exerts antiinflammatory and antifibrotic effects | |
| Vascular smooth muscle cells | Exerts antiatherogenic effects | |
| Vascular endothelial cells | Exerts antihypertension effects | |
| Macrophages | Exerts antiinflammatory effects | |
| TGR5 | Liver sinusoidal endothelial cells | Vasodilation |
| Cholangiocytes | Exerts antiapoptotic and antiinflammatory effects | |
| Macrophages | Exerts antiinflammatory effects | |
| Gallbladder | Gallbladder filling | |
| Pancreas | Stimulates insulin secretion | |
| Skeletal muscle and brown adipose tissue | Increases energy expenditure | |
| Intestinal L cells | Stimulates GLP-1 secretion | |
| Enteric neurons | Promotes motility or peristalsis | |
| Primary spinal afferent and sensory neurons | Induces neuronal hyper excitability and release of GRP (gastrin-releasing peptide) |
Thus, FXR functions as a sensor of metabolic signals and plays an important role in the regulation of bile acids synthesis and transport, lipid metabolism and glucose homeostasis (Fig. 2) [32–34].
Fig. 2.
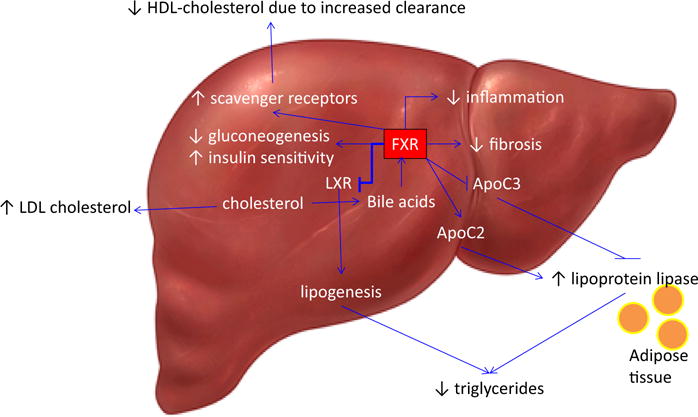
The Role of FXR on Lipid Metabolism and glucose homeostasis. FXR is activated by bile acids and has several downstream effects including inhibition of lipogenesis, decreased gluconeogenesis, increased insulin sensitivity and with recent data, a significant decrease in inflammation and fibrosis. FXR has the effect of increasing lipoprotein lipase by positively stimulating its activator, Apolipoprotein C2 (ApoC2), and repressing the inhibitor of lipoprotein lipase, Apolipoprotein C3 (ApoC3). The FLINT trial has shown an increase in Low-Density Lipoprotein (LDL) and a decrease in High-Density Lipoprotein (HDL)
TGR5
Takeda G-protein coupled receptor clone 5 (TGR5), also known as GPBAR1, M-BAR or BG37, was first discovered in 2002 by two independent research groups in Japan [35, 36]. TGR5 is encoded by a single exon gene that maps to chromosome position 1c3 in mice and to chromosome 2q35 in humans. The coding region of the human TGR5 gene consisting of 993 bp encodes 330 amino acids and the protein shares 82, 83, 86 and 90 % homology with the rat, mouse, bovine and rabbit, respectively [37]. TGR5 is a member of the rhodopsin-like (class A) subfamily of GPCRs and is ubiquitously expressed with varied degrees of expression in different organs such as the spleen, placenta, gall bladder, liver, kidney, small intestine, colon, heart, skeletal muscles and pancreas, etc. (Table 1) [38]. The bile acid receptor TGR5 is activated by different bile acids, with taurine-conjugated lithocholic acid (TLCA) being the most potent endogenous agonist with an EC50 value of 0.33 μM. The other bile acids that activate TGR5 include LCA, DCA, CDCA and CA with EC50s of 0.53, 1.01, 4.43 and 7.72 μM, respectively [39, 40]. In addition to their cognate receptors, bile acids are also known to modulate the activity of formyl-peptide receptors (FPR), muscarinic receptors and S1PR2 (sphingosine-1 phosphate receptor 2) [41, 42].
Steroids such as progesterone, androgen, estrogens, oxysterols and pregnenolone also stimulate TGR5. However, the concentrations of these steroids required to activate TGR5 are much higher than physiologically relevant concentrations and hence they are not considered true in vivo agonists of TGR5 [35]. This may be an artifact secondary to bile acids and steroids sharing cholesterol as a precursor molecule.
Functions of FXR that can be leveraged for therapeutics
FXR has numerous targets and through recent investigations has broadened the once limited role of bile acids on physiologic processes. Bile acids, via hepatic FXR activation, facilitate decreased triglyceride synthesis and secretion and are an important regulator of lipid metabolism (Fig. 2) [31, 32]. Additionally, the role of FXR in glucose homeostasis via the regulation of gluconeogenesis and glycogenolysis has been demonstrated [43]. The activation of FXR decreases gluconeogenesis and increases glycogen synthesis, the combined effect of which reduces hyperglycemia. FXR also plays an important role in skeletal muscle and adipose tissue by increasing insulin sensitivity and is thus a potential therapeutic target for treating metabolic syndrome [31–34] and its liver manifestation, nonalcoholic fatty liver disease. Cardiovascular causes of death are the leading cause of death in those with nonalcoholic liver disease, and animal studies using FXR ligands have demonstrated a reduction of atherosclerosis with treatment [44]. Hence, this may be an attractive target to reduce mortality secondary to cardiovascular disease. The dual FXR/TGR5 agonist, INT-767, was also found to reduce atherosclerotic plaque formation and aortic inflammation via NF-KB inactivation in mouse models [45]. However, recent human trials indicate an increase in LDL cholesterol by 8–10 mg/dl in those receiving an FXR agonist [46]. On the other hand, it is now increasingly appreciated that LDL cholesterol alone is neither a very sensitive nor specific parameter to assess cardiovascular risk. Thus, it is yet unknown if these animal data will be translatable to humans.
Obeticholic acid, also known as INT-747 and 6α-ethyl-chenodeoxycholic acid (6-ECDCA), is a potent agonist for FXR. It is a derivative of CDCA, the natural agonist for FXR. Recent studies in rat cirrhosis models demonstrate obeticholic acid’s ability to improve portal pressure via decreasing intrahepatic vascular resistance without the complication of systemic hypotension [47]. Thus, FXR agonists have a potential role in the treatment of portal hypertension and its clinical consequences of variceal bleeding, ascites and hepatic encephalopathy. Furthermore, rodent liver fibrosis models (porcine serum and bile duct ligation) have demonstrated the antifibrotic effects of 6-ECDCA on the liver, while the endogenous ligand of FXR, CDCA and ursodeoxycholic acid, an epimer of CDCA with no meaningful FXR activity, provided no protection from fibrosis [48]. Hence, there is a potential role for fibrosis prevention and even reversal with potent FXR agoinsts.
There is a human trial using FXR agonists in the setting of nonalcoholic fatty liver disease performed by the NIDDK NASH Clinical Research Network that is in phase 2b, the Farnesoid X Receptor Ligand Obeticholic Acid in Nonalcoholic Steatohepatitis Treatment (FLINT). The protocol of the study stipulated an interim analysis of histology and safety parameters after half of the patients had finished treatment and had an end-of-treatment liver biopsy. Due to the interim analysis, the study was terminated early as it was determined that the primary endpoint of the decrease in histological disease severity was met with treatment with obeticholic acid (OCA). OCA improved steatosis, inflammation and histologic ballooning [49]. Of note, there was a significant improvement in fibrosis, which has not been definitively demonstrated with other treatments for NASH in a single study. However, given the increase in LDL cholesterol, larger long-term trials are now needed to establish the therapeutic utility of OCA for NASH [46].
TGR5 agonists
Bile acids play an important role in glucose homeostasis. Studies have demonstrated that bile acids via activation of TGR5 induce glucagon-like peptide-1 (GLP-1) secretion from the intestinal L cells [50]. INT-777, a specific TGR5 agonist, stimulates GLP-1 secretion in mouse enteroendocrine cells, STC-1, as well as in human intestinal NCI-H716 cells [51]. GLP-1 is known to regulate postprandial glycemic response by stimulating glucose-dependent insulin secretion from the pancreatic β cells and inhibition of glucagon secretion from pancreatic α cells [52]. TGR5, in addition to inducing insulin secretion, also increases insulin sensitivity. Insulin induced by TGR5 increases glucose uptake in the skeletal muscle as well as adipose tissue [53, 54]. The possible mechanisms involve an increase in glucose transporter 4 (GLUT-4) expression, an insulin-regulated glucose transporter, leading to the uptake of glucose (postprandial glycemic control) and its oxidative phosphorylation [51]. Thus, the involvement of TGR5 in glucose homeostasis was investigated in vivo using both gain of function and loss of function genetic approaches and was further elucidated by treatment with a selective TGR5 ligand and/or INT-777, a specific TGR5 ligand [51]. These studies were performed in mice, and there are currently no human trial data using pure TGR5 agonists.
Conclusion
Bile acids and their derivatives have emerged as potential therapeutics for the management of many liver diseases, especially nonalcoholic fatty liver disease. The targets of these bile acids and their derivatives are beyond the previous scope of their traditional mechanisms of cholesterol homeostasis and lipid digestion. Bile acids also act as signaling molecules via nuclear and plasma membrane receptors and have effects on atherosclerosis, insulin sensitization, glucose utilization and relieving portal hypertension. With human trials afoot, we will soon be able to understand the dynamic impact of bile acid receptor targets on the physiologic, biochemical and histologic aspects of human liver diseases.
Acknowledgments
This work is funded by NIH grants T32 07150-37 (AJS and AA). Dr. Sanyal is a consultant to Abbvie, Bristol Myers-Squibb, Genfit, Exhalenz, Islet Pharmaceuticals, Nimbus, Nitto-Denko and Tobira. His institution receives research funding from Gilead, Galectin, Novartis, Salix, Cumberland and Intercept Pharmaceuticals. He has also served as a non-remunerated consultant to Immuron and Echosens.
Footnotes
Conflict of interest Dr. Amon Asgharpour declares that he has no conflict of interest. Dr. Divya Kumar declares that she has no conflict of interest.
References
- 1.Dawson PA, Shneider BL, Hofmann AF. Physiology of the gastrointestinal tract. 4th. San Diego: Elsevier; 2006. Bile formation and the enterohepatic circulation; pp. 1437–1462. [Google Scholar]
- 2.Agellon LB. Biochemistry of lipids, lipoproteins and membranes. 5th. Amsterdam: Elsevier; 2008. Metabolism and function of bile acids; pp. 423–440. [Google Scholar]
- 3.Schiff ER, Dietschy JM. Current concepts of bile acid absorption. Am J Clin Nutr. 1969;22:273–278. doi: 10.1093/ajcn/22.3.273. [DOI] [PubMed] [Google Scholar]
- 4.Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J. 2006;25:1419–1425. doi: 10.1038/sj.emboj.7601049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofmann AF. The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci. 2009;14:2584–2598. doi: 10.2741/3399. [DOI] [PubMed] [Google Scholar]
- 6.Keitel V, Kubitz R, Haussinger D. Endocrine and paracrine role of bile acids. World J Gastroenterol. 2008;14:5620–5629. doi: 10.3748/wjg.14.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 9.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–983. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 11.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Dietschy JM. Mechanisms for the intestinal absorption of bile acids. J Lipid Res. 1968;9:297–309. [PubMed] [Google Scholar]
- 13.Hofmann AF, Mysels KJ. Bile acid solubility and precipitation in vitro and in vivo: the role of conjugation, pH, and Ca2+ ions. J Lipid Res. 1992;33:617–626. [PubMed] [Google Scholar]
- 14.Chiang JY. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr Rev. 2002;23:443–463. doi: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 15.Li T, Chiang JY. Bile Acid signaling in liver metabolism and diseases. J Lipids. 2012;2012:754067. doi: 10.1155/2012/754067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 17.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann AF. Biliary secretion and excretion in health and disease: current concepts. Ann Hepatol. 2007;6:15–27. [PubMed] [Google Scholar]
- 19.Weinman SA, Jalil S. Textbook of gastroenterology. Oxford: Blackwell; 2008. Bile secretion and cholestasis; pp. 401–421. [Google Scholar]
- 20.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50:2340–2357. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.St-pierre MV, Kullak-ublick GA, Hagenbuch B, Meier PJ. Transport of bile acids in hepatic and non-hepatic tissues. J Exp Biol. 2001;204:1673–1686. doi: 10.1242/jeb.204.10.1673. [DOI] [PubMed] [Google Scholar]
- 22.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24:1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang DQH, Neuschwander-Tetri BA, Portincasa P. The biliary system. In Colloquium series on integrated systems physiology: from molecule to function. Morgan & Claypool. 2012:109–145. [Google Scholar]
- 24.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 25.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;21:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 27.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, Mackenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protect against liver toxicity. Proc Natl Acad Sci USA. 2000;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA. 2001;98:3370–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 30.Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368:17–29. doi: 10.1016/j.mce.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. FXR, a multipurpose nuclear receptor. Trends Biochem Sci. 2006;31:572–580. doi: 10.1016/j.tibs.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Claudel T, Staels B, Kuipers F. The farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol. 2005;25:2020–2030. doi: 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- 33.Teodoro JS, Rolo AP, Palmeira CM. Hepatic FXR: key regulator of whole-body energy metabolism. Trends Endocrinol Metab. 2011;22:458–466. doi: 10.1016/j.tem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Fiorucci S, Cipriani S, Baldelli F, Mencarelli A. Bile acid-activated receptors in the treatment of dyslipidemia and related disorders. Prog Lipid Res. 2010;49:171–185. doi: 10.1016/j.plipres.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 36.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 37.Tiwari A, Maiti P. TGR5: an emerging bile acid G-protein-coupled receptor target for the potential treatment of metabolic disorders. Drug Discov Today. 2009;14:523–530. doi: 10.1016/j.drudis.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis. 2014;46:302–312. doi: 10.1016/j.dld.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieu T, Jayaweera G, Bunnett NW. GPBA: a GPCR for bile acids and an emerging therapeutic target for disorders of digestion and sensation. Br J Pharmacol. 2014;171:1156–1166. doi: 10.1111/bph.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bunnett NW. Neuro-humoral signaling by bile acids and the TGR5 receptor in the gastrointestinal tract. J Physiol. 2014;592:2943–2950. doi: 10.1113/jphysiol.2014.271155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferrari C, Macchiarulo A, Costantino G, Pellicciari R. Pharmacophore model for bile acids recognition by the FPR receptor. J Comput Aided Mol Des. 2006;20:295–303. doi: 10.1007/s10822-006-9055-1. [DOI] [PubMed] [Google Scholar]
- 42.Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62–68. doi: 10.1016/j.steroids.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1119. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hambruch, et al. J Pharmacol Exp Ther. 2012;343(3):556–567. doi: 10.1124/jpet.112.196519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyazaki-Anzai S, Masuda M, Levi M, Keenan AL, Miyazaki M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS One. 2014;9(9):e108270. doi: 10.1371/journal.pone.0108270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, nonalcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2014;(14):S0140–6736. 61933–61934. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verbeke L, Farre R, Trebicka J, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59:2286–2298. doi: 10.1002/hep.26939. [DOI] [PubMed] [Google Scholar]
- 48.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127(5):1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 49.NIDDKwebsite:http://www.niddk.nih.gov/news/for-reporters/Pages/interim-results-FLINT-clinical-trial.aspx
- 50.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 51.Thomas C, Gioliello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doyle ME, Egan JM. Mechanisms of action of GLP-1 in the pancreas. Pharmacol Ther. 2008;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5-connecting nutrition and metabolism. Thyroid. 2008;18:167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Lou G, Meng Z, Huang W. TGR5: a novel target for weight maintenance and glucose metabolism. Exp Diabetes Res. 2011:853501–803506. doi: 10.1155/2011/853501. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]