

Abstract
Oxidative stress and enhanced lipid peroxidation are linked to many chronic inflammatory diseases, including age-related macular degeneration (AMD). AMD is the leading cause of blindness in Western societies, but its aetiology remains largely unknown. Malondialdehyde (MDA) is a common lipid peroxidation product that accumulates in many pathophysiological processes, including AMD. Here we identify complement factor H (CFH) as a major MDA-binding protein that can block both the uptake of MDA-modified proteins by macrophages and MDA-induced proinflammatory effects in vivo in mice. The CFH polymorphism H402, which is strongly associated with AMD, markedly reduces the ability of CFH to bind MDA, indicating a causal link to disease aetiology. Our findings provide important mechanistic insights into innate immune responses to oxidative stress, which may be exploited in the prevention of and therapy for AMD and other chronic inflammatory diseases.
Increased oxidative stress has been implicated in the pathogenesis of many different diseases1. As a consequence of oxidative stress, proteins, lipids and DNA can be damaged, often resulting in structural changes. For example, when membrane phospholipids undergo lipid peroxidation, MDA and other reactive decomposition products are generated2. These can in turn modify endogenous molecules, generating novel oxidation-specific epitopes (OSEs), which are also present on the surface of apoptotic cells and blebs released from them3. Many of these OSEs are recognized as danger signals by innate immune receptors4. Elucidating the molecular mechanisms by which oxidative damage challenges the immune system would pave the road for new diagnostic and therapeutic approaches in several pathologies.
MDA and its condensation products are reliable markers for oxidative stress and have been associated with many disorders, including atherosclerosis1,4 and AMD, a degenerative disease affecting the retina that leads to irreversible vision loss5,6. AMD is the most common cause of blindness in the elderly in Western societies7. A hallmark of developing AMD is the accumulation of extracellular deposits, termed drusen, which have been shown to contain MDA8. MDA-modified proteins are known to induce inflammatory responses and are recognized by innate immunity9–11. We recently demonstrated that OSEs in general are a major target of innate natural antibodies both in mice and humans and that ~15% of all immunoglobulin M (IgM) natural antibodies bound MDA-type adducts, suggesting a great need to defend against this specific modification12. However, the abundance of MDA and the danger associated with it suggests that additional, evolutionary conserved innate defence mechanisms exist.
CFH binds MDA modifications
We used an unbiased proteomic approach to identify plasma proteins binding to MDA modifications. Because normal plasma contains high titres of MDA-specific natural antibodies12, we purified MDA-binding proteins from plasma of atherosclerotic Rag−/−Ldlr−/− mice that lack immunoglobulins. Pooled plasma was incubated with beads coupled to either malondialdehyde-acetaldehyde (MAA)-modified or unmodified polylysine, respectively. MAA is an advanced MDA-lysine adduct the structure of which is shown in Supplementary Fig. 1 (see also ref. 13). Bound proteins were eluted and identified by mass spectrometry. As many as 45 unique peptides were found exclusively in MAA-polylysine pull-downs, of which >55% could be attributed to CFH (Supplementary Fig. 2 and Supplementary Table 1). CFH is a major regulator of the complement system and protects host tissues from complement-mediated damage14. Immunoblot analysis revealed the presence of CFH on MAA-coated beads but not on control beads (Fig. 1a). This finding was confirmed using human plasma (Fig. 1b). Interestingly, the anti-CFH antibody also detected lower molecular weight bands, which may represent CFH-related proteins (CFHRs) that share high sequence homology with CFH.
Figure 1. CFH specifically binds to MDA modifications.
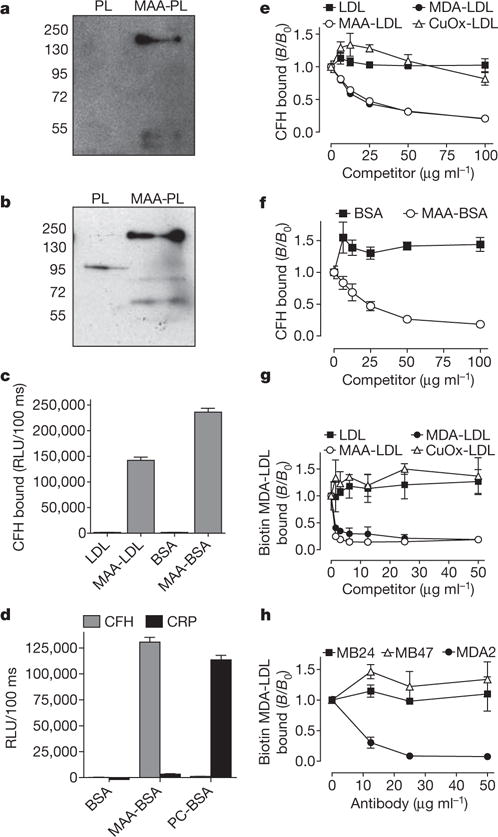
a, b, Immunoblot for CFH (molecular weight: 150 kDa) using eluates from either polylysine (PL) or MAA-polylysine (MAA-PL) beads incubated with Ldlr−/−Rag−/− mouse plasma (a) or human plasma (b). c, ELISA for binding of purified CFH (5 μg ml−1) to coated native LDL, MAA-LDL, BSA and MAA-BSA. Values are mean ± s.d. relative light units (RLU) per 100 ms of triplicate determinations. d, ELISA for binding of purified CFH or CRP (5 μg ml−1) to coated BSA, MAA-BSA and PC-BSA. Values are mean ± s.d. RLU per 100 ms of triplicate determinations. e–h, Competition immunoassays. e, f, Binding of purified CFH (e) or binding of plasma CFH (f) to coated MAA-BSA in the presence of increasing concentrations of LDL, MDA-LDL, MAA-LDL and Cu2+-oxidized (CuOx)-LDL, or BSA and MAA-BSA. g, Binding of biotinylated MDA-LDL to coated CFH in the presence of increasing concentrations of LDL, MDA-LDL, MAA-LDL and CuOx-LDL. h, Binding of biotinylated MDA-LDL to coated CFH in the presence of increasing concentrations of monoclonal antibodies specific for ApoB100 (MB24 and MB47) or MDA (MDA2). Data are expressed as a ratio of binding in the presence of competitor divided by the binding in the absence of competitor (B/B0) and represent the mean ± s.d. of triplicate determinations. As an estimate for the affinity, the dissociation constants Kd were calculated as 6.4 × 10−8 mol l−1 for the binding of CFH to coated MAA-BSA and 1.6 × 10−9 mol l−1 for the binding of MAA-BSA to coated CFH.
Using enzyme-linked immunosorbent assay (ELISA), we demonstrated that CFH bound to MDA directly and independently of the protein carrying the adducts. Purified CFH bound in a calcium-independent manner to both MAA-modified low density lipoprotein (MAA-LDL) and MAA-modified bovine serum albumin (MAA-BSA), but not to unmodified proteins (Fig. 1c and Supplementary Fig. 3). Moreover, we tested binding of CFH to the oxidation-specific modifications phosphocholine-BSA (PC-BSA), which is bound by C reactive protein (CRP), as well as carboxyethylpyrrole-BSA (CEP-BSA) and 4-hydroxynonenal-BSA (4-HNE-BSA). None of these modifications were bound by CFH (Fig. 1d and Supplementary Fig. 4A, B). CRP and C3 were also detected in the MAA-polylysine pull-downs (Supplementary Table 1), but neither of them bound to coated MAA-BSA (Fig. 1d and Supplementary Fig. 4C).
To characterize the specificity of the binding of CFH to MDA, we performed competition assays. Only MDA- and MAA-modified LDL competed in a concentration-dependent manner for the binding of CFH to coated MAA-BSA. Neither native LDL nor the negatively charged Cu2+-oxidized LDL showed inhibition, thereby excluding non-specific interactions mediated by charge effects (Fig. 1e). To demonstrate a dose-dependent interaction in plasma, the binding of CFH to coated MAA-BSA was tested in different plasma dilutions (Supplementary Fig. 5). Consistent with the notion that CFH is a major MDA-binding protein in plasma, binding of CFH to coated MAA-BSA was competed by soluble MAA-BSA with similar efficiency in whole plasma (Fig. 1f). In a reciprocal experiment, binding of biotinylated MDA-LDL to immobilized purified CFH was fully competed by either MDA- or MAA-modified LDL, even at very low competitor concentrations (Fig. 1g). In the same assay, the MDA-lysine-specific monoclonal antibody MDA2 fully inhibited binding of MDA-LDL to CFH. In contrast, the apoB-100-specific monoclonal antibodies MB47 and MB24, which bind apoB-100 of MDA-LDL (Supplementary Fig. 6), did not inhibit this interaction (Fig. 1h). Using surface plasmon resonance, we observed a concentration-dependent binding of CFH to coated MAA-BSA (Supplementary Fig. 7). Taken together, these findings prove that CFH binds specifically to MDA modifications.
The CFH H402 variant has impaired MDA binding
To map the binding site for MDA on CFH, we performed binding studies using recombinantly expressed CFH fragments (Fig. 2a). CFH is composed of 20 globular short consensus repeats (SCRs)15. Only fragments containing either SCR7 or SCR20 bound to coated MDA (Fig. 2a). Reciprocally, soluble MDA-LDL only bound to immobilized fragments containing either SCR7 or SCR20, respectively (Supplementary Fig. 8). Importantly, these domains have also been identified as clustering points of various disease-related mutations15.
Figure 2. The SCR7 domain of CFH is critical for MDA binding.
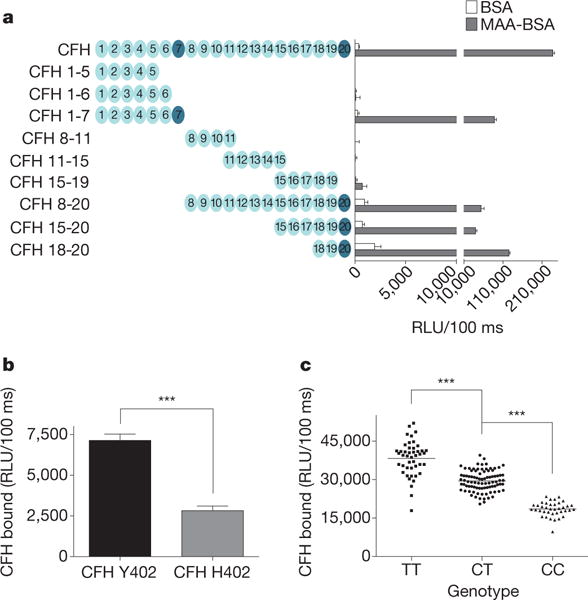
a, ELISA for binding of CFH and recombinantly expressed CFH fragments to coated BSA (white bars) or MAA-BSA (black bars). The length of CFH fragments is indicated by schematic representations with each circle depicting one SCR. Values are mean ± s.d. RLU per 100 ms of triplicate determinations. b, ELISA for binding of purified CFH variant Y402 and CFH variant H402 (both at 1 μg ml−1) to coated MAA-BSA. Values are mean ± s.d. RLU per 100 ms of triplicate determinations. c, ELISA for binding of plasma CFH to coated MDA-LDL in plasma of subjects homozygous for the H402 risk allele (CC, n = 38), heterozygous for the H402 risk allele (CT, n = 88) or homozygous for the Y402 allele (TT, n = 45). The association of rs1061170 with CFH binding to MDA was calculated with P = 1.29−40 using an additive model. Symbols represent individual subject samples with horizontal bars indicating the mean of each group. Values are mean ± s.d. RLU per 100 ms of triplicate determinations (***P < 0.001).
One of the most widely studied single nucleotide polymorphisms (SNPs) in CFH is the prevalent rs1061170 SNP, which causes an amino acid switch on position 402 (YRH) in SCR7. To determine the effect of the H402 substitution, we purified CFH from plasma of homozygous individuals expressing either CFH Y402 or CFH H402, respectively, and tested the binding to MDA. Compared to the common Y402 variant, the CFH variant H402 exhibited significantly impaired binding to MAA-BSA (Fig. 2b). The H402 variant has been associated with a significant risk for the development of AMD16–19. Therefore, we analysed the binding of CFH to coated MDA-LDL in plasma samples of AMD patients with the respective genotypes. Compared to the extent of CFH binding to MDA-LDL using plasma of individuals homozygous for the protective allele, binding in plasma of heterozygous subjects was reduced by 23% (P < 0.001), and by 52% (P < 0.001) in plasma of subjects homozygous for the H402 risk allele (Fig. 2c), irrespective of the total plasma CFH levels (Supplementary Fig. 9A). Moreover, plasma levels of MDA-specific IgG and IgM antibodies were similar in all groups (Supplementary Fig. 9B, C).
The genetic deletion of CFHR1 and CFHR3 has been reported to protect from AMD and could influence CFH binding to MDA20. Less than 25% of individuals in this study carried deletions at these loci and their removal from our analysis did not alter the significance of the association of rs1061170 with MDA binding (Supplementary Fig. 9D). Taken together, the impaired ability of the risk variant to bind MDA suggested an important role for this interaction in AMD pathogenesis.
CFH binds cellular debris via MDA epitopes
Owing to constant light exposure, the retina provides an environment that facilitates lipid peroxidation7. We detected MDA epitopes by immunohistochemistry in the eyes of subjects with and without AMD. MDA epitopes were detectable throughout the choroid and Bruch’s membrane (Fig. 3a, d). In eyes without AMD, labelling for MDA was stronger in the outer than inner Bruch’s membrane (Fig. 3a). In eyes with AMD, MDA staining was seen diffusely throughout Bruch’s membrane (Fig. 3d). Staining for CFH followed a similar pattern (Fig. 3b, e). In addition, strong CFH labelling was seen in the retinal pigment epithelium (RPE) and choriocapillaris basement membranes. Moreover, the presence of C3d, a cleavage product of iC3b, indicated co-factor activity at the same sites (Fig. 3c, f). We further demonstrated by confocal microscopy the presence of MDA epitopes on the surface of in vitro-generated necrotic RPE cells, a major cell type affected in AMD. Moreover, CFH co-localized with MDA epitopes, suggesting that MDA mediates recognition of dying cells by CFH (Fig. 3j). To demonstrate this directly, we used flow cytometry to assess the binding of CFH to apoptotic blebs from Jurkat T cells in the presence of MAA-BSA as competitor. Consistent with the presence of MDA epitopes on only a subgroup of apoptotic blebs, we found that CFH bound between 5–45% of apoptotic blebs (Supplementary Fig. 10). Importantly, MAA-BSA competed for this binding by more than 60%, whereas unmodified BSA did not (Fig. 3k). Thus, MDA adducts present in several retinal compartments and on the surface of necrotic RPE cells represent in vivo ligands for CFH. In addition, we predicted that CFH might also bind to MDA adducts in other tissues and confirmed this in atherosclerotic lesions (Supplementary Fig. 11).
Figure 3. CFH binds to MDA epitopes present in AMD lesions, on necrotic cells and apoptotic blebs.
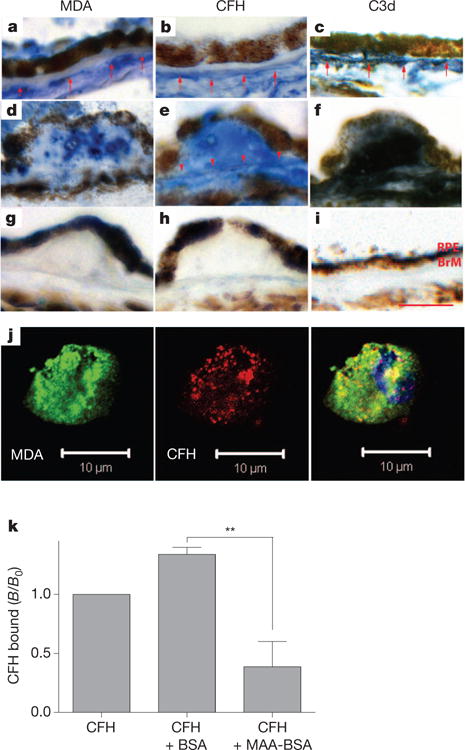
a–i, Immunohistochemistry of MDA (left), CFH (middle) and C3d (right) localization in human maculas. a–c, Eye of a 72-year-old subject heterozygous for the H402 SNP without AMD. d–f, Eye of a 93-year-old subject homozygous for the H402 SNP with AMD. g–i, IgG control immunostains. Red arrows indicate positive labelling of choriocapillaris basement membrane and arrowheads indicate labelling of Bruch’s membrane (BrM). Scale bar, 25 μm. Sections are representative for 7 donors (5 AMD, 2 controls). j, Confocal immunofluorescent photograph of necrotic RPE cells stained with the MDA-specific IgM natural antibody EO14 (green) and CFH (red), respectively. The right panel shows a merged picture indicating co-localization of CFH binding with the presence of MDA epitopes (yellow). k, Competition assay for the binding of CFH to apoptotic blebs from Jurkat T cells either alone or in the presence of BSA or MAA-BSA assessed by flow cytometry. Values are expressed as mean ± s.e.m. CFH binding (B/B0) based on mean fluorescence intensities of four independent experiments (**P < 0.01).
CFH inactivates complement on MDA-bearing surfaces
An important regulatory activity of CFH lies with its capacity to act as co-factor for serine protease factor I, thereby promoting the degradation of C3b into inactive iC3b fragments. Deposition of iC3b on apoptotic cells increases their clearance in an anti-inflammatory manner21,22. We therefore tested whether CFH induces iC3b generation when bound to MDA. Indeed, CFH promoted the formation of iC3b in a dose- and time-dependent manner when bound to coated MAA-BSA (Supplementary Fig. 12). When comparing the co-factor activity of the 402 variants on MDA-decorated surfaces, we discovered a strong functional difference in that impaired MDA-binding of the risk variant resulted in severely reduced factor-I-mediated C3 cleavage (Fig. 4a). This activity of CFH may represent an important protective mechanism in conditions in which MDA is continuously generated, for example, on the surface of dying cells.
Figure 4. CFH inactivates complement on MDA-bearing surfaces.
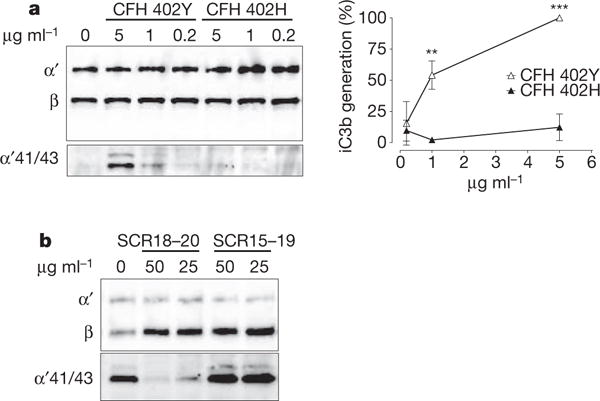
a, Immunoblot of C3b cleavage products induced by three concentrations of CFH variants bound to coated MAA-BSA. α41/43 indicates iC3b cleavage products of the C3b α-chain. β indicates the C3b β-chain that remains uncleaved and served as a loading control. α41/43 was densitometrically quantified, and data are presented as percentage of iC3b generation achieved with 5 μg ml−1 CFH Y402 (= 100%). Shown is the mean ± s.e.m. of three independent experiments. b, Immunoblot of C3b degradation products induced by CFH bound to coated MAA-BSA in the presence of either CFH 18–20 or CFH 15–19.
Importantly, other members of the CFH family such as CFHR1/CFHR3 show homology with the carboxy terminus of CFH and therefore contain a potential MDA-binding site without possessing co-factor activity. Deletions of CFHR1/3 have been reported to be protective in AMD20, suggesting a negative role of these proteins in this pathology. To demonstrate the potential capacity of MDA-binding CFHR to inhibit the beneficial co-factor activity of CFH, we tested whether C-terminal CFH fragments could compete for the co-factor activity by binding to MDA. Indeed, the MDA-binding fragment SCR18–20 prevented CFH from inducing iC3b generation, whereas a non-binding fragment containing SCR15–19 had no effect (Fig. 4b). These data point towards a complex regulation of complement activation on MDA-decorated surfaces.
CFH neutralizes proinflammatory effects of MDA
The inflammatory process in AMD lesions has been suggested to be propagated by the secretion of cytokines including IL-8 (ref. 23). Stimulation of RPE cells (ARPE-19) with MAA-BSA induced the expression of IL-8 and caused an antioxidant response as indicated by upregulation of NAD(P)H dehydrogenase and hemoxygenase-1 (Fig. 5a). We then tested the effect of CFH on MAA-LDL binding to macrophages, another cell type involved in AMD pathogenesis24. In a cell-based ELISA, CFH inhibited binding of MAA-LDL to thioglycollate-elicited macrophages in a dose-dependent manner (Fig. 5b). This indicates that CFH binds the same epitope on MAA-LDL that is necessary for its recognition by macrophages. Similar to ARPE-19 cells, monocytic THP-1 cells exhibited a robust expression of IL-8 following MAA-BSA stimulation. In addition, MAA-BSA induced the expression of TNF-α and IL-1β, but not IL-12β (Supplementary Fig. 13). Importantly, MAA-BSA-induced IL-8 secretion was inhibited by physiological concentrations of CFH in a dose-dependent manner (Fig. 5c). In contrast, CFH had no effect on IL-8 production induced by phorbol myristate acetate (Supplementary Fig. 14).
Figure 5. CFH neutralizes proinflammatory effects of MDA.
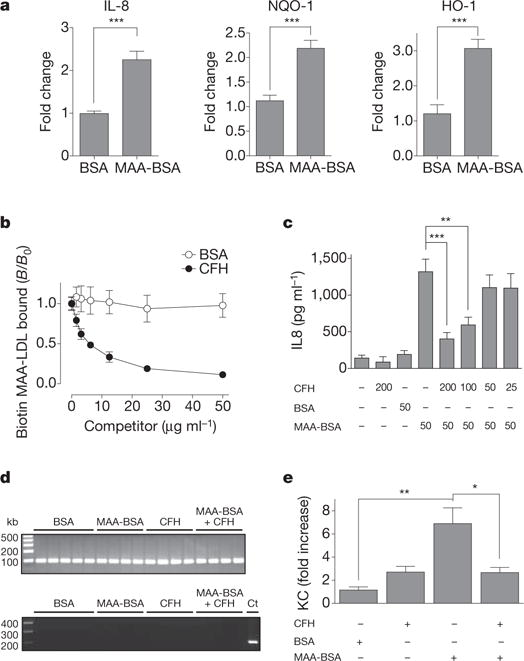
a, Expression of indicated genes in ARPE-19 cells stimulated for 24 h with 50 μg ml−1 BSA compared to MAA-BSA as determined by quantitative RT–PCR. Data represent the mean ± s.e.m. of three independent experiments. b, Cell-based ELISA for the binding of biotinylated MAA-LDL to thioglycollate-elicited macrophages in the presence of BSA or CFH. Data are expressed as B/B0 and represent mean ± s.d. of triplicate determinations. c, Secretion of IL-8 by THP-1 cells stimulated for 12 h with BSA or MAA-BSA in the absence or presence of CFH. Numbers below indicate concentrations of CFH, BSA and MAA-BSA in μg ml−1. Error bars represent mean ± s.e.m. of three independent experiments. d, e, Intravitreal injection of BSA, MAA-BSA and/or CFH in mice (n = 4–5 per group). Six hours after injection, RPE/choroid was isolated. d, RT–PCR for Rpe65 and Rho in RPE/choroid fractions. cDNA isolated from neurosensory retina was used as a control (Ct). e, Expression of KC in RPE/choroid as assessed by quantitative RT–PCR. Error bars represent mean ± s.e.m. expression normalized to the BSA-injected group (*P < 0.05, **P < 0.01, ***P < 0.001).
To evaluate the importance of this interaction in vivo, we examined the effect of MAA adducts in a murine model. First we validated that MAA-BSA could induce secretion of KC, the mouse orthologue to human IL-8, in murine macrophages (Supplementary Fig. 15). To test the proinflammatory effect of MAA-BSA as well as the scavenging capacity of CFH in an AMD-relevant site, we performed intravitreal microinjections of MAA-BSA with or without CFH. After six hours mice were killed, RPE/choroid was isolated from each eye after enucleation, and RNA was extracted. The purity of the preparation was confirmed by the expression of the RPE-specific gene Rpe65 and the lack of rhodopsin (Rho) expression as a marker for the neurosensory retina in all samples (Fig. 5d). MAA-BSA injection led to a sevenfold upregulation of KC expression in these RPE preparations, whereas BSA injection had no effect. Importantly, addition of CFH completely inhibited the effect induced by MAA-BSA (Fig. 5e). Thus, MAA adducts promote inflammatory responses in different cell types involved in AMD in vitro and in the eye in vivo, and CFH specifically neutralizes this property.
Discussion
We report the identification of CFH as a hitherto unrecognized innate defence protein against MDA, which is a ubiquitously generated proinflammatory product of lipid peroxidation1,9,10. Our discovery of CFH as a major MDA-binding protein demonstrates that innate immunity has a pivotal role in providing homeostatic responses against endogenous oxidation-specific danger-associated molecular patterns4. This parallels the innate immune response to another OSE, the PC headgroup of oxidized phosphatidylcholine, mediated by the macrophage scavenger receptors CD36 and SR-B1, the murine IgM natural antibody EO6/T15 and the acute phase reactant CRP4,25. In an analogous manner, MDA is recognized by macrophage scavenger receptor SR-A11, several germline IgM natural antibodies12 and—as we now demonstrate—by CFH.
CFH is one of the most abundant plasma proteins (~100–700 μg ml−1) and the major regulator of complement activation14. It mediates anti-inflammatory housekeeping functions by protecting self cells from complement activation14, which is especially important for dying cells that lose other surface-associated complement regulators26,27. A number of potential ligands for CFH on host cells have been studied, including glycosaminoglycans28, as well as annexin A2, DNA and histones on apoptotic cells29. We now identify MDA as a major ligand for CFH on apoptotic/necrotic cells and show that MDA epitopes provide a surface for CFH to allow local generation of anti-inflammatory iC3b fragments. This becomes relevant in situations when large amounts of cellular debris are generated. Of note, necrotic and, under certain conditions, apoptotic cells are proinflammatory per se30,31. In this regard, the interaction of CFH with MDA-modified cellular compounds is also important because CFH limits MDA-induced IL-8 secretion. This provides an explanation for the ability of CFH to reduce endothelial IL-8 secretion in response to apoptotic blebs32. Thus, MDA epitopes are responsible for the recruitment of CFH to the surface of apoptotic cells, where it neutralizes their pro-inflammatory properties and halts complement activation.
We demonstrate that SCR7 and SCR20 mediate the binding of CFH to MDA. These two domains are clustering sites for mutations associated with AMD, but also other diseases14. The most prominent example is the H402 exchange in SCR7, which has a frequency of 35%, and may be responsible for over half of all AMD cases33,34. However, direct evidence for functional consequences of this polymorphism remained elusive. Here we show that the H402 variant exhibits severely impaired binding to MDA in a gene-dosage-dependent manner, which correlates well with the H402-associated risk for developing AMD. The H402 variant has been suggested to favour local complement activation as a result of reduced binding to glycosaminoglycans in the eye35. However, in contrast to glycosaminoglycans, MDA is enriched in the membranes of dying cells, which are continuously generated in the retina and need to be efficiently removed36. By demonstrating that the H402 variant has a reduced capacity to generate anti-inflammatory iC3b fragments on MDA-bearing surfaces, we provide a functional explanation for its strong disease association. It remains to be seen whether other genetic variations of CFH family members also affect MDA binding and thereby contribute to disease pathogenesis.
Consistent with an earlier report, we found MDA epitopes throughout the choroid and Bruch’s membrane including drusen of AMD lesions8. However, even under physiological conditions, oxidized phospholipids are formed as a result of photic stimulation of retinal photoreceptors and subsequently scavenged by several processes including clearance via CD36 (ref. 37). As one of the major degradation products of peroxidized phospholipids, MDA is continuously generated. Several lipid peroxidation products, including MDA, can cause RPE damage38. Therefore, physiological housekeeping mechanisms are critically needed to prevent their accumulation and adverse reactions mediated by them. Our immunohistochemical results support a role for CFH, because CFH is found in the same locations as MDA in eyes with and without AMD. We show that MDA adducts, similar to what has been shown following the ingestion of oxidized photoreceptors23, have the capacity to induce IL-8 secretion in RPE, which can be blocked by CFH. Increased IL-8 expression correlates with higher incidence of AMD, underlining its important pathogenic role39. Therefore, neutralization of MDA adducts by CFH has the potential to limit several pathogenic events in AMD.
Undoubtedly, there are multiple defences against the ubiquitous MDA adducts. The described homeostatic response may be particularly limiting in the eye, as opposed to other sites where MDA adducts accumulate such as the vascular wall40. Future studies need to evaluate the contribution of this newly found interaction in the pathogenesis of other inflammatory diseases. The findings described here may lead to novel approaches exploiting endogenous defence mechanisms for the prevention and therapy of chronic inflammation in general.
METHODS SUMMARY
Subjects and clinical diagnosis
The patient cohort used in this study has been described41.
Protein modifications
The MAA modifications of LDL, BSA or polylysine were performed as described13.
Intravitreal injection
Intravitreal injections of BSA, MAA-BSA and/or CFH were performed in male and female C57BL/6 mice. Six hours later, the RPE/choroid was isolated and expression of target genes assessed by quantitative RT–PCR.
Bead coupling and pull-down procedure
Mouse or human plasma was incubated with polylysine or MAA-polylysine beads. Bound proteins were analysed by LC-MSMS. The interaction was verified by immunoblotting and characterized by ELISA and Biacore.
Flow cytometry and immunohistochemistry
The presence of MDA epitopes and CFH was visualized by flow cytometry on apoptotic Jurkat T-cell microparticles and by immunohistochemistry for histological specimens.
Co-factor assay
CFH bound to coated MAA-BSA was incubated with C3b and factor I and the generation of iC3b fragments was visualized by immunoblotting.
Cell culture
Following stimulation with BSA or MAA-BSA in the presence or absence of CFH, gene expression was determined by quantitative RT–PCR and/or IL-8/KC secretion was quantified by ELISA. Binding of biotinylated MAA-LDL to peritoneal macrophages was assessed in the presence or absence of competitors as described42.
Statistical analysis
Data are presented as mean ± s.d. or mean ± s.e.m. where indicated. Results were analysed by one-way analysis of variance and Student’s unpaired t-test.
Supplementary Material
Acknowledgments
We are indebted to M. Ozsvar-Kozma for technical assistance, S. Hälbich for performing the surface plasmon resonance analysis, A. Hartmann for purification of CFH variants from patient plasma, C. Mannhalter for help with genotyping, and E. N. Montano. This work was supported by the Austrian Academy of Sciences, a BRIDGE grant from the Austrian Research Promotion Agency, the SFB Lipotox F30 of the Austrian Science Fund (C.J.B.); NIH grants HL088093 (C.J.B., S.T., J.L.W.), RO1 HL086599 (K.H., J.L.W.), EY14005, EY019044 (J.T.H.); the Edward N. & Della L. Thome Memorial Foundation Awards Program in AMD Research, Research to Prevent Blindness (Wilmer Eye institute) (J.T.H.); the Deutsche Forschungsgemeinschaft (P.F.Z., C.S.); the ProRetina Foundation (N.L., P.F.Z., C.S.); the Fondation Leducq (C.J.B., S.T., J.L.W.); the Wynn-Gund Translational Research Acceleration Program Enhanced Research and Clinical Training Award, National Neurovision Research Institute – Foundation Fighting Blindness, Macular Degeneration Research Award, American Health Assistance Foundation (H.P.N.S.); European Commission and the Seventh European Community Framework Program, Marie Curie Intra-European Fellowship (P.C.I.). K.H. was supported by the Scientist Development Grant 0630228N of the American Heart Association. J.T.H. is the Robert Bond Welch Professor. We thank all patients for participation.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions D.W. and C.J.B. conceived the project and designed and analysed the experiments with contributions from J.L.W., P.F.Z., J.T.H., G.S.-F. and C.S.; D.W. performed most of the experiments, with contributions from K.H., N.L., K.L.B., M.C., S.T. and H.B.; H.P.N.S. and P.C.I. obtained and provided AMD plasmasamples; D.W. and C.J.B. wrote the manuscript with contributions from J.L.W., P.F.Z. and J.T.H.
The authors declare no competing financial interests.
References
- 1.Chou MY, et al. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med. 2008;263:479–488. doi: 10.1111/j.1365-2796.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 2.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 3.Chang MK, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci USA. 1999;96:6353–6358. doi: 10.1073/pnas.96.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller YI, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollyfield JG, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nature Med. 2008;14:194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki M, et al. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol Vis. 2007;13:772–778. [PMC free article] [PubMed] [Google Scholar]
- 7.Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 8.Schutt F, Bergmann M, Holz FG, Kopitz J. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:3663–3668. doi: 10.1167/iovs.03-0172. [DOI] [PubMed] [Google Scholar]
- 9.Thiele GM, et al. Malondialdehyde-acetaldehyde (MAA) modified proteins induce pro-inflammatory and pro-fibrotic responses by liver endothelial cells. Comp Hepatol. 2004;3(suppl. 1):S25. doi: 10.1186/1476-5926-2-S1-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shanmugam N, et al. Proinflammatory effects of advanced lipoxidation end products in monocytes. Diabetes. 2008;57:879–888. doi: 10.2337/db07-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shechter I, et al. The metabolism of native and malondialdehyde-altered low density lipoproteins by human monocyte-macrophages. J Lipid Res. 1981;22:63–71. [PubMed] [Google Scholar]
- 12.Chou MY, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119:1335–1349. doi: 10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu D, et al. Epitope characterization of malondialdehyde-acetaldehyde adducts using an enzyme-linked immunosorbent assay. Chem Res Toxicol. 1997;10:978–986. doi: 10.1021/tx970069t. [DOI] [PubMed] [Google Scholar]
- 14.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nature Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 15.Józsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol. 2008;29:380–387. doi: 10.1016/j.it.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 17.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards AO, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 19.Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hughes AE, et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nature Genet. 2006;38:1173–1177. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 21.Takizawa F, Tsuji S, Nagasawa S. Enhancement of macrophage phagocytosis upon iC3b deposition on apoptotic cells. FEBS Lett. 1996;397:269–272. doi: 10.1016/s0014-5793(96)01197-0. [DOI] [PubMed] [Google Scholar]
- 22.Amarilyo G, et al. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-κB-dependent blockade. Eur J Immunol. 2010;40:699–709. doi: 10.1002/eji.200838951. [DOI] [PubMed] [Google Scholar]
- 23.Higgins GT, Wang JH, Dockery P, Cleary PE, Redmond HP. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptorouter segments. Invest Ophthalmol Vis Sci. 2003;44:1775–1782. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- 24.de Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355:1474–1485. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- 25.Binder CJ, et al. Innate and acquired immunity in atherogenesis. Nature Med. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 26.Flierman R, Daha MR. The clearance of apoptotic cells by complement. Immunobiology. 2007;212:363–370. doi: 10.1016/j.imbio.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Trouw LA, et al. C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J Biol Chem. 2007;282:28540–28548. doi: 10.1074/jbc.M704354200. [DOI] [PubMed] [Google Scholar]
- 28.Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA. 1990;87:3982–3986. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leffler J, et al. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J Biol Chem. 2010;285:3766–3776. doi: 10.1074/jbc.M109.045427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nature Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 31.Chang MK, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mihlan M, Stippa S, Jozsi M, Zipfel PF. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. 2009;16:1630–1640. doi: 10.1038/cdd.2009.103. [DOI] [PubMed] [Google Scholar]
- 33.Thakkinstian A, et al. Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Hum Mol Genet. 2006;15:2784–2790. doi: 10.1093/hmg/ddl220. [DOI] [PubMed] [Google Scholar]
- 34.Donoso LA, Vrabec T, Kuivaniemi H. The role of complement factor H in age-related macular degeneration: a review. Surv Ophthalmol. 2010;55:227–246. doi: 10.1016/j.survophthal.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Clark SJ, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. 2006;281:24713–24720. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- 36.Green WR, Enger C. Age-related macular degeneration histopathologic studies. (The 1992 Lorenz E Zimmerman Lecture).Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- 37.Sun M, et al. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: a potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J Biol Chem. 2006;281:4222–4230. doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaemmerer E, Schutt F, Krohne TU, Holz FG, Kopitz J. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Invest Ophthalmol Vis Sci. 2007;48:1342–1347. doi: 10.1167/iovs.06-0549. [DOI] [PubMed] [Google Scholar]
- 39.Goverdhan SV, et al. Interleukin-8 promoter polymorphism −251A/T is a risk factor for age-related macular degeneration. Br J Ophthalmol. 2008;92:537–540. doi: 10.1136/bjo.2007.123190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sofat R, et al. Genetic variation in complement factor H and risk of coronary heart disease: eight new studies and a meta-analysis of around 48,000 individuals. Atherosclerosis. 2010;213:184–190. doi: 10.1016/j.atherosclerosis.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 41.Scholl HP, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binder CJ, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nature Med. 2003;9:736–743. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.