

Abstract
Background
The immune system orchestrates the repair of infarcted myocardium. Imaging of the cellular inflammatory response by 18F-FDG PET/MRI in the heart has been demonstrated in preclinical and clinical studies. However, the clinical relevance of post-MI 18F-FDG uptake in the heart has not been elucidated. The objective of this study was to explore the value of 18F-FDG-PET/MRI in patients after AMI as a biosignal for left ventricular functional outcome.
Methods and Results
We prospectively enrolled 49 patients with STEMI and performed 18F-FDG-PET/MRI 5 days after PCI and follow-up cardiac MRI after 6–9 months. In a subset of patients, 99mTc-sestamibi-SPECT was performed with tracer injection prior to revascularization. Cellular innate immune response was analyzed at multiple time points. Segmental comparison of 18F-FDG-uptake and LGE showed substantial overlap (κ=0.66), while quantitative analysis demonstrated that 18F-FDG extent exceeded LGE extent (33.2±16.2 %LV vs. 20.4±10.6 %LV, p<0.0001) and corresponded to the area-at-risk (r=0.87, p<0.0001). The peripheral blood count of CD14high/CD16+ monocytes correlated with the infarction size and 18F-FDG signal extent (r=0.53, p<0.002 and r=0.42, p<0.02, respectively). 18F-FDG uptake in the infarcted myocardium was highest in areas with transmural scar and the SUVmean was associated with left ventricular functional outcome independent of infarct size (ΔEF: p<0.04, ΔEDV: p<0.02, ΔESV: p<0.005).
Conclusions
In the current study, the intensity of 18F-FDG uptake in the myocardium after AMI correlated inversely with functional outcome at 6 months. Thus, 18F-FDG uptake in infarcted myocardium may represent a novel biosignal of myocardial injury.
Keywords: myocardial infarction, inflammation, PET/MRI, 18F-FDG, outcome, monocytes
Coronary artery disease and myocardial infarction claim more lives worldwide than any other disease. Advances in clinical cardiology have improved survival after AMI.1 Despite optimal therapy, heart failure is a persisting problem developing soon after myocardial infarction in almost 25% of patients, with numbers increasing over time.1, 2
While the role of the immune system in the pathogenesis of myocardial disease has been established for pathogen-driven diseases such as viral myocarditis, research more recently has focused on the cardiac inflammatory response after AMI as a risk factor for adverse remodeling. Peripheral blood monocyte counts have been linked to the development of heart failure in patients after AMI3, making the immune response after myocardial infarction a potential therapeutic target. High levels of presumably pro-inflammatory monocytes have been related to impaired infarct healing.4–6
The underlying biological processes have mostly been studied in rodent models.7, 8 Myocardial infarction triggers cellular and molecular changes which attract neutrophils and inflammatory monocytes expressing high levels of the CC chemokine receptor 2 (CCR2) to the site of injury. After a few days, the injured heart switches to CX3CL1-mediated recruitment of non-inflammatory monocytes.9 The early monocyte subset expresses proinflammatory mediators and proteases for wound debridement, while the late subset and M2 macrophages support neoangiogenesis and extracellular matrix synthesis.10 The emerging picture of inflammation biology positions leukocytes as both protective and harmful post AMI. An optimal repair after AMI requires a balance between coordinated cell recruitment that removes dead cell debris and strengthens the wound so that the heart performs its vital function.11
18F-FDG PET has been used to image inflammation in multiple clinical and research settings as activated inflammatory cells show increased expression of glucose transporters and thus increased 18F-FDG uptake.12 For instance, imaging of macrophages using 18F-FDG PET has been successfully performed in atherosclerotic plaques.13 A recent preclinical study in mice presented evidence that, under certain conditions, post-MI 18F-FDG uptake reflects the monocytic inflammatory response in the myocardium.14 Transient suppression of the physiologic 18F-FDG uptake into cardiomyocytes is required to image monocytes or macrophages, which has previously been achieved in humans by low-glucose diet, fasting, and administration of heparin prior to PET imaging.15, 16
Initial experience with myocardial fasting 18F-FDG PET in patients after AMI has been presented by us and other groups14, 17, 18, but the clinical relevance of this novel imaging approach has not yet been tested.
Therefore, the aim of the present study was to characterize post-ischemic myocardium in patients with a first AMI by fasting 18F-FDG PET/MRI in relation to the cellular immune response and the functional outcome after 6 months.
METHODS
Please see Supplemental Method Section for additional information.
Patient sample
Eligible patients admitted to the hospital with a first AMI between May 2013 and October 2014 were prospectively enrolled in the study after PCI and giving written informed consent. The study was approved by the local ethics committee and was performed in agreement with the Declaration of Helsinki.
PET/MR imaging
Patients were imaged 5.0±1.3 days after PCI by simultaneous PET/MRI using a hybrid system (Biograph mMR, Siemens Healthcare GmbH, Erlangen, Germany), which allows PET imaging in combination with a detailed characterization of the infarcted heart including scar size, scar transmurality and LV function in one examination.17
To suppress physiological myocardial 18F-FDG uptake, patients received a low-carbohydrate diet the day prior to imaging followed by a 12-hour fasting period. Unfractionated heparin (50 I.U./kg body weight IV) was administered 30 minutes before 18F-FDG injection.
Follow-up imaging by MRI only was performed on the identical system 252±68 days after PCI.
Regional analysis
The AHA-17-segment model19 was applied to LGE and 18F-FDG images aligned by the MunichHeart/m3p software.20, 21 LGE transmurality22 and 18F-FDG uptake were visually scored for each segment with scores ranging from 0 to 4 (see Suppl. Figure 1B). Results were additionally analyzed for mere absence or presence of LGE or 18F-FDG signals. Analysis was performed blinded to the respective other modality. Furthermore, the degree of wall motion abnormality in each segment was evaluated using a 5-point scale as previously described22: normal wall motion=0, mild to moderate hypokinesia=1, severe hypokinesia=2, akinesia=3, and dyskinesia=4. A functional improvement of wall motion was defined as a decrease of the wall motion score of at least 1 point score. Two experienced observers performed all analyses.
Quantitative analysis
In order to determine the 18F-FDG extent, a region grow algorithm with a threshold of 50% of the maximal uptake was performed (Syngo MMWP [workstation], Syngo TrueD [software]; Siemens Healthcare). Within this volume of interest (VOI), the average tracer uptake was derived and normalized to the lean body mass and injected tracer dose yielding SUVmean.23 The volume with tracer uptake above the mentioned threshold was finally expressed as %LV (LV myocardial volume derived from MRI) and defined as 18F-FDG extent (i.e. the volume of the significant 18F-FDG signal normalized to the total myocardial volume of the LV). SUVmean in remote myocardium was determined in manually drawn regions in the myocardial wall opposing the infarct on 3 consecutive slices.
The LGE extent of the LV myocardium was determined by manual delineation on short axis images using the MunichHeart/MR software and expressed as percentage of the left ventricle.
SPECT imaging
A subgroup of patients (n=23) underwent 99mTc-sestamibi SPECT with tracer injection prior to PCI and image acquisition after PCI for assessment of the area at risk.
99mTc-sestamibi SPECT images were analyzed using MunichHeart/NM.24, 25 The method for assessment of the area at risk (as percentage of the left ventricle) has been described previously.26
Blood analysis
Blood was drawn daily for up to 6 days after admission for differential blood count, creatine kinase (and MB fraction) and Troponin T. For analyses of monocyte subpopulations, blood was drawn at least once early (day 0–3) and late (day 4–6) during hospitalization. If more than one sample was available for one time point, average values were used. Furthermore, a differential blood count and the different monocyte subpopulations were determined at the follow-up scan.
Flow cytometry analysis was performed on a Cytomics FC 500 flow cytometer (Beckman Coulter, CA, USA). Data were analyzed with Kaluza 1.2 Analysis Software (Beckman Coulter, CA, USA). Red blood cells were lysed, leukocytes stained with anti-human monoclonal fluorochrome-conjugated antibodies, and immediately analyzed by flow-cytometry. After gating for CD45 and forward/side scatter, gating was performed similar to previously described strategies. Inflammatory, intermediate, and non-inflammatory monocyte subsets were defined by the expression of CD14, CD16 and/or CCR2 (see Suppl. Fig. 2A).4–6, 27
Statistics
Results are shown as mean ± standard deviation. P-values smaller than 0.05 were considered significant. For comparison of unmatched, continuous variables the 2-tailed unpaired Student t-test was used and the χ2 test or the Fisher’s exact test (in case of small numbers) to compare nominal variables. To compare matched continuous variables the 2-tailed paired Student t-test was applied. The association between continuous variables was investigated using the Pearson correlation coefficient. A stepwise multivariable regression analysis was performed to correct for the influence by different covariates, i.e. in a first step a unadjusted analysis was performed to detect associated variables (p<0.10), which then entered a subsequent multivariable analysis. In order to detect any outliers regarding functional outcome measures (ΔEF, ΔEDV and ΔESV), a Grubbs double-sided test was performed to check the most extreme value at either side. Categorical intermethod agreement between 18F-FDG uptake in PET and LGE in MRI was assessed by the Cohen κ (κ<0, no agreement; κ=0–0.20, poor agreement; κ=0.20–0.40, slight agreement; κ=0.40–0.60, moderate agreement; κ=0.60–0.80, substantial agreement; κ=0.80–1.00, very good agreement). To compare categorical measures or binary parameters under the consideration of within-patient correlations the generalized estimating equation (GEE) method with exchangeable correlation matrix was used. For statistical analyses, MedCalc (version 15.8; MedCalc Software) for Windows (Microsoft) and SPSS Statistics for Windows (Version 22.0 IBM Corp. Released 2013. Armonk, NY: IBM Corp.) were used.
RESULTS
Patient characteristics
A study flow chart is depicted in Figure 1. In total, 49 patients were initially enrolled in the study and imaged by 18F-FDG PET/MRI. Ten subjects were excluded from the initial analysis. Follow-up imaging was obtained in 29 patients. For this follow-up group initial 18F-FDG PET and MRI data, follow-up MRI data, leukocyte and monocyte counts were all available. All patients were treated with statins, betablockers and ACE-inhibitors or ARBs before 18F-FDG PET/MR imaging. The characteristics of the initial imaging sample and the different investigated subgroups are described in Table 1.
Figure 1. Study flow chart.
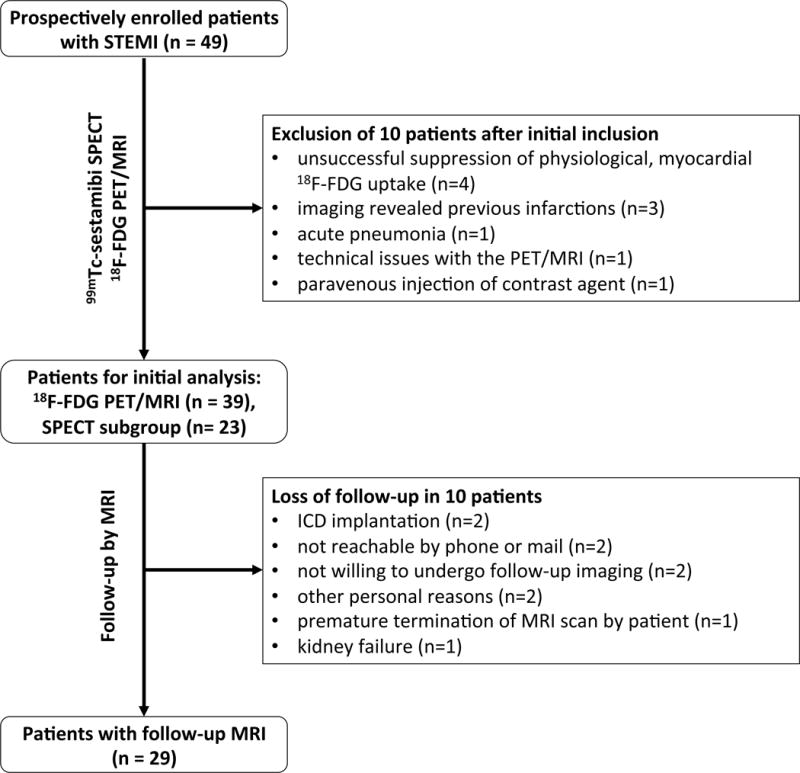
A total of 49 patients were initially enrolled in the study. From this sample 10 subjects had to be excluded after 18F-FDG PET/MRI for various reasons. 39 subjects were included in the initial analysis of PET/MRI and blood parameters. A subgroup of 23 patients underwent 99mTc-sestamibi SPECT with tracer injection prior to PCI to determine the area at risk.
At follow-up, MR imaging could not be obtained in 10 more patients, resulting in a follow-up group of 29 patients.
Table 1.
Patient characteristics undergoing initial 18F-FDG PET/MR and of the different investigated subgroups
| All included patients (n=39) |
Subgroup with 99mTc-sestamibi SPECT (n=23) |
Subgroup with follow-up MRI (n=29) |
Subgroup without follow-up MRI (n=10) |
p* | |
|---|---|---|---|---|---|
| Male | 35 (90) | 22 (96) | 27 (93) | 8 (80) | 0.27 |
| Age, yrs | 59.5 ± 12.7 | 60.4 ± 12.4 | 59.2 ± 13.2 | 60.6 ± 11.7 | 0.76 |
| Infarct vessel | |||||
| LM | 1 (3) | 0 (0) | 1 (3) | 0 (0) | 1.00 |
| LAD | 21 (54) | 12 (52) | 15 (52) | 6 (60) | |
| RCX | 5 (13) | 1 (4) | 4 (14) | 1 (10) | |
| RCA | 12 (31) | 10 (43) | 9 (31) | 3 (30) | |
| No. of diseased vessels | 2.1 ± 0.9 | 2.2 ± 0.9 | 2.1 ± 0.9 | 2.0 ± 0.9 | 0.75 |
| 1-vessel disease | 13 (33) | 7 (30) | 9 (31) | 4 (40) | 0.90 |
| 2-vessel-disease | 10 (26) | 5 (22) | 8 (28) | 2 (20) | |
| 3-vessel-disease | 16 (41) | 11 (48) | 12 (41) | 4 (40) | |
| Presence of non-culprit lesions requiring PCI | 19 (49) | 12 (52) | 15 (52) | 4 (40) | 0.72 |
| LVEF, % | 46 ± 11 | 45 ± 13 | 47 ± 10 | 43 ± 15 | 0.27 |
| Pain-to-balloon time, hrs | 8.3 ± 10.5 [1.5 – 55.7] |
7.8 ± 12.5 [1.5 – 55.7] |
9.3 ± 11.7 [2.1 – 55.7] |
5.5 ± 4.9 [1.5 – 16.9] |
0.35 |
| Pain-to-scan time, hrs | 130.5 ± 36.4 [73.5 – 217.3] |
119.9 ± 33.9 [73.5 – 202.1] |
124.9 ± 33.9 [73.5 – 209.3] |
141.4 ± 40.4 [97.7 – 217.3] |
0.24 |
| Balloon-to-scan time, hrs | 123.5 ± 35.8 [68.5 – 214.0] |
115.1 ± 34.5 [68.5 – 193.8] |
117.5 ± 33.3 [68.5 – 193.8] |
136.7 ± 38.6 [94.3 – 214.0] |
0.14 |
| CK max, U/l | 2292 ± 1955 | 2303 ± 2295 | 2450 ± 2055 | 1833 ± 1640 | 0.40 |
| CKMB max, U/l | 262 ± 183 | 261 ± 214 | 271 ± 199 | 235 ± 128 | 0.62 |
| TnT max, ng/ml | 3.6 ± 4.3 | 4.5 ± 5.3 | 3.8 ± 4.7 | 3.1 ± 3.0 | 0.69 |
| Maximum leukocyte count, G/l | 13.1 ± 4.2 | 12.5 ± 3.3 | 12.8 ± 3.2 | 14.0 ± 6.4 | 0.44 |
| at day | 0.6 ± 0.8 | 0.4 ± 0.6 | 0.4 ± 0.6 | 1.2 ± 1.0 | <0.001 |
| Maximum monocyte count, G/l | 1.2 ± 0.5 | 1.1 ± 0.3 | 1.2 ± 0.4 | 1.4 ± 0.5 | 0.16 |
| at day | 1.8 ± 1.1 | 1.7 ± 1.0 | 1.8 ± 1.1 | 1.7 ± 0.8 | 0.85 |
Values are n (%) or mean ± SD.
p-values for patients with follow-up MRI vs. patients without follow-up MRI
18F-FDG uptake and localization in relation to LGE
LGE and 18F-FDG uptake was observed within the infarcted myocardial region. Two different patient examples are shown in Figure 2. In all patients, both the LGE signal intensity and the 18F-FDG uptake intensity were at least 5 standard deviations higher in the ischemically compromised myocardium compared to remote myocardium (SUVmean infarct vs. remote: 2.2±0.4 vs. 0.7±0.2, p<0.0001; Suppl. Fig. 1A). There was no correlation between the 18F-FDG uptake in the post-ischemic myocardium and remote myocardium (r=0.25, p=0.13). The post-ischemic 18F-FDG uptake in the infarct area did not correlate with pain-to-balloon (r=0.02, p=0.89), pain-to-scan (r=0.10, p=0.56) or balloon-to-scan time (r=0.01, p=0.93).
Figure 2. 18F-FDG PET/MR images of two patients shortly after AMI.
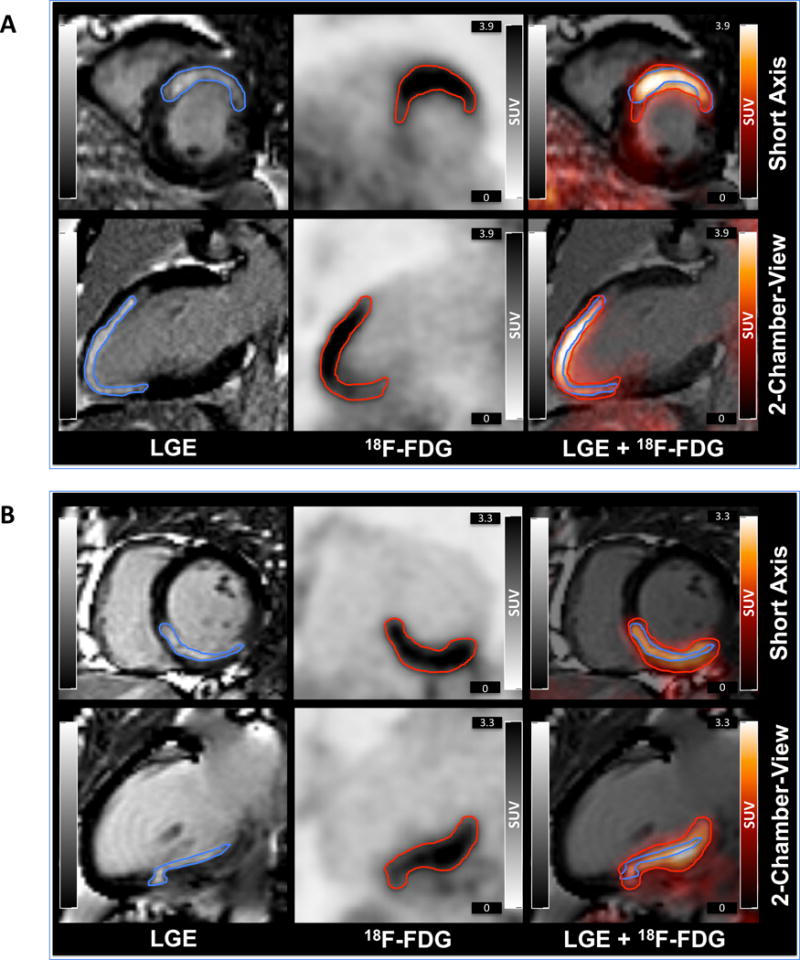
Short and long axis views of LGE MR images (left), 18F-FDG PET images (middle), and overlay (right) of patients with anterior (A) or inferior (B) myocardial infarction.
In 15 (38%) patients microvascular obstruction (MVO) was observed. There was a trend towards a higher FDG uptake in patients with MVO, which did not reach statistic significance (2.4±0.4 vs. 2.1±0.4, p=0.09).
A regional analysis showed substantial agreement for the presence or absence of LGE and 18F-FDG uptake (κ=0.66). A detailed analysis of LGE transmurality and 18F-FDG uptake intensity (score 0 – 4) still showed substantial agreement between both modalities (κ=0.63; Suppl. Figure 1 B) and a strong correlation between the summed 18F-FDG and LGE score was found (r=0.82, p<0.0001; Suppl. Figure 1C). High 18F-FDG scores were significantly more often present in segments with transmural infarction as compared to segments with non-transmural infarction (GEE: p<0.001; Suppl. Figure 1D). Consequently, the strongest 18F-FDG uptake was observed in areas with most transmural LGE extent.
Quantification of 18F-FDG extent, LGE extent, and area at risk
In the regional analysis, 18F-FDG uptake was present in more segments than LGE (372/663 segments, 56.1% vs. 292/663 segments, 44.0%, GEE: p<0.001). Quantitative comparison of cardiac 18F-FDG and LGE extent confirmed this finding. While both modalities showed close correlation (r=0.78, p<0.0001; Figure 3A, left and middle panel), the 18F-FDG extent (i.e. the volume of the 18F-FDG signal expressed as %LV) significantly exceeded the LGE extent (33.2±16.2 %LV vs. 20.4±10.6 %LV, p<0.0001; Figure 3A, right panel).
Figure 3. Comparison of 18F-FDG extent, LGE extent, and area at risk.
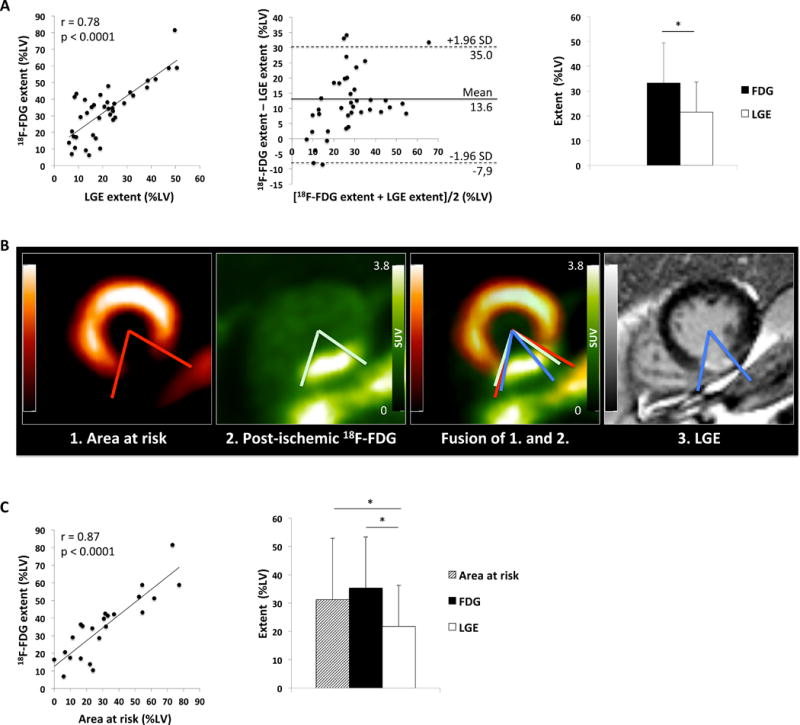
A: Correlation between quantitative 18F-FDG extent (%LV) and LGE extent (%LV) (left). Bland-Altman-Plot of the difference between 18F-FDG extent (%LV) and LGE extent (%LV) against the mean of both measures (middle). Comparison of 18F-FDG uptake area and LGE area in all 39 patients with (*p<0.0001) (right).
B: Short axis views of 99mTc-sestamibi SPECT depicting the area at risk, 18F-FDG PET, fusion of the two and LGE MR images of a patient with inferior myocardial infarction. The area at risk nicely matches the post-ischemic 18F-FDG uptake, while both measures are larger than the LGE extent.
C: Correlation between quantitative 18F-FDG extent (%LV) and area at risk (%LV) (left). Comparison of 18F-FDG uptake extent, LGE extent and area at risk in 23 patients with 99mTc-sestamibi-SPECT (right, 18F-FDG vs. LGE, *p<0.0001; Area at risk vs. LGE *p<0.004).
In the patient subset with 99mTc-sestamibi SPECT imaging (sample in Figure 3B), 18F-FDG uptake similarly exceeded the LGE extent (35.4±18.1 %LV vs. 21.8±14.5 %LV; p<0.0001, Figure 3C, right panel) and showed a high correlation with the area at risk (r=0.87, p<0.0001, Figure 3C, left panel). Accordingly, the area at risk was larger than the LGE extent (31.3±21.6 %LV vs. 21.8±14.5 %LV; p<0.004), but did not differ significantly from the 18F-FDG extent (31.3±21.6 %LV vs. 35.4±18.1 %LV; p=0.08) (Figure 3C, right panel).
Comparison of PET/MRI with circulating leukocytes and monocytes
Both the LGE and the 18F-FDG extent correlated with peak counts of leukocytes from peripheral blood (r=0.38, p<0.02 and r=0.47, p<0.003, Figure 4A, left and middle panel). Also, the 18F-FDG and the LGE extent correlated with CCR2+ monocytes (LGE: r=0.41, p<0.02, FDG: r=0.40, p<0.02, Figure 4A, right panel) and with CD14high/CD16+ (also referred to as intermediate) monocytes (LGE: r=0.53, p<0.002, 18F-FDG: r=0.42, p<0.02, Figure 4B, middle column) released early (during the first 3 days) after infarction, indicating that leukocyte and monocyte release and migration to the heart may be dependent on the infarct size. The CD14high/CD16− (inflammatory) and CD14low/CD16+ (non-inflammatory or reparative) monocytes released early after infarction were not significantly related to the LGE extent or the 18F-FDG extent in our patient sample (Figure 4B, left and right column).
Figure 4. Peak leukocytes, CCR2+ monocytes and monocyte subpopulations in relation to LGE and 18F-FDG extent.
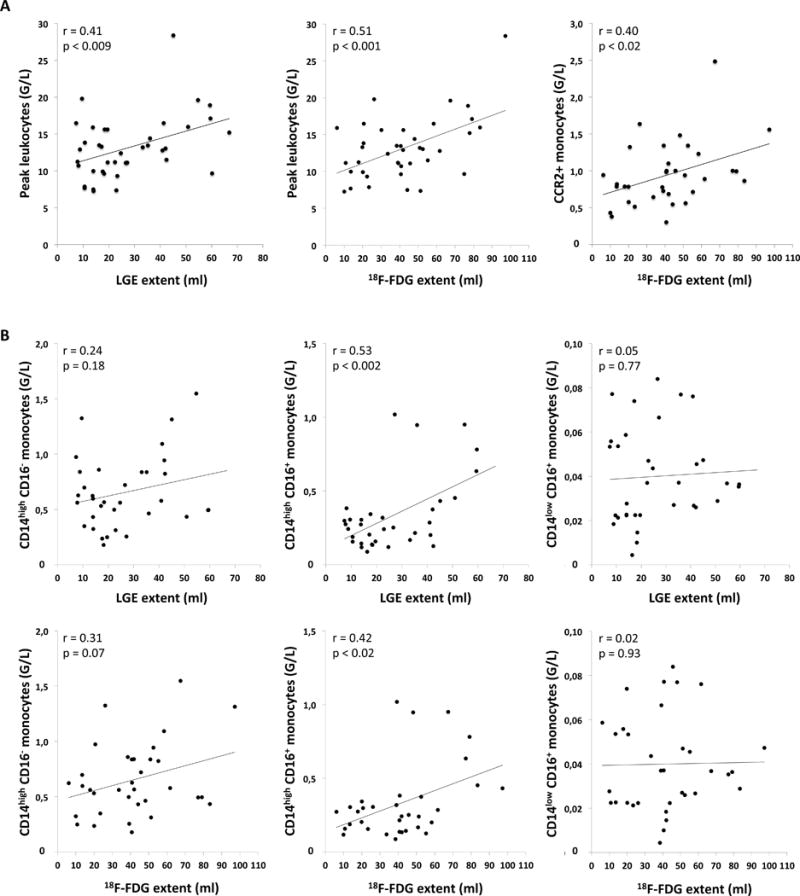
A: Correlation between infarct size and peak leukocytes after infarction (left), 18F-FDG extent and peak leukocytes (middle), and 18F-FDG extent and CCR2+ monocytes early after myocardial infarction (right). (G/L = 109 cells per liter).
B: CD14highCD16− (inflammatory), CD14highCD16+ (intermediate), and CD14lowCD16+ (reparative) monocyte subpopulations determined during the first 3 days after AMI in relation to the LGE extent and 18F-FDG extent. (G/L = 109 cells per liter).
Interestingly, there was no correlation between the SUVmean (i.e. the mean intensity of the 18F-FDG signal) in the postischemic myocardium and the LGE extent, initial ejection fraction (EF), peak counts of leukocytes, or monocyte subpopulations released early after infarction (Suppl. Figure 2B and 2C).
18F-FDG uptake in bone marrow and aorta after myocardial infarction
A significant correlation between the 18F-FDG uptake intensity in the infarct and the 18F-FDG uptake in the aorta (r=0.38, p<0.02) was found, indicating a connection between the inflammatory processes in the heart and vessels after infarction. There was no relationship, however, between the 18F-FDG uptake in the infarct area and the bone marrow (r=0.09, p=0.61) or between the bone marrow and the aorta (r=0.20, p=0.21).
To further elucidate this point, correlations of the 18F-FDG signals in the aorta and the bone marrow with peak counts of leukocytes/monocytes and with counts of monocyte subsets were investigated. The 18F-FDG signal of the bone marrow and the aortic wall did not correlate with peak leukocyte counts or peak monocyte counts. While counts of CD14high/CD16− (inflammatory) and CD14high/CD16+ (intermediate) monocyte subsets did not correlate with the 18F-FDG uptake in the bone marrow or the aortic wall, CD14low/CD16+ (non-inflammatory or reparative) showed an inverse correlation with the 18F-FDG uptake in the aortic wall (r=−0.46, p<0.008).
Follow-up MRI
The LGE extent declined between initial and follow-up imaging in almost all patients (20.0±11.2 % vs. 14.9±10.0%, p<0.0001, Suppl. Figure 3A). In contrast, in average there was no change in the global EF at follow-up (47.4±9.7% vs. 46.4±11.8%, p=0.56, Suppl. Figure 3B). There was no correlation between the post-ischemic 18F-FDG uptake in the infarct area or remote myocardium and the decrease of LGE (ΔLGE) (r=0.10, p=0.62 and r=0.24, p=0.21, respectively).
As expected, the change in EF, enddiastolic volume (EDV), and endsystolic volume (ESV) between initial and follow-up imaging were associated with the infarct size (ΔEF: r=−0.39, p<0.05; ΔEDV: r=0.41, p<0.03; ΔESV: r=0.59, p<0.002, Figure 5A). No outliers were detected in the functional outcome measures (ΔEF, ΔEDV and ΔESV). Interestingly, the change in EF, EDV, and ESV was inversely correlated to the post-ischemic FDG uptake (SUVmean) in the heart 5 days after infarction (ΔEF: r= −0.47, p<0.02; ΔEDV: r=0.51, p<0.007; ΔESV: r=0.60, p<0.0008, Figure 5B). There was no correlation between the 18F-FDG uptake in remote myocardium and the changes in EF, ESV or EDV (ΔEF: r=−0.29, p=0.12; ΔEDV: r=0.03, p=0.90; ΔESV: r=0.18, p=0.35).
Figure 5. Association of post-ischemic 18F-FDG uptake and infarct size with left ventricular functional outcome.
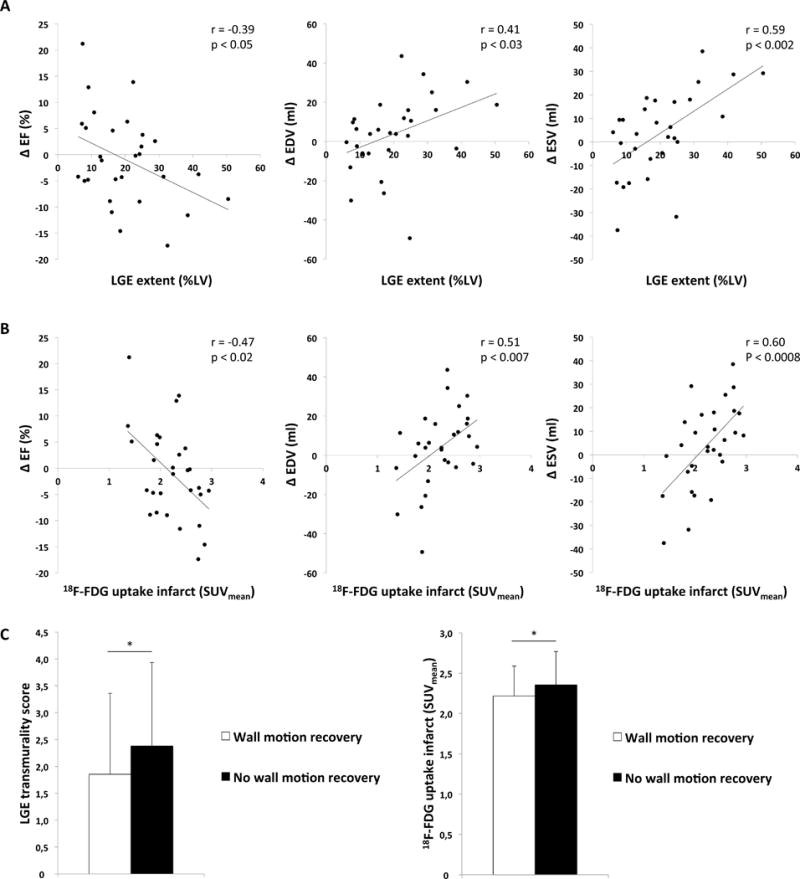
A: Correlation between LGE extent and ΔEF (EF at follow-up – EF at initial imaging), ΔEDV (EDV at follow-up – EDV at initial imaging), and ΔESV (ESV at follow-up – ESV at initial imaging).
B: Correlation between post-ischemic 18F-FDG uptake in the infarct area (SUVmean) and ΔEF, ΔEDV and ΔESV.
C: Segmental analysis of the wall motion recovery at follow-up. Comparison of LGE transmurality and 18F-FDG uptake in the infarct (SUVmean).
To further elucidate the significance of the post-ischemic 18F-FDG uptake, a stepwise multiple regression analysis with the following factors was performed: LGE extent, 18F-FDG extent, SUVmean, peak leucocyte count, peak monocyte count, counts of the different subsets and the pain-to-balloon time. Only the 18F-FDG extent, the LGE extent and the post-ischemic 18F-FDG uptake (SUVmean) showed in the preceding unadjusted analysis an association (p<0.10) with at least one of the measures of functional outcome ΔEF, ΔEDV or ΔESV (Table 2). In the subsequent multivariable analysis the SUVmean remained an independent marker for left ventricular functional outcome independent from LGE or 18F-FDG extent (Table 3).
Table 2.
Unadjusted analysis of the change in global left ventricular function (ΔEF, ΔEDV, ΔESV) after myocardial infarction.
| Factor | Unadjusted Ps | ||
|---|---|---|---|
| ΔEF | ΔEDV | ΔESV | |
| LGE extent (%LV) | 0.04 | 0.03 | 0.001 |
| FDG extent (%LV) | 0.57 | 0.18 | 0.10 |
| SUVmean (infarct) | 0.01 | 0.006 | <0.001 |
| SUVmean (remote) | 0.14 | 0.86 | 0.37 |
| Peak leukocytes | 0.61 | 0.61 | 0.36 |
| Peak monocytes | 0.17 | 0.57 | 0.69 |
| CCR2+ monocytes | 0.17 | 0.78 | 0.67 |
| CD14high/CD16− | 0.12 | 0.49 | 0.47 |
| CD14high/CD16+ | 0.19 | 0.97 | 0.39 |
| CD14low/CD16+ | 0.89 | 0.48 | 0.57 |
| Pain-to-balloon | 0.86 | 0.80 | 0.67 |
Table 3.
Multivariable analysis of the change in global left ventricular function (ΔEF, ΔEDV, ΔESV) after myocardial infarction.
| Factor | Multivariable Ps | ||
|---|---|---|---|
| ΔEF | ΔEDV | ΔESV | |
| LGE extent (%LV) | 0.14 | 0.10 | 0.01 |
| FDG extent (%LV) | – | – | 0.38 |
| SUVmean (infarct) | 0.04 | 0.02 | 0.005 |
Of the 29 patients with follow-up MRI, a total of 493 myocardial segments (according to the AHA 17-segment model) were analyzed regarding wall motion abnormalities. At baseline 247 of the analyzed segments (247/493, 50.1%) showed wall motion abnormalities with a mean wall motion abnormality score of 1.7±0.8. At follow-up, 139 segments (139/493, 28.2%) still demonstrated any wall motion abnormality and the wall motion abnormality score decreased to 1.0±1.1 (p<0.0001). Segments with an improved wall motion (160/247, 64.8%) had both a lower LGE transmurality score (1.9±1.5 vs. 2.4±1.6, p=0.01) as well as a lower SUVmean (2.2±0.4 vs. 2.4±0.4, p<0.008) compared to segments without wall motion improvement (Figure 5C). A multivariable regression analysis with LGE transmurality score and SUVmean as variables for the association with regional wall motion improvement demonstrated that both measures independently correlated with regional wall motion improvement (LGE: p<0.03, SUVmean: p<0.02).
DISCUSSION
Despite advances in cardiovascular medicine, myocardial infarction and subsequent loss of cardiomyocytes still results in adverse remodeling and the development of heart failure in many patients. While regenerative approaches aiming for cardiomyocyte replacement are still under investigation, the immune system has been identified as a key player of endogenous cardiac repair and scar formation. Different leukocytes invade the heart after ischemic injury and orchestrate the removal of cell debris, the formation of new vessels, and the turnover of extracellular matrix. Of note, both an overshooting and abolished inflammatory response after myocardial infarction appear to have deleterious effects on outcome in animal models and observational studies, leading to the concept of a parabolic relationship between inflammatory response and healing quality.28 Pro- and anti-inflammatory monocyte and macrophage subtypes have been proposed to be balancing forces in this complex and tightly regulated process.9, 29 Furthermore, altered levels of these cellular subtypes have been associated with impaired infarct healing in patients4, 5, making the immune system a potential therapeutic target for the prevention of heart failure after myocardial infarction. While several immune modulatory strategies led to an improved infarct healing in preclinical trials, most clinical pilot trials have shown less promising results so far30, suggesting that possibly not all patients with AMI may benefit from these therapies. A recent pilot study suggested colchicine as a beneficial anti-inflammatory agent after AMI resulting in a decreased infarct size31, but further studies will be needed to validate these findings.
18F-FDG-PET/MRI has been proposed as a tool to measure the cellular immune response in the myocardium in a preclinical study in mice.14 Successful in vivo imaging of immune cells in the heart after myocardial infarction in patients may help to guide novel therapeutic approaches. Therefore, we sought to explore the value of 18F-FDG-PET/MRI in patients with myocardial infarction in relation to the cellular immune response and functional outcome.
Our study showed two key findings: (i) The size of the 18F-FDG-PET signal and the numbers of leukocytes released to peripheral blood – particularly CD14highCD16+ monocytes – showed a significant correlation with the size of the infarction. (ii) The intensity of the 18F-FDG-PET signal was associated with MRI measures of left ventricular global and regional functional outcome, independent of infarction size and peripheral white blood cell counts.
The preclinical work by Lee et al. suggested that 18F-FDG-PET could be used to monitor CD11b+ monocytes and macrophages in the heart.14 Translating this concept to the clinical setting, our study demonstrated that simultaneous cardiac 18F-FDG-PET/MRI after myocardial infarction reproducibly labeled cells within and surrounding the infarcted myocardium. The SUVmean of the 18F-FDG-PET signal in the infarcted area measured 5 days after myocardial infarction correlated with an increase in EDV and ESV and a decrease of EF after 6 months. These relationships were still significant when the influence of the infarction size was taken into account. No correlation was found between the 18F-FDG uptake in remote myocardium and left ventricular functional outcome. This lack of correlation may be attributed to the early imaging time point after MI in our study, as the infiltration of remote myocardium by inflammatory cells occurs later according to the aforementioned preclinical study.14
The post-ischemic 18F-FDG uptake in the infarct (SUVmean) showed no correlation with infarction size, initial EF or peripheral blood laboratory parameters, suggesting that the imaging strategy added an additional level of prognostic information. The study by Lee et al. showed a correlation between the FDG uptake in the infarct area and the number of CD11+ cells in the murine myocardium.14 At first sight, it may appear puzzling that the post-ischemic 18F-FDG uptake did not correlate with peripheral blood monocyte counts in our study. However, this lack of correlation does not exclude the possibility that immune cells were labeled by 18F-FDG, as the post-ischemic 18F-FDG uptake in the myocardium is likely dependent on leukocyte recruitment to the myocardium, on monocyte differentiation into different macrophage subtypes and on macrophage activation – mechanisms that are possibly independent from peripheral blood cell counts. It is likely that monocyte/macrophage recruitment into the myocardium is regulated by additional factors such as local adhesion molecule and chemokine expression, and does not passively follow blood monocyte levels. From this perspective, it seems less surprising that the post-ischemic 18F-FDG uptake did not correlate with peripheral blood monocyte numbers. This lack of correlation rather demonstrates the additional value of 18F-FDG PET/MRI over circulating biomarkers to non-invasively measure the inflammatory state in the infarct area after myocardial infarction.
Nevertheless, the use of 18F-FDG as a tracer for inflammatory cells has limitations. As a glucose analogue tracer, 18F-FDG is taken up by a variety of cells and has been used to monitor cardiomyocyte viability32 and metabolism in response to hypoxia.33 In the present study, 18F-FDG uptake of normal cardiomyocytes was suppressed by patient preparation including high fat diet, fasting, and heparin injection prior to the scan – methods that have been developed to image inflammation in coronary arteries and sarcoidosis.15, 16 However, it has not been shown whether these methods reliably suppress 18F-FDG uptake in viable post-ischemic cardiomyocytes, as acutely ischemic and hibernating myocardium shifts its metabolic use of fatty acids towards glucose with depletion of glycogen stores34 and upregulation of glucose transporters 1 (GLUT1)35, a state termed “ischemic memory”.32, 33, 36 Nevertheless, using this technique mean SUV values in the infarcted tissue were approximately three times lower when compared to healthy remote myocardium in conventional 18F-FDG PET imaging indicating that this signal is not originating from vital cardiomyocytes.37 Another limitation of 18F-FDG PET is that it may not be able to distinguish between different leukocyte subpopulations that play opposing roles in the complex process of infarct healing. In the light of these limitations, it seems even more remarkable that the level of 18F-FDG uptake in the heart after myocardial infarction could still be associated with adverse global and regional functional outcome after 6 months.
Thus, the association of a higher intensity of 18F-FDG uptake in the infarct area with signs of adverse remodeling suggested a prevalent 18F-FDG uptake by a cell type, which can adversely affect infarct healing if it is abundantly present or shows strong metabolic activity. Interestingly, macrophages have been shown to exhibit increased 18F-FDG uptake in vitro in response to inflammatory cytokines as well as hypoxia.38–40 Furthermore, we found the strongest 18F-FDG signal in areas with most transmural LGE extent, i.e. least viable myocardium, supporting the idea that the predominant fraction of the 18F-FDG signal came from inflammatory cells in this area. This finding appears to be in line with post-mortem analyses of the hearts from patients who died from myocardial infarctions, showing increasing monocyte numbers in the infarct core at 5–14 days after myocardial infarction.41 The area of the myocardial 18F-FDG uptake strongly correlated with the infarction size measured by LGE. In detail, the 18F-FDG uptake area was slightly larger than the LGE area in most patients and more closely resembled the area at risk determined in a subset of patients by 99mTc-sestamibi SPECT with tracer injection prior to PCI. This is in line with the finding of a recent study, showing a close relationship between the 18F-FDG signal extent and myocardial edema by T2-weighted cardiac MRI.18 This may be attributed to immune cells in the border zone of the infarction, to 18F-FDG uptake in “ischemic memory” cardiomyocytes, or to methodical differences between the imaging modalities. The peak circulating levels of leukocytes after myocardial infarction were also associated with the spatial extent of myocardial injury and 18F-FDG uptake in our patient sample. Intermediate (CD14highCD16+) monocytes mobilized during the first 3 days after AMI showed a positive correlation with infarction size and the 18F-FDG uptake area, supporting the idea that the 18F-FDG signal reflected mobilization and migration of monocytes to a relevant degree. Expression of CCR2 (corresponding to murine Ly-6Chigh) was sufficient to identify a monocyte population correlating with infarction size, suggesting a direct neuronal or humoral pathway from the infarcted heart to the spleen and bone marrow that signals the precise demand for CCR2+ cells. This seems of particular interest in the context of recent publications that show a role of Ly-6Chigh monocytes in orchestrating both pro- and anti-inflammatory macrophages and track the release of these monocytes back to a subset of CCR2+ hematopoietic stem and progenitor cells in the mouse.42, 43 The association of CCR2+ (and particularly intermediate) monocyte counts with impaired outcome after myocardial infarction in patients seems to be in line with our observation, although an association of monocytes subtypes with outcome independent of infarction size has been proposed in some studies.5, 6
As described before, we also observed a decrease in LGE after MI between baseline imaging and 6-month follow-up.44 As the decline in LGE after myocardial infarction is mainly attributed to a decrease in edematous tissue, it is not surprising that no correlation with the post-ischemic FDG uptake in the heart was found.
AMI is a severe traumatic event that leads to immune responses in multiple organ systems. A substantial number of the monocytes initially recruited to the heart derive from a reservoir in the spleen. The first few days after infarction leukocyte recruitment remains at high levels. To meet this demand, cell production increases in the bone marrow and the spleen. Several reports demonstrated a higher 18F-FDG uptake within the spleen, the bone marrow and the carotid arteries in AMI patients compared to controls45. The focus of this study was to investigate the relevance of post-ischemic 18F-FDG uptake in the myocardium regarding LV functional outcome after six months. Therefore – and to limit the duration in the scanner for the post-MI patients – the imaging protocol was focused to image the heart; other organs (e.g. the spleen) were not fully covered in most patients and were therefore not studied. Furthermore, the inflammatory processes in spleen and bone marrow are known to follow distinct temporal dynamics, differing from the dynamics in heart. Therefore, imaging the 3 organ systems at the same time point was not considered ideal for the analysis of their interactions. Consistently, we did not find strong relationships between circulating cells, 18F-FDG uptake in the heart and 18F-FDG uptake in the bone marrow.
However, we found a correlation between the 18F-FDG uptake in the post-ischemic myocardium and the aortic wall, supporting the idea that the systemic immune response after myocardial infarction may affect atherosclerosis. Interestingly, we also found a significant inverse correlation between the CD14low/CD16+ (non-inflammatory or reparative) monocyte subset and the 18F-FDG uptake in the aortic wall, indicating a possible protective role of these cells regarding arterial inflammation following acute MI. However, it has to be acknowledged, that the release of leukocytes and monocytes after AMI is highly dynamic and a single imaging session might not be sufficient to clarify these questions.
This study was performed using a clinical hybrid PET/MRI. It has to be acknowledged that all obtained data could have also been acquired using separate devices (such as standalone MRI and PET or PET/CT). This would have resulted, however, in an increased complexity of the workflow (regarding the need for two independent imaging appointments) and would have increased stress and radiation dose for the patients due to the necessity of an additional scan for attenuation correction using PET/CT or standalone PET. Furthermore, data analysis would be complicated using separate scanners because ideal coregistration is only guaranteed when performing a simultaneous scan using hybrid PET/MRI. Still, it has to be pointed out that centers without access to a hybrid PET/MRI scanner may use standalone PET and MRI to further investigate this novel imaging approach.
This study has some limitations. We planned this observational study with a relatively small sample size, due to the unknown variance of the readout. However, additional patients were lost to MRI follow-up. The small patient number does also not allow association with hard endpoints. Due to the small patient number and the explorative nature of our study, validation of our findings in a larger cohort will be necessary. The prospective nature of our study, the detailed assessment of clinical and laboratory parameters, and the inclusion of a functional follow-up are strengths of our study. The time point of imaging was extrapolated from small animal studies, as serial PET imaging in patients did not seem to be justified. Serial imaging in large animals may help to optimize the timing of this novel imaging strategy.
In conclusion, the results of the present study suggest that 18F-FDG-PET/MRI may offer additional prognostic information regarding adverse functional outcome in patients with myocardial infarction. The paucity of beneficial results from early clinical therapeutic trials targeting the immune system in myocardial infarction may partly have been caused by an unspecific patient selection in combination with potentially harmful side effects. We believe, that this imaging approach may be able to identify patients at risk for an excessive immune response who are more likely to benefit from immune modulatory therapies that target specific cell subsets. Furthermore, PET/MRI could be used to monitor and guide these therapies, particularly in clinical trials. Taken together, there is accumulating evidence from ours and other studies that fasting 18F-FDG PET reliably measures inflammation in the heart. Nevertheless, we believe that more specific PET tracers for cellular subsets of the immune response will yield more relevant readouts.
Supplementary Material
Clinical Perspective.
The cardiac inflammatory response following myocardial infarction represents an emerging target for future therapeutic approaches in the treatment of myocardial infarction. While inflammation is required for proper infarct healing, a dysregulated immune response is known to increase the risk for adverse cardiac remodeling and the development of heart failure. Nevertheless, no approved immune-modulatory therapies for myocardial infarction are available to date, and quantification of the immune response in patients largely relies on measurements from peripheral blood. In this study, combined PET/MR imaging after suppression of physiological myocardial glucose uptake was used to visualize the 18F-FDG consumption within the heart five days after ST-elevation MI as a novel biosignal for post-ischemic myocardial inflammation. The observed 18F-FDG signal labeled the post-ischemic myocardial area, exceeded the infarct area and corresponded to the myocardial area-at-risk. Follow-up functional cardiac MRI studies revealed an inverse correlation between the 18F-FDG signal intensity after 5 days and cardiac function after 6 months (assessed by change of EDV, ESV, and EF), independent of infarction size and with a stronger correlation. Thus, the signal obtained by this novel imaging strategy presumably visualizes migrated immune cells to the heart and appears to be of prognostic significance. This imaging approach may be useful for the identification of patients at risk for the development of post-MI heart failure and may be ideal for patient selection and monitoring of future immune-modulatory therapies.
Acknowledgments
We gratefully acknowledge the excellent technical assistance of Sylvia Schachoff, Anna Winter and Claudia Meisinger, logistical support of Gitti Dzewas and the radiochemistry team headed by Michael Herz. We would also like to thank Petra Hoppmann und Robert Byrne for their help in patient recruitment and Alexander Goedel and Daniel Sinnecker for helpful discussions.
Sources of Funding
This study was supported by the German Research Foundation (DFG: initiative on major instrumentation (funding for the installation of the PET/MR scanner); Research Unit 923, La 1238 3-1/4-1 to K.-L.L.)), by the European Research Council (ERC Grant MUMI, ERC-2011-ADG_20110310 to M.S. and ERC 261053 to K.-L.L.), the DZHK (German Centre for Cardiovascular Research), partner site Munich Heart Alliance, and the National Institutes of Health (NIH grant R01 HL096576 to M.N.). K.G. was funded by the German Research Foundation (DFG: FOR 2033 GO 713/2-1 and SFB 1243) and the DJCLS R 14/16.
Abbreviations and acronyms
- AMI
acute myocardial infarction
- 18F-FDG
18-fluorodeoxyglucose
- LGE
late gadolinium enhancement
- MRI
magnetic resonance imaging
- PCI
percutaneous coronary intervention
- PET
positron emission tomography
- SPECT
single photon emission computed tomography
Footnotes
Disclosures
None.
References
- 1.Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D’Agostino RB, Levy D, Kannel WB, Vasan RS. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–62. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hellermann JP, Jacobsen SJ, Redfield MM, Reeder GS, Weston SA, Roger VL. Heart failure after myocardial infarction: clinical presentation and survival. European journal of heart failure. 2005;7:119–25. doi: 10.1016/j.ejheart.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nature reviews Cardiology. 2014;11:255–65. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Laan AM, Hirsch A, Robbers LF, Nijveldt R, Lommerse I, Delewi R, van der Vleuten PA, Biemond BJ, Zwaginga JJ, van der Giessen WJ, Zijlstra F, van Rossum AC, Voermans C, van der Schoot CE, Piek JJ. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST-segment elevation myocardial infarction: monocytes and myocardial infarction. Am Heart J. 2012;163:57–65. e2. doi: 10.1016/j.ahj.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, Grosse-Dunker G, Heisel I, Hornof F, Jeken J, Rebling NM, Ulrich C, Scheller B, Bohm M, Fliser D, Heine GH. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. Journal of the American College of Cardiology. 2012;60:1512–20. doi: 10.1016/j.jacc.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 6.Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, Kitabata H, Okochi K, Arita Y, Ishibashi K, Komukai K, Kataiwa H, Nakamura N, Hirata K, Tanaka A, Akasaka T. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. Journal of the American College of Cardiology. 2009;54:130–8. doi: 10.1016/j.jacc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 7.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Lahn AM, Swirski FK, Weissleder R, Nahrendorf M. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circulation research. 2014;115:284–95. doi: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol. 2010;134:33–46. doi: 10.1016/j.clim.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009;206:2141–9. doi: 10.1084/jem.20090866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–6. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love C, Tomas MB, Tronco GG, Palestro CJ. FDG PET of infection and inflammation. Radiographics: a review publication of the Radiological Society of North America, Inc. 2005;25:1357–68. doi: 10.1148/rg.255045122. [DOI] [PubMed] [Google Scholar]
- 13.Tarkin JM, Joshi FR, Rudd JH. PET imaging of inflammation in atherosclerosis. Nature reviews Cardiology. 2014;11:443–57. doi: 10.1038/nrcardio.2014.80. [DOI] [PubMed] [Google Scholar]
- 14.Lee WW, Marinelli B, van der Laan AM, Sena BF, Gorbatov R, Leuschner F, Dutta P, Iwamoto Y, Ueno T, Begieneman MP, Niessen HW, Piek JJ, Vinegoni C, Pittet MJ, Swirski FK, Tawakol A, Di Carli M, Weissleder R, Nahrendorf M. PET/MRI of inflammation in myocardial infarction. Journal of the American College of Cardiology. 2012;59:153–63. doi: 10.1016/j.jacc.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogers IS, Nasir K, Figueroa AL, Cury RC, Hoffmann U, Vermylen DA, Brady TJ, Tawakol A. Feasibility of FDG imaging of the coronary arteries: comparison between acute coronary syndrome and stable angina. JACC Cardiovasc Imaging. 2010;3:388–97. doi: 10.1016/j.jcmg.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Ishimaru S, Tsujino I, Takei T, Tsukamoto E, Sakaue S, Kamigaki M, Ito N, Ohira H, Ikeda D, Tamaki N, Nishimura M. Focal uptake on 18F-fluoro-2-deoxyglucose positron emission tomography images indicates cardiac involvement of sarcoidosis. European heart journal. 2005;26:1538–43. doi: 10.1093/eurheartj/ehi180. [DOI] [PubMed] [Google Scholar]
- 17.Rischpler C, Nekolla SG, Dregely I, Schwaiger M. Hybrid PET/MR imaging of the heart: potential, initial experiences, and future prospects. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2013;54:402–15. doi: 10.2967/jnumed.112.105353. [DOI] [PubMed] [Google Scholar]
- 18.Wollenweber T, Roentgen P, Schafer A, Schatka I, Zwadlo C, Brunkhorst T, Berding G, Bauersachs J, Bengel FM. Characterizing the inflammatory tissue response to acute myocardial infarction by clinical multimodality noninvasive imaging. Circulation Cardiovascular imaging. 2014;7:811–8. doi: 10.1161/CIRCIMAGING.114.001689. [DOI] [PubMed] [Google Scholar]
- 19.Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, Verani MS, American Heart Association Writing Group on Myocardial S and Registration for Cardiac I Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation. 2002;105:539–42. doi: 10.1161/hc0402.102975. [DOI] [PubMed] [Google Scholar]
- 20.Metz S, Ganter C, Lorenzen S, van Marwick S, Herrmann K, Lordick F, Nekolla SG, Rummeny EJ, Wester HJ, Brix G, Schwaiger M, Beer AJ. Phenotyping of tumor biology in patients by multimodality multiparametric imaging: relationship of microcirculation, alphavbeta3 expression, and glucose metabolism. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2010;51:1691–8. doi: 10.2967/jnumed.110.077719. [DOI] [PubMed] [Google Scholar]
- 21.Reeps C, Bundschuh RA, Pellisek J, Herz M, van Marwick S, Schwaiger M, Eckstein HH, Nekolla SG, Essler M. Quantitative assessment of glucose metabolism in the vessel wall of abdominal aortic aneurysms: correlation with histology and role of partial volume correction. The international journal of cardiovascular imaging. 2013;29:505–12. doi: 10.1007/s10554-012-0090-9. [DOI] [PubMed] [Google Scholar]
- 22.Kim RJ, Wu E, Rafael A, Chen EL, Parker MA, Simonetti O, Klocke FJ, Bonow RO, Judd RM. The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. The New England journal of medicine. 2000;343:1445–53. doi: 10.1056/NEJM200011163432003. [DOI] [PubMed] [Google Scholar]
- 23.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2009;50(Suppl 1):122S–50S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marinelli M, Martinez-Moller A, Jensen B, Positano V, Weismuller S, Navab N, Landini L, Schwaiger M, Nekolla SG. Registration of myocardial PET and SPECT for viability assessment using mutual information. Medical physics. 2010;37:2414–24. doi: 10.1118/1.3395554. [DOI] [PubMed] [Google Scholar]
- 25.Castellani M, Colombo A, Giordano R, Pusineri E, Canzi C, Longari V, Piccaluga E, Palatresi S, Dellavedova L, Soligo D, Rebulla P, Gerundini P. The role of PET with 13N-ammonia and 18F-FDG in the assessment of myocardial perfusion and metabolism in patients with recent AMI and intracoronary stem cell injection. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2010;51:1908–16. doi: 10.2967/jnumed.110.078469. [DOI] [PubMed] [Google Scholar]
- 26.Kastrati A, Schomig A, Dirschinger J, Mehilli J, Dotzer F, von Welser N, Neumann FJ. A randomized trial comparing stenting with balloon angioplasty in small vessels in patients with symptomatic coronary artery disease. ISAR-SMART Study Investigators. Intracoronary Stenting or Angioplasty for Restenosis Reduction in Small Arteries. Circulation. 2000;102:2593–8. doi: 10.1161/01.cir.102.21.2593. [DOI] [PubMed] [Google Scholar]
- 27.Shantsila E, Wrigley B, Tapp L, Apostolakis S, Montoro-Garcia S, Drayson MT, Lip GY. Immunophenotypic characterization of human monocyte subsets: possible implications for cardiovascular disease pathophysiology. Journal of thrombosis and haemostasis: JTH. 2011;9:1056–66. doi: 10.1111/j.1538-7836.2011.04244.x. [DOI] [PubMed] [Google Scholar]
- 28.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R, Nahrendorf M. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. Journal of the American College of Cardiology. 2010;55:1629–38. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. Journal of the American College of Cardiology. 2014;63:1593–603. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Deftereos S, Giannopoulos G, Angelidis C, Alexopoulos N, Filippatos G, Papoutsidakis N, Sianos G, Goudevenos J, Alexopoulos D, Pyrgakis V, Cleman MW, Manolis AS, Tousoulis D, Lekakis J. Anti-Inflammatory Treatment With Colchicine in Acute Myocardial Infarction: A Pilot Study. Circulation. 2015;132:1395–403. doi: 10.1161/CIRCULATIONAHA.115.017611. [DOI] [PubMed] [Google Scholar]
- 32.Camici P, Araujo LI, Spinks T, Lammertsma AA, Kaski JC, Shea MJ, Selwyn AP, Jones T, Maseri A. Increased uptake of 18F-fluorodeoxyglucose in postischemic myocardium of patients with exercise-induced angina. Circulation. 1986;74:81–8. doi: 10.1161/01.cir.74.1.81. [DOI] [PubMed] [Google Scholar]
- 33.Wijns W, Melin JA, Leners N, Ferrant A, Keyeux A, Rahier J, Cogneau M, Michel C, Bol A, Robert A, Pouleur H, Charlier A, Beckers C. Accumulation of polymorphonuclear leukocytes in reperfused ischemic canine myocardium: relation with tissue viability assessed by fluorine-18-2-deoxyglucose uptake. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 1988;29:1826–32. [PubMed] [Google Scholar]
- 34.Schwaiger M, Schelbert HR, Ellison D, Hansen H, Yeatman L, Vinten-Johansen J, Selin C, Barrio J, Phelps ME. Sustained regional abnormalities in cardiac metabolism after transient ischemia in the chronic dog model. Journal of the American College of Cardiology. 1985;6:336–47. doi: 10.1016/s0735-1097(85)80169-8. [DOI] [PubMed] [Google Scholar]
- 35.Egert S, Nguyen N, Schwaiger M. Myocardial glucose transporter GLUT1: translocation induced by insulin and ischemia. Journal of molecular and cellular cardiology. 1999;31:1337–44. doi: 10.1006/jmcc.1999.0965. [DOI] [PubMed] [Google Scholar]
- 36.Taegtmeyer H, Dilsizian V. Imaging myocardial metabolism and ischemic memory. Nature clinical practice Cardiovascular medicine. 2008;5(Suppl 2):S42–8. doi: 10.1038/ncpcardio1186. [DOI] [PubMed] [Google Scholar]
- 37.Heusch P, Buchbender C, Beiderwellen K, Nensa F, Hartung-Knemeyer V, Lauenstein TC, Bockisch A, Forsting M, Antoch G, Heusner TA. Standardized uptake values for [(1)(8)F] FDG in normal organ tissues: comparison of whole-body PET/CT and PET/MRI. Eur J Radiol. 2013;82:870–6. doi: 10.1016/j.ejrad.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Deichen JT, Prante O, Gack M, Schmiedehausen K, Kuwert T. Uptake of [18F]fluorodeoxyglucose in human monocyte-macrophages in vitro. Eur J Nucl Med Mol Imaging. 2003;30:267–73. doi: 10.1007/s00259-002-1018-8. [DOI] [PubMed] [Google Scholar]
- 39.Paik JY, Lee KH, Choe YS, Choi Y, Kim BT. Augmented 18F-FDG uptake in activated monocytes occurs during the priming process and involves tyrosine kinases and protein kinase C. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2004;45:124–8. [PubMed] [Google Scholar]
- 40.Folco EJ, Sheikine Y, Rocha VZ, Christen T, Shvartz E, Sukhova GK, Di Carli MF, Libby P. Hypoxia but not inflammation augments glucose uptake in human macrophages: Implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. Journal of the American College of Cardiology. 2011;58:603–14. doi: 10.1016/j.jacc.2011.03.044. [DOI] [PubMed] [Google Scholar]
- 41.van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsch A, Lavaei M, Nahrendorf M, Horrevoets AJ, Niessen HW, Piek JJ. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. European heart journal. 2014;35:376–85. doi: 10.1093/eurheartj/eht331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dutta P, Sager HB, Stengel KR, Naxerova K, Courties G, Saez B, Silberstein L, Heidt T, Sebas M, Sun Y, Wojtkiewicz G, Feruglio PF, King K, Baker JN, van der Laan AM, Borodovsky A, Fitzgerald K, Hulsmans M, Hoyer F, Iwamoto Y, Vinegoni C, Brown D, Di Carli M, Libby P, Hiebert SW, Scadden DT, Swirski FK, Weissleder R, Nahrendorf M. Myocardial Infarction Activates CCR2(+) Hematopoietic Stem and Progenitor Cells. Cell Stem Cell. 2015;16:477–87. doi: 10.1016/j.stem.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circulation research. 2014;114:1611–22. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ibrahim T, Hackl T, Nekolla SG, Breuer M, Feldmair M, Schomig A, Schwaiger M. Acute myocardial infarction: serial cardiac MR imaging shows a decrease in delayed enhancement of the myocardium during the 1st week after reperfusion. Radiology. 2010;254:88–97. doi: 10.1148/radiol.09090660. [DOI] [PubMed] [Google Scholar]
- 45.Kim EJ, Kim S, Kang DO, Seo HS. Metabolic activity of the spleen and bone marrow in patients with acute myocardial infarction evaluated by 18f-fluorodeoxyglucose positron emission tomograpic imaging. Circulation Cardiovascular imaging. 2014;7:454–60. doi: 10.1161/CIRCIMAGING.113.001093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.