

Abstract
Optogenetics refers to the ability to control cells that have been genetically modified to express light-sensitive ion channels. The introduction of optogenetic approaches has facilitated the dissection of neural circuits. Optogenetics allows for the precise stimulation and inhibition of specific sets of neurons and their projections with fine temporal specificity. These techniques are ideally suited to investigating neural circuitry underlying motor and cognitive dysfunction in animal models of human disease. Here, we focus on how optogenetics has been used over the last decade to probe striatal circuits that are involved in Parkinson disease, a neurodegenerative condition involving motor and cognitive abnormalities resulting from degeneration of midbrain dopaminergic neurons. The precise mechanisms underlying the striatal contribution to both cognitive and motor dysfunction in Parkinson disease are unknown. Although optogenetic approaches are somewhat removed from clinical use, insight from these studies can help identify novel therapeutic targets and may inspire new treatments for Parkinson disease. Elucidating how neuronal and behavioral functions are influenced and potentially rescued by optogenetic manipulation in animal models could prove to be translatable to humans. These insights can be used to guide future brain-stimulation approaches for motor and cognitive abnormalities in Parkinson disease and other neuropsychiatric diseases.
Keywords: animal model, dopamine, optogenetics, Parkinson disease, striatum
Abstract
La optogenética se refiere a la capacidad de controlar células que han sido modificadas genéticamente para expresar canales iónicos sensibles a la luz. La introducción de las estrategias optogenéticas ha facilitado la disección de los circuitos neurales. La optogenética permite precisar la estimulación e inhibición de conjuntos específicos de neuronas y sus proyecciones con una alta especificidad temporal. Estas técnicas idealmente están adaptadas para investigar los circuitos neurales que subyacen a la disfunción motora y cognitiva en modelos animales de la enfermedad humana. Este artículo se enfoca en cómo se ha empleado la optogenética durante la última década para explorar los circuitos neurales que están involucrados en la Enfermedad de Parkinson, una condición neurodegenerativa que incluye alteraciones motoras y cognitivas resultantes de la degeneración de neuronas dopaminérgicas del mesencéfalo. Aunque las estrategias optogenéticas están algo alejadas del empleo clínico, el conocimiento a partir de estos estudios puede ayudar a identificar nuevos blancos terapéuticos y puede inspirar nuevos tratamientos para la Enfermedad de Parkinson. El esclarecer cómo las mediciones neurales y conductuales son influenciadas y potencialmente recuperadas por la manipulación optogenética podría llegar a ser traducible a los humanos. Estos conocimientos pueden ser empleados para guiar futuras estrategias de estimulación cerebral para anormalidades motoras y cognitivas en la Enfermedad de Parkinson y otras enfermedades neuropsiquiátricas.
Abstract
L'optogénétique est une méthode permettant de contrôler des cellules qui ont été préalablement génétiquement modifiées pour exprimer des canaux ioniques sensibles à la lumière. Son utilisation a ouvert la voie à l'analyse des circuits neuronaux car elle permet la stimulation et l'inhibition précises de groupes spécifiques de neurones et de leurs projections avec une excellente spécificité temporale. Ces techniques sont parfaitement adaptées à l'examen des circuits neuronaux sous-tendant une dysfonction motrice et cognitive dans des modèles animaux de pathologies humaines. Cet article met l'accent sur la façon dont l'optogénétique a été utilisée ces 10 dernières années pour examiner les circuits striataux impliqués dans la maladie de Parkinson, une maladie neurodégénérative dont les troubles moteurs et cognitifs résultent d'une dégénérescence des neurones dopaminergiques du mésencéphale. Les mécanismes précis sous-tendant la contribution du striatum au dysfonctionnement moteur et cognitif de la maladie de Parkinson sont encore méconnus. Bien que l'optogénétique soit quelque peu éloignée de l'usage clinique, les connaissances issues de ces études peuvent aider à identifier de nouvelles cibles thérapeutiques et suggérer de nouveaux traitements pour la maladie de Parkinson. Une fois élucidés, les mécanismes par lesquels les manipulations optogénétiques peuvent influencer et potentiellement restaurer les fonctions neuronales et comportementales pourraient être transposés chez l'homme. Ces connaissances pourraient alors être utilisées pour mener de futures stratégies de stimulation cérébrale dans les anomalies motrices et cognitives de la maladie de Parkinson et d'autres maladies neuropsychiatriques.
Introduction
The advent of optogenetic technologies has advanced the field of neuroscience by allowing the manipulation of specific neuronal populations with millisecond resolution using light. Optogenetics allows researchers to stimulate or inhibit brain cells defined by a specific promoter and to specifically target a particular brain region. Opsins, or light-sensitive proteins, are genetically expressed in the neurons of a model organism and can then be activated with ~1-ms temporally precise delivery of specific wavelengths of light Figure 1.1,2 Optogenetic viruses, constructs, and equipment are now readily available, making this technology accessible for various applications.
Figure 1. Optogenetic tools for modulating membrane voltage potential. Stimulating the neurons expressing the nonselective cation channel Channelrhodopsin-2 (ChR2) using blue light immediately depolarizes the neuron and triggers an action potential. Sometimes it is desirable to inhibit neuronal signaling instead of triggering it. Light stimulation of halorhodopsin (NpHR), a chloride pump, hyperpolarizes neurons and inhibits spikes in response to yellow light. Recent variants (named eNpHR2.0 and eNpHR3.0) exhibit improved membrane targeting in mammalian cells and consequently, photocurrents. Light-driven proton pumps such as archaerhodopsin-3 (Arch), Mac, bacteriorhodopsin (eBR), and rhodopsin-3 (GtR3) can also be used to hyperpolarize neurons and block signaling. Ca2+, calcium; ChETA, channelrhodopsin-2 mutant E12ET; mV, millivolts; Na+, sodium; nm, nanometer; SFO, step-function opsin; VChR1, Volvox-derived channelrhodopsin-1. Reproduced from reference 2: Pastrana E. Optogenetics: controlling cell function with light. Nat Methods. 2011;8(1):24-25. Copyright © Nature America, Inc. 2011.
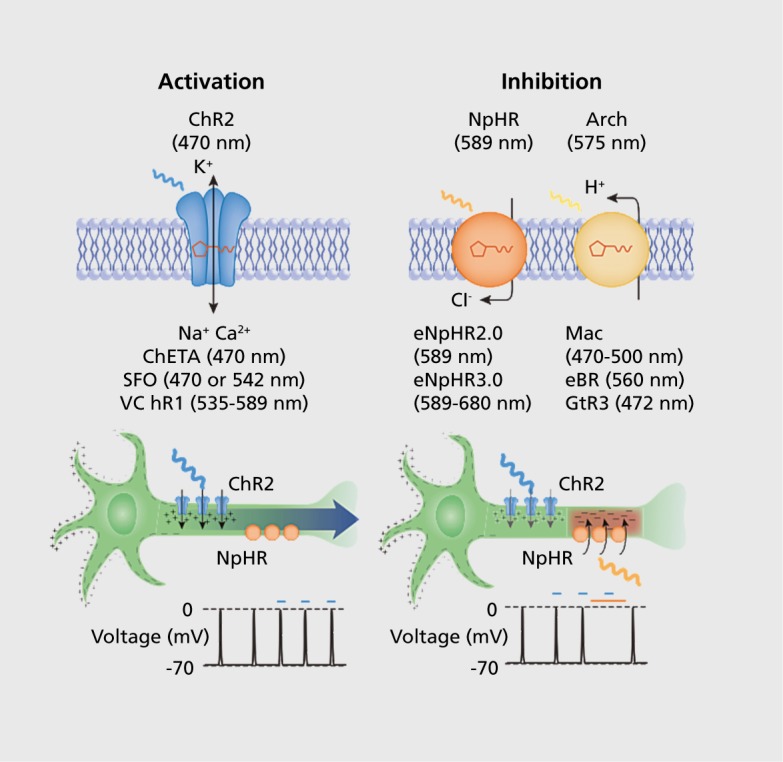
The first light-sensitive protein was isolated in the 1970s from Halobacterium halobium, though it took many years for its bioengineering potential to be realized.1,3 In 2003, initial work was published by Ernst Bamberg, describing the use of Channelrhodopsin-2 (ChR2) to drive neuronal activity with light.4 In a 2005 paper, Boyden et al from Karl Deisseroth's laboratory further characterized ChR2's potential for fast, precise, and dynamic stimulation of neurons and made significant improvements in the ease of genetic expression and efficacy.5 Furthermore, optogenetic stimulation has almost no measurable side effects for the host tissue, proving to be minimally invasive for use in vivo in mammals.6 Optogenetic proteins have also been engineered for the targeted inhibition of neuronal populations (Figure).The two primary inhibitory opsins are NpHR, a halorhodopsin, and Arch, an archaerhodopsin.7,5 NpHR, a chloride pump, requires constant light to move through its photocycle and has slower dynamics than Arch, a proton pump, which has more potent inactivation effects.7,9,10
Genetic technology facilitates cell-type specificity for targeting optogenetic proteins to individual classes of neurons. Lentiviruses or the more commonly used adeno-associated viral (AAV) vectors can be used to express a given construct in all neurons, excitatory pyramidal neurons, inhibitory interneurons, astrocytes, or oligodendrocytes, depending on the genetic promoter used.6,11-14 The use of transgenic Cre-recombinase mouse lines affords definitive cell-type specificity. In combination with Cre-dependent AAV vectors or transgenic mice, robust expression can be attained in a cell type restricted by a chosen Cre-driving promoter.15 This technique allows researchers to use various existing transgenic Cre mouse lines and to ask precise, directed questions that can be used to gather information about the actions of neuronal subclasses.16,17 Another advantage presented by optogenetic technologies is the ability to target not only a specific cellular class, but also modulate the projections of that cell type to a structure of interest. Optical cannulae can be implanted in downstream structures receiving projections from the injection location as a way to target only efferent projections Figure 2.1-20
Figure 2. Targeting optogenetic tools in vivo. (Top) Direct stimulation of neuronal cell bodies is achieved by injecting virus at the target region and then implanting a light-delivery device above the injected region. Even this simple experiment can provide specificity with viruses that will not transduce afferent axons and fibers of passage. Additional cell-type specificity is attained either by cell-type-specific promoters in the viral vector or via a recombinase-dependent virus, injected in a transgenic animal expressing a recombinase such as Cre in specific cells, leading to specific expression of the transgene only in defined cell types. (Bottom) Projection (axonal) targeting is achieved by viral injection at the region harboring cell bodies, followed by implantation of a light-delivery device above the target region containing neuronal processes from the virally transduced region; in this way, cell types are targeted by virtue of their projections. Reproduced from reference 19: Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13(4):251-266. Copyright © Nature Publishing Group, 2012. Originally published in reference 20: Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71(1):9-34. Copyright © Elsevier, Inc. 2011.

Because of their versatility, the various optogenetic proteins and stimulation techniques present a rich tool-box for a multitude of research questions. For comprehensive reviews on the use of optogenetics for a wide range of applications, see Yizhar et al,20 Zhang et al,21 and Deisseroth.1 Manipulating neuronal activity with the high level of temporal and spatial precision, cell type specificity, and control that optogenetics allows has led to major advancements in our understanding of basic neural circuitry. Additionally, pairing these methods with multichannel and multisite neuronal recordings has the potential to reveal how these circuits are disrupted in neuro psychiatric diseases such as Parkinson disease (PD).
Parkinson disease neural circuitry and current treatments
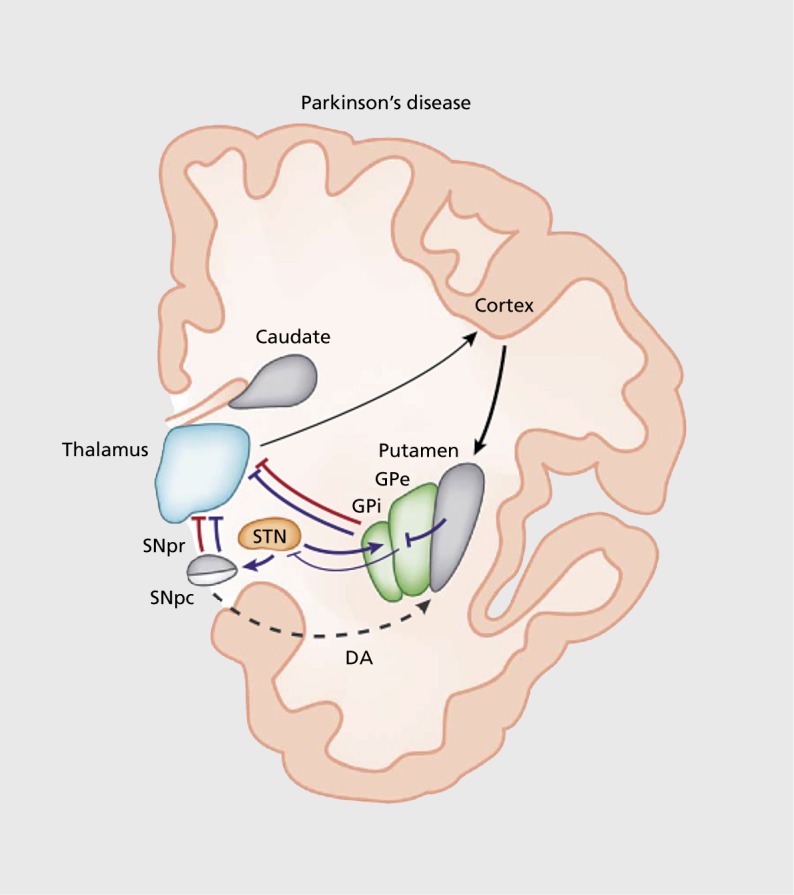
PD is a neurodegenerative disorder characterized by the gradual death of midbrain dopaminergic neurons in the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA).22-24 The cardinal symptoms of PD in humans are resting tremor, muscle rigidity, and akinesia (difficulty in initiating movements)—of note, PD can also lead to nonmotor symptoms.25,26 PD symptoms result from striatal dysfunction. The striatum integrates projections from the cortex and thalamus to promote action selection Figure 3.27-29 It mainly consists of medium spiny neurons (MSNs) expressing two main types of dopamine receptors: D1-type and D2-type.30>-33 These populations are expressed in two main output pathways: D1 in the striatonigral “direct” pathway, and D2 in the striatopallidal or “indirect” pathway.31,32 The striatonigral pathway directly inhibits the globus pallidus interna (Gpi) and substantia nigra pars reticulata (SNr). The striatopallidal pathway indirectly excites the GPi/SNr via disinhibition (Figure 3).29,34 The striatonigral (direct) pathway has been speculated to promote motor actions, whereas the striatopallidal (indirect) pathway suppresses actions. Imbalances between neural activities in the direct and indirect pathways in the basal ganglia caused by a loss of dopaminergic input result in profound motor deficits among patients with PD.35,36
Figure 3. In Parkinson disease, dopamine arising from the substantia nigra pars compacta is thought to activate D1-expressing striatal medium spiny neurons of the direct pathway (red lines) and to inhibit D2-expressing striatal neurons of the indirect pathway (blue lines). The output nuclei globus pallidus interna and substantia nigra pars reticulata project to the thalamus, which in turn sends efferents that complete the cortico-basal ganglia-thalamo-cortical loop. In Parkinson disease, degeneration of nigral neurons reduces dopamine-receptor stimulation in striatal medium spiny neurons. The imbalance between direct and indirect pathways results in abnormal activation of output nuclei and overinhibition of thalamic neurons projecting to the cortex. DA, dopamine; GPe, globus pallidus externa; GPi, globus pallidus interna; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus. Reproduced from reference 29: Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17(8):1022-1030. Copyright © Nature Publishing Group, 2014.
Optogenetics to probe striatal circuits and motor symptoms in animal models of PD
In animal models of PD, optogenetic approaches have been applied to answer a variety of fundamental research questions. There are two primary toxin-based animal models of PD, both of which are induced by toxic pharmacological lesion. The first involves a 6-hydroxydopamine (6-OHDA) injection into the substantia nigra, medial forebrain bundle (MFB), or striatum. The second requires repeatedly administering 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intraperitoneally.37-40 Although 6-OHDA and MPTP animal models do not replicate the progressive loss of dopaminergic neurons, they model catecholamine dysfunction in PD. Other models involve α-synuclein overexpression or mutations; to our knowledge, optogenetics has not been explored in these models.41-43
Optogenetics has provided details of striatal MSNs, interneurons, and afferents that could have implications for PD and other neuropsychiatric diseases (for complete review, see ref 44). As a result of the efficacy of subthalamic nucleus (STN) deep brain stimulation (DBS) on the motor symptoms of PD, Gradinaru et al12 investigated STN optogenetic inhibition and excitation as a method to mimic the effects of DBS in animals with 6-OHDA injections in the MFB Figure 4 12,19,45 While optogenetic excitation and inhibition of STN neurons had no effect, high-frequency stimulation of afferent fibers projecting from the motor cortex to the STN ameliorated motor symptoms. It is important to note that infusions of muscimol and lidocaine in the STN have been demonstrated to relieve some PD symptoms in monkeys treated with MPTP46 Changes in STN activity following stimulation could influence downstream structures, including the striatum. These results illustrate how optogenetics can be used to precisely define neural circuitry and describe the influence of stimulation and inhibition on behavioral measures.
Figure 4. A schematic diagram (top panel) shows key neural projections that are involved in parkinsonian behavior and treatment. Data in the bottom left panel are from a study that used a constitutively expressing ChR2 mouse line (Thy1::ChR2) to identify a mechanistic explanation for the therapeutic effects of deep brain stimulation. By illuminating and recording in the subthalamic nucleus, this paper showed that afferent fibers entering the subthalamic nucleus, rather than local cell bodies themselves, are likely to be the direct target of deep brain stimulation in the correction of parkinsonian motor activity. High-frequency stimulation of the afferent fibers into subthalamic nucleus potently silenced the structure as shown and reversibly abolished the parkinsonian symptoms. By contrast, low-frequency stimulation of the afferents simply added spikes on top of endogenous spikes and worsened parkinsonian symptoms.12 Data in the bottom right panel are from a study that used a Cre-AAV to selectively express ChR2 in either D1 dopamine receptor (D1R)::Cre or D2 dopamine receptor (D2R)::Cre mice to examine the differential contributions of the direct and indirect pathways with respect to motor output. Activation of D1R-expressing neurons silenced local basal ganglia activity and increased ambulation, whereas activation of D2R-expressing neurons increased this activity and enhanced immobile or bradykinetic (slow) behavior.45 Black bars indicate the duration of illumination. AAV, adeno-associated viral; ChR2, Channelrhodopsin-2; HFS, high-frequency stimulation; LFS, low-frequency stimulation; M1, primary motor cortex; μV, microvolts; D1, D1-type dopamine receptor; D2, D2-type dopamine receptor; GABA, γ-aminobutyric acid; GP, globus pallidus; Hz, hertz; ms, milliseconds; s, seconds; SNr, substantia nigra pars reticulata; STN, subthalamic nuclus. Reproduced from reference 19: Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13(4):251-266. © 2012, Nature Publishing Group. Bottom left image group originally published in reference 12: Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324(5925):354-359. © 2009, American Association for the Advancement of Science. Bottom right image group originally published in reference 45: Kravitz AV, Freeze BS, Parker PRL, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010; 466(7306): 622-626. Copyright © Nature Publishing Group, 2010.

As previously described, it is thought that hyperactivity in the indirect pathway and insufficient activity in the direct pathways contribute to the motor symptoms of PD.35 To investigate this hypothesis, Kravitz et al45 used optogenetics to selectively probe these pathways in mice expressing D1-Cre or D2-Cre recombinasedependent ChR2 in MSNs. They found that bilateral stimulation of the D2 neurons of the indirect pathway induced freezing and bradykinesia and decreased locomotion, whereas stimulation of the D1 neurons of the direct pathway reduced freezing and increased locomotion. Additionally, they reported that optogenetic stimulation of the direct pathway rescued motor abnormalities induced by 6-OHDA lesions in the MFB (Figure 4). These data indicate the importance of the opposing influences of the direct and indirect pathways and indicate potential therapeutic strategies to ameliorate the motor deficits associated with PD.45
Optogenetics has also been used to probe the mechanisms underlying the efficacy of grafting human pluripotent stem cell-derived dopaminergic neurons in disease models. Animals with striatal 6-OHDA lesions exhibit motor impairments that are robustly reversed following engraftment of dopaminergic neurons in the lesioned area.47 To probe the mechanisms underlying the efficacy of this effect, Steinbeck et al48 optogenetically inhibited engrafted dopamine stem cells after motor recovery. They report that inhibiting dopamine neurons resulted in the reappearance of motor deficits, indicating the essential role of dopaminergic neurons at the lesion site.
The studies described here provide evidence for the importance of optogenetic exploration of striatal function in animal models of PD. These results could inform the design of novel drug- and stimulation-based therapies to rescue dopaminergic dysfunction and motor symptoms in PD.
Optogenics to probe neural circuitry and cognitive dysfunction in animal models of PD
In addition to the characteristic motor symptoms, non-motor symptoms are also present in PD including depression, psychosis, and cognitive dysfunction.49 Cognitive impairment is a serious component of PD for many patients. Though initially controversial, cognitive deficits are now an integral part of PD symptomatology. These cognitive symptoms can be diverse, including visuospatial dysfunction, working-memory deficits, and executive dysfunction.50 Despite its prevalence, the underlying basis of cognitive dysfunction in PD is poorly understood. Whereas treatments exist that successfully alleviate motor symptoms of PD, their cognitive benefits are not established.51-55
Cognitive dysfunction in PD patients may involve the prefrontal cortex, a key input structure to the striatum.56-61 Frontostriatal circuits are the primary neural pathways responsible for cognitive dysfunction in PD. The frontal cortex receives dopaminergic projections from dopaminergic neurons in the VTA. Dopaminergic neurons in the VTA also die during PD and therefore could play a role in disease-related cognitive abnormalities.22 Given the critical role of the dopaminergic system in the development of PD, its significance in cognitive symptoms of PD has also been studied.62,63
As described earlier in this review, animal models are a critical component of PD research. However, the relevance of animal models is less obvious when dealing with complicated internal processes like “cognitive dysfunction.” When trying to approach cognitive deficits of PD in an animal model, it is essential to find a behavioral assay that is simple, relies on the structures involved in the disease, and is translatable to human patients. Temporal processing tasks meet these criteria. Interval-timing tasks require subjects to make a motor estimation of the passage of an interval of time. Interval timing can be studied in both rodents and humans.64,65 Tasks that rely on time estimation are known to rely on dopaminergic systems.66-70 Our group has shown that interval-timing tasks are impaired in PD and in rodents administered 6-OHDA injections in the VTA or in the frontal cortex.71 Specifically, we found that disrupting dopamine synthesis also impaired interval timing. We used optogenetics to specifically implicate prefrontal D1 dopamine receptors and demonstrated that stimulating prefrontal D1 neurons could enhance performance of timing tasks.72 Additionally, PD patients and rodent PD models had diminished frontal, low-frequency, cue-related activity and single-neuron ramping activity, indicating conserved mechanisms for timing and cognitive function.73
Optogenetics provides a novel way to probe frontostriatal interactions in animals performing the interval-timing task and can be tailored specifically to D1 dopamine receptors using Cre-dependent expression of ChR2 or other opsins. These techniques can be used in animal models of PD with depleted D1 dopamine to try to ameliorate aberrant frontal and striatal neuronal activity and rescue performance on behavioral tasks. The same techniques could be used to probe cognitive flexibility and frontostriatal circuitry in other paradigms including reversal learning, attentional set-shifting, and task switching. These tasks are impaired in PD and can all be explored in animal models.74 Elucidating the neural mechanisms of cognitive impairment in neuropsychiatric disease may identify novel sites for DBS or transcranial magnetic stimulation in PD, schizophrenia, and other neuropsychiatric diseases that share dysfunctional D1-dopamine and striatal abnormalities. 75>-78 Results from these proposed studies have the potential to illuminate the neural mechanisms of cognitive impairment and help identify novel biomarkers and novel therapeutic targets for the D1 dopamine system.
Future directions
This review summarizes the powerful contribution that optogenetics has made to our understanding of striatal circuitry, abnormalities in PD, and the potential for striatal modulation as a novel therapeutic target in PD. Yet, it is clear that if we aim to inspire new treatments for PD, there is a great need for further optogenetic exploration of striatal circuitry and function in animal models. Looking forward, optogenetics can be used to pave the way for emerging technologies to adaptively stimulate brain circuitry in diseases of impaired motor and cognitive function. If we can map specific neuronal abnormalities and show that optogenetic stimulation or inhibition successfully rescues motor and/or cognitive impairments in animal models of PD, an online, closed-loop design could be used to stimulate aberrant circuits. In patients with PD, cognitive and motor function could be restored using concomitant electroencephalography to detect neural abnormalities and deep brain or transcranial stimulation to adaptively rescue specific patterns of neuronal activity.79
Selected abbreviations and acronyms
- ChR2
Channelrhodopsin-2
- DBS
deep brain stimulation
- MFB
medial forebrain bundle
- MSN
medium spiny neurons
- PD
Parkinson disease
- STN
subthalamic nucleus
- VTA
ventral tegmental area
Contributor Information
Krystal L. Parker, Department of Neurology, University of Iowa, Iowa City, Iowa, USA.
Youngcho Kim, Department of Neurology, University of Iowa, Iowa City, Iowa, USA.
Stephanie L. Alberico, Department of Neurology, University of Iowa, Iowa City, Iowa, USA.
Eric B. Emmons, Department of Neurology, University of Iowa, Iowa City, Iowa, USA.
Nandakumar S. Narayanan, Department of Neurology, University of Iowa, Iowa City, Iowa, USA.
REFERENCES
- 1.Deisseroth K. Optogenetics. Nat Methods. 2011;8(1):26–29. doi: 10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pastrana E. Optogenetics: controlling cell function with light. Nat Methods. 2011;8(1):24–25. [Google Scholar]
- 3.Oesterhelt D., Stoeckenius W. Functions of a new photoreceptor membrane. Proc Natl Acad Sci U S A. 1973;70(10):2853–2857. doi: 10.1073/pnas.70.10.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagel G., Szellas T., Huhn W., et al Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100(24):13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyden ES., Zhang F., Bamberg E., Nagel G., Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8(9):1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 6.Aravanis AM., Wang LP., Zhang F., et al An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng. 2001;4(3):S143. doi: 10.1088/1741-2560/4/3/S02. [DOI] [PubMed] [Google Scholar]
- 7.Chow BY., Han X., Dobry AS., et al High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463(7277):98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gradinaru V., Zhang F., Ramakrishnan C., et al Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141(1):154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okazaki A., Takagi S. An optogenetic application of proton pump ArchT to C. elegans cells. Neurosci Res. 2013;75(1):29–34. doi: 10.1016/j.neures.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Witten IB., Lin SC., Brodsky M., et al Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330(6011):1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diester I., Kaufman MT., Mogri M., et al An optogenetic toolbox designed for primates. Nat Neurosci. 2011;14(3):387–397. doi: 10.1038/nn.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gradinaru V., Mogri M., Thompson KR., Henderson JM., Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324(5925):354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawlor PA., Bland RJ., Mouravlev A., Young D., During MJ. Efficient gene delivery and selective transduction of glial cells in the mammalian brain by AAV serotypes isolated from nonhuman primates. Mol Ther. 2009;17(10):1692–1702. doi: 10.1038/mt.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathanson JL., Jappelli R., Scheeff ED., et al Short promoters in viral vectors drive selective expression in mammalian inhibitory neurons, but do not restrict activity to specific inhibitory cell-types. Front Neural Circuits. 2009;3:19. doi: 10.3389/neuro.04.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atasoy D., Aponte Y., Su HH., Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28(28):7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cardin JA. Dissecting local circuits in vivo: integrated optogenetic and electrophysiology approaches for exploring inhibitory regulation of cortical activity. J Physiol Paris. 2012;106(3-4):104–111. doi: 10.1016/j.jphysparis.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cardin JA., Carlen M., Meletis K., et al Targeted optogenetic stimulation and recording of neurons in vivo using cell-type-specific expression of Channelrhodopsin-2. Nat Protoc. 2010;5(2):247–254. doi: 10.1038/nprot.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hira R., Ohkubo F., Tanaka YR., et al In vivo optogenetic tracing of functional corticocortical connections between motor forelimb areas. Front Neural Circuits. 2013;7:55. doi: 10.3389/fncir.2013.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tye KM., Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13(4):251–266. doi: 10.1038/nrn3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yizhar O., Fenno LE., Davidson TJ., Mogri M., Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71(1):9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Zhang F., Aravanis AM., Adamantidis A., de Lecea L., Deisseroth K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8(8):577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]
- 22.Alberico SL., Cassell MD., Narayanan NS. The vulnerable ventral tegmental area in Parkinson's disease. Basal Ganglia. 2015;5(2-3):51–55. doi: 10.1016/j.baga.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Damier P., Hirsch EC., Agid Y., Graybiel AM. The substantia nigra of the human brain II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999;122(8):1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 24.Zold CL., Kasanetz F., Pomata PE., et al Striatal gating through up states and oscillations in the basal ganglia: implications for Parkinson's disease. J Physiol Paris. 2012;106(1-2):40–46. doi: 10.1016/j.jphysparis.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhuri KR., Healy DG., Schapira AH. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. 2006;5(3):235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- 26.Parker KL., Lamichhane D., Caetano MS., Narayanan NS. Executive dysfunction in Parkinson's disease and timing deficits. Front Integr Neurosci. 2013;7:75. doi: 10.3389/fnint.2013.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolam JP., Hanley JJ., Booth PA., Bevan MD. Synaptic organisation of the basal ganglia. J Anat. 2000;196(Pt 4):527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freemon FR. The synaptic organisation of the brain. JAMA. 1980;243(18):1850–1850. [Google Scholar]
- 29.Calabresi P., Picconi B., Tozzi A., Ghiglieri V., Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17(8):1022–1030. doi: 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- 30.Gerfen CR. Molecular effects of dopamine on striatal-projection pathways. Trends Neurosci. 2000;23:S64–S70. doi: 10.1016/s1471-1931(00)00019-7. [DOI] [PubMed] [Google Scholar]
- 31.Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- 32.Smith Y., Bevan MD., Shink E., Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998;86(2):353–387. doi: 10.1016/s0306-4522(98)00004-9. [DOI] [PubMed] [Google Scholar]
- 33.Surmeier DJ., Ding J., Day M., Wang Z., Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30(5):228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Obeso JA., Rodriguez-Oroz MC., Rodriguez M., et al Pathophysiology of the basal ganglia in Parkinson's disease. doi:http://dx.doi.org/10.1016/51471-1931(00)00028-8. Trends Neurosci. 2000;23(suppl 1):S8–S19. doi: 10.1016/s1471-1931(00)00028-8. [DOI] [PubMed] [Google Scholar]
- 35.Albin RL., Young AB., Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 36.Graybiel AM., Aosaki T., Flaherty AW., Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265(5180):1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- 37.Fahn S., Marsden CD. World Congress of Neurology Carnforth, Lanes, UK: Park Ridge, NJ, USA: Parthenon Pub Group: 1990. http://catalog.hathitrust.org/Record/002235224. The Assessment and Therapy of Parkinsonism: The Proceedings of a Symposium Held at the XlVth World Congress of Neurology, New Delhi, October 1989. [Google Scholar]
- 38.Kaakkola S., Teräväinen H. Animal models of parkinsonism. Pharmacol Toxicol. 1990;67(2):95–100. doi: 10.1111/j.1600-0773.1990.tb00792.x. [DOI] [PubMed] [Google Scholar]
- 39.Schober A. Classic toxin-induced animal models of Parkinson's disease: 6-OHD A and MPTP. Cell Tissue Res. 2004;318(1):21 5–224. doi: 10.1007/s00441-004-0938-y. [DOI] [PubMed] [Google Scholar]
- 40.Ungerstedt U., Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24(3):485–493. doi: 10.1016/0006-8993(70)90187-3. [DOI] [PubMed] [Google Scholar]
- 41.Chesselet MF., Fleming S., Mortazavi F., Meurers B. Strengths and limitations of genetic mouse models of Parkinson's disease. Parkinsonism Relat Disord. 2008;14(supp 12):S84–S87. doi: 10.1016/j.parkreldis.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee MK., Stirling W., Xu Y., et al Human α-synuclein-harboring familial Parkinson's disease-linked Ala- 53 ” Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA. 2002;99(13):8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Decressac M., Kadkhodaei B., Mattsson B., Laguna A., Perlmann T., Björklund A. α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci TransI Med. 2012;4(163):163ra156. doi: 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- 44.Lenz JD., Lobo MK. Optogenetic insights into striatal function and behavior. Behav Brain Res. 2013;255:44–54. doi: 10.1016/j.bbr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 45.Kravitz AV., Freeze BS., Parker PRL., et al Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466(7306):622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levy R., Lang AE., Dostrovsky JO., et al Lidocaine and muscimol microinjections in subthalamic nucleus reverse Parkinsonian symptoms. Brain. 2001;124(Pt 10):2105–2118. doi: 10.1093/brain/124.10.2105. [DOI] [PubMed] [Google Scholar]
- 47.Kriks S., Shim JW., Piao J., et al Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480(7378):547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steinbeck JA., Choi SJ., Mrejeru A., et al Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson's disease model. Nat Biotechnol. 2015;33(2):204–209. doi: 10.1038/nbt.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaudhuri KR., Schapira AH. Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8(5):464–474. doi: 10.1016/S1474-4422(09)70068-7. [DOI] [PubMed] [Google Scholar]
- 50.Dubois B., Pillon B. Cognitive deficits in Parkinson's disease. J Neurol. 1997;244(1):2–8. doi: 10.1007/pl00007725. [DOI] [PubMed] [Google Scholar]
- 51.Deuschl G., Schade-Brittinger C., Krack P., et al A randomized trial of deep-brain stimulation for Parkinson's disease. N Engl J Med. 2006;355(9):896–908. doi: 10.1056/NEJMoa060281. [DOI] [PubMed] [Google Scholar]
- 52.Morrison CE., Borod JC., Brin MF., Hälbig TD., Olanow CW. Effects of levodopa on cognitive functioning in moderate-to- severe Parkinson's disease (MSPD) J Neural Transm (Vienna). 2004;111(10-11):1333–1341. doi: 10.1007/s00702-004-0145-8. [DOI] [PubMed] [Google Scholar]
- 53.Pascual-Sedano B., Kulisevsky J., Barbanoj M., et al Levodopa and executive performance in Parkinson's disease: a randomized study. J Int Neuropsychol Soc. 2008;14(5):832–841. doi: 10.1017/S1355617708081010. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez-Oroz MC., Obeso JA., Lang AE., et al Bilateral deep brain stimulation in Parkinson's disease: a multicentre study with 4 years follow-up. Brain. 2005;128(10):2240–2249. doi: 10.1093/brain/awh571. [DOI] [PubMed] [Google Scholar]
- 55.Weaver FM., Follett K., Stern M., et al Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63–73. doi: 10.1001/jama.2008.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cools R., Stefanova E., Barker RA., Robbins TW., Owen AM. Dopaminergic modulation of high-level cognition in Parkinson's disease: the role of the prefrontal cortex revealed by PET. Brain. 2002;125(Pt 3):584–594. doi: 10.1093/brain/awf052. [DOI] [PubMed] [Google Scholar]
- 57.Gotham AM., Brown RG., Marsden CD. 'Frontal' cognitive function in patients with Parkinson's disease 'on' and 'off' levodopa. Brain. 1988;111(Pt 2):299–321. doi: 10.1093/brain/111.2.299. [DOI] [PubMed] [Google Scholar]
- 58.Lewis SJ., Dove A., Robbins TW., Barker RA., Owen AM. Cognitive impairments in early Parkinson's disease are accompanied by reductions in activity in frontostriatal neural circuitry. J Neurosci. 2003;23(15):6351–6356. doi: 10.1523/JNEUROSCI.23-15-06351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Narayanan NS., Rodnitzky RL., Uc EY. Prefrontal dopamine signaling and cognitive symptoms of Parkinson's disease. Rev Neurosci. 2013;24(3):267–278. doi: 10.1515/revneuro-2013-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Owen AM. Cognitive fysfunction in Parkinson's disease: The role of frontostriatal circuitry. Neuroscientist. 2004;10(6):525–537. doi: 10.1177/1073858404266776. [DOI] [PubMed] [Google Scholar]
- 61.Zgaljardic DJ., Borod JC., Foldi NS., et al An examination of executive dysfunction associated with frontostriatal circuitry in Parkinson's disease. J Clin Exp Neuropsychol. 2006;28(7):1127–1144. doi: 10.1080/13803390500246910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jokinen P., Karrasch M., Brück A., Johansson J., Bergman J., Rinne JO. Cognitive slowing in Parkinson's disease is related to frontostriatal dopaminergic dysfunction. J Neurol Sci. 2013;329(1-2):23–28. doi: 10.1016/j.jns.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 63.Sawamoto N., Piccini P., Hotton G., Pavese N., Thielemans K., Brooks DJ. Cognitive deficits and striato-frontal dopamine release in Parkinson's disease. Brain. 2008;131(5):1294–1302. doi: 10.1093/brain/awn054. [DOI] [PubMed] [Google Scholar]
- 64.Balci F., Papachristos EB., Gallistel CR., Brunner D., Gibson J., Shumyatsky GP. Interval timing in genetically modified mice: a simple paradigm. Genes Brain Behav. 2008;7(3):373–384. doi: 10.1111/j.1601-183X.2007.00348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rakitin BC., Gibbon J., Penney TB., Malapani C., Hinton SC., Meek WH. Scalar expectancy theory and peak-interval timing in humans. J Exp Psychol Anim Behav Process. 1998;24(1):15–33. doi: 10.1037//0097-7403.24.1.15. [DOI] [PubMed] [Google Scholar]
- 66.Buhusi CV., Meek WH. Relativity theory and time perception: single or multiple clocks? PLoS One. 2009;4(7):e6268. doi: 10.1371/journal.pone.0006268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lake JI., Meek WH. Differential effects of amphetamine and haloperidol on temporal reproduction: dopaminergic regulation of attention and clock speed. Neuropsychologia. 2013;51(2):284–292. doi: 10.1016/j.neuropsychologia.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 68.Malapani C., Rakitin B., Levy R., et al Coupled temporal memories in Parkinson's disease: a dopamine-related dysfunction. J Cogn Neurosci. 1998;10(3):316–331. doi: 10.1162/089892998562762. [DOI] [PubMed] [Google Scholar]
- 69.Maricq AV., Church RM. The differential effects of haloperidol and methamphetamine on time estimation in the rat. Psychopharmacology. 1983;79(1):10–15. doi: 10.1007/BF00433008. [DOI] [PubMed] [Google Scholar]
- 70.Parker KL., Alberico SL., Miller AD., Narayanan NS. Prefrontal D1 dopamine signaling is necessary for temporal expectation during reaction time performance. Neuroscience. 2013;255:246–254. doi: 10.1016/j.neuroscience.2013.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parker KL., Chen KH., Kingyon JR., Cavanagh JF., Naryanan NS. Medial frontal ~4 Hz activity in humans and rodents is attenuated in PD patients and in rodents with cortical dopamine depletion. J Neurophysiol. 2015;114(2):1310–1320. doi: 10.1152/jn.00412.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Narayanan NS., Land BB., Solder JE., Deisseroth K., DiLeone RJ. Prefrontal D1 dopamine signaling is required for temporal control. Proc Natl Acad Sci U S A. 2012;109(50):20726–20731. doi: 10.1073/pnas.1211258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parker KL., Chen K-H., Kingyon JR., Cavanagh JF., Narayanan NS. D1-dependent 4 Hz oscillations and ramping activity in rodent mediaI frontal cortex during interval timing. J Neurosci. 2014;34:16774–16783. doi: 10.1523/JNEUROSCI.2772-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kehagia AA., Murray GK., Robbins TW. Learning and cognitive flexibility: frontostriatal function and monoaminergic modulation. Curr Opin Neurobiol. 2010;20(2):199–204. doi: 10.1016/j.conb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 75.Abi-Dargham A., Mawlawi O., Lombardo I., et al Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22(9):3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brunelin J., Fecteau S., Suaud-Chagny MF. Abnormal striatal dopamine transmission in schizophrenia. Curr Med Chem. 2013;20(3):397–404. doi: 10.2174/0929867311320030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corson PW., Nopoulos P., Miller DD., Arndt S., Andreasen NC. Change in basal ganglia volume over 2 years in patients with schizophrenia: typical versus atypical neuroleptics. Am J Psychiatry. 1999;156(8):1200–1204. doi: 10.1176/ajp.156.8.1200. [DOI] [PubMed] [Google Scholar]
- 78.Weinberger DR., Berman KF., Zee RF. Physiologic dysfunction of dorso-lateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry. 1986;43(2):114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- 79.Grosenick L., Marshel JH., Deisseroth K. Closed-loop and activity-guided optogenetic control. Neuron. 2015;86(1):106–139. doi: 10.1016/j.neuron.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]