

Summary
The E3 ubiquitin ligase Smurf1 targets the master regulator of osteoblast differentiation Runx2, for degradation yet the function of Smurf1 if any during osteoblast differentiation in vivo is ill-defined. Here we show that Smurf1 prevents osteoblast differentiation by decreasing Runx2 accumulation in osteoblasts. Remarkably, mice harboring a substitution-mutation at serine 148 (S148) in Smurf1 that prevents its phosphorylation by AMPK (Smurf1ki/ki) display an equally severe premature osteoblast differentiation phenotype as Smurf1−/− mice, a high bone mass and are also hyperinsulinemic and hypoglycemic. Consistent with the fact that Smurf1 targets the insulin receptor for degradation, there is in Smurf1ki/ki mice an increase in insulin signaling in osteoblasts that triggers a rise in the circulating levels of osteocalcin, a hormone that favors insulin secretion. These results identify Smurf1 as a determinant of osteoblast differentiation during development, of bone formation and glucose homeostasis post-natally and demonstrate the necessity of S148 for these functions.
Introduction
The transcription factor Runx2 has many attributes of a master regulator of osteoblast differentiation. In its absence, there are no osteoblasts anywhere in the skeleton and its haplo-insufficiency, by delaying osteoblast differentiation in bone forming through intramembranous ossification, results in a cleidocranial dysplasia (CCD), a disease characterized by open fontanelles and short clavicles (Ducy et al., 1997; Komori et al., 1997; Lee et al., 1997; Mundlos et al., 1997; Otto et al., 1997). Conversely, an increase in Runx2 activity as seen in mice and humans lacking one allele of Twist, results in craniosynostosis because of a premature osteoblast differentiation in the skull leading to early closure of the sutures (Bialek et al., 2004).
Not surprisingly given the paramount importance of this transcription factor for skeletogenesis, the mechanisms regulating the accumulation of Runx2 in cells of the osteoblast lineage have been intensively studied. Runx2 accumulation in osteoblast progenitor cells and differentiated osteoblasts is regulated in part by ubiquitination and several E3 ubiquitin ligases have been implicated in targeting Runx2 for degradation (Jones et al., 2006; Kaneki et al., 2006; Zhao et al., 2003). One of them Smurf1, interacts with Runx2 and other proteins regulating bone mass accrual such as Smad1, Smad5, MEKK2 and the insulin receptor (InsR) (Wei et al., 2014; Yamashita et al., 2005; Zhu et al., 1999). Consistent with these biochemical findings, forced expression of Smurf1 in osteoblasts inhibits, whereas deletion of Smurf1 in all cells favors bone formation in adult mice (Yamashita et al., 2005; Zhao et al., 2004). It has been proposed that Smurf1 achieves these functions in part by targeting MEKK2 for degradation (Yamashita et al., 2005). Surprisingly given the biochemical evidence indicating that Smurf1 favors the degradation of Runx2, no loss of function study has yet addressed the role of Smurf1 as a regulator of osteoblast differentiation in vivo.
A second important question regarding Smurf1 biology is to identify a domain if not a single amino acid that would confer to this protein the ability to target Runx2 for degradation in vivo. This question is even more relevant in view of the demonstration that in vitro, Smurf1 must be phosphorylated by AMPK on serine 148 (S148) in order to trigger the degradation of Runx2 (Wei et al., 2015). This raises the question of the biological importance of this residue in the functions of Smurf1.
We have addressed the aforementioned questions by analyzing Smurf1−/− mice and mice harboring a mutated form of Smurf1 in which S148 is mutated to alanine (Smurf1ki/ki). We show here that Smurf1 inhibits osteoblast differentiation through its ability to target Runx2 for degradation and that this function requires the presence of S148. Remarkably Smurf1ki/ki mice are also hypoglycemic and hyperinsulinemic because S148 is needed for Smurf1 ability to target the InsR for degradation. As a result, there is an accumulation of the InsR in bones of Smurf1ki/ki mice. This leads to an increase in the circulating levels of the bioactive form of the bone-derived hormone osteocalcin that favors insulin secretion and can cause hypoglycemia (Ferron et al., 2010a; Lee et al., 2007). These results define critical functions of Smurf1 in cells of the osteoblast lineage throughout life and highlight the importance of S148 for Smurf1 ability to target Runx2 and InsR for degradation.
Results
Smurf1 regulates osteoblast differentiation
To determine whether Smurf1 affects osteoblast differentiation in vivo, we analyzed in P4 and P10 Smurf1−/− mice two parameters that reflect the activity of Runx2 in cells of the osteoblast lineage during development: the closure of sutures in the skull and the length of clavicles.
Alcian blue/alizarin red staining of skeletal preparations showed that clavicles of Smurf1−/− mice were significantly longer than those of WT mice (Figure 1A) and sagittal sutures of Smurf1−/− skulls showed evidence of craniosynostosis (Figure 1B–C). Next, we analyzed by in situ hybridization the expression of Bone sialoprotein (Bsp), a biomarker of osteoblast differentiation (Bialek et al., 2004) in calvarial bones of E14.5 WT and Smurf1−/− embryos. The expression of Bsp was stronger in calvarial bones of E14.5 Smurf1−/− than in those of WT embryos (Figure 1D). The same was true for the expression of Bsp and Osteocalcin (Ocn), another osteoblast differentiation marker, in femurs of E14.5 Smurf1−/− embryos whether this was assayed by qPCR or in situ hybridization (Figure 1E–G). As a negative control in this experiment, we analyzed the expression of α1(I) Collagen (α1(I)Col), that is not regulated by Runx2 in vivo (Wei et al., 2015).
Figure 1. Regulation of osteoblast differentiation by Smurf1 in vivo.
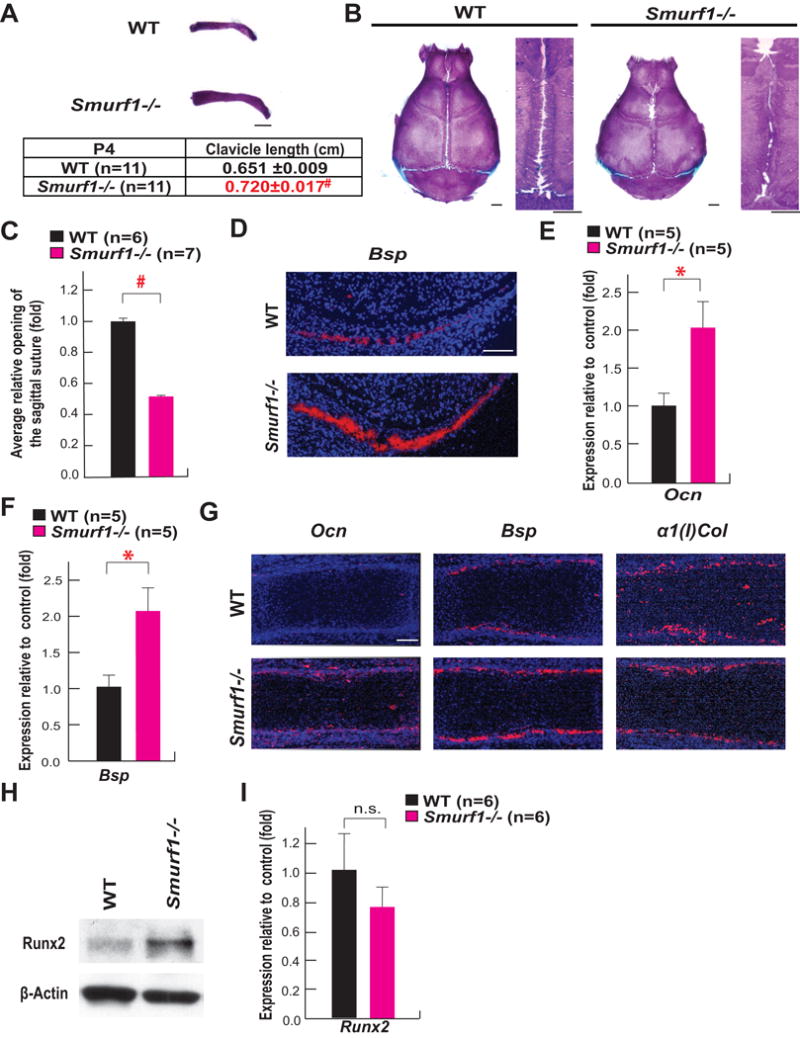
A–B. Alcian blue/alizarin red staining of clavicles and skulls of P4 and P10 WT and Smurf1−/− mice, respectively (scale bar, 1mm).
C. Opening of sagittal sutures in P10 Smurf1−/− and WT mice.
D. In situ hybridization analysis of Bsp expression in calvarial bones of E14.5 Smurf1−/− and WT embryos (scale bar, 100 μm).
E to F. qPCR analysis of Ocn (E) and Bsp (F) expression in femurs of E14.5 WT and Smurf1−/− embryos (n=5).
G. In situ hybridization analysis of Ocn, Bsp and α1(I)Col expression in femurs of E14.5 Smurf1−/− and WT embryos (scale bar, 100 μm).
H. Runx2 accumulation in skulls of P10 WT and Smurf1−/− mice.
I. Expression of Runx2 in the femurs of P10 WT and Smurf1−/− mice (n=6). N.S., not significant.
Mean ± SEM in all panels. p values (* P ≤ 0.05, # P≤ 0.005) are from Student’s t tests.
To link this premature osteoblast differentiation to an increase in Runx2 accumulation, we analyzed the abundance of this transcription factor in the skulls of Smurf1−/− and WT littermates and observed that Runx2 was more abundant in Smurf1−/− than WT skulls (Figure 1H). This latter result explains the increase in the expression of Ocn, a target of Runx2 (Ducy et al., 1997) (Figure 1E, 1G and 1I). Taken together, these results established that Smurf1 is a negative regulator of osteoblast differentiation in vivo, and suggested that this function occurs in part by targeting Runx2 for degradation.
Serine 148 is necessary for Smurf1 ability to inhibit osteoblast differentiation and bone formation
In vitro the phosphorylation of Smurf1 by AMPK at serine 148 (S148) is needed for its ability to target Runx2 for degradation (Wei et al., 2015). Thus, we tested the importance of this residue for Smurf1 ability to inhibit osteoblast differentiation by analyzing mutant mice harboring a S148A mutation in Smurf1 (Smurf1ki/ki) (Figure S1A–S1C). A western blot analysis verified that the phosphorylation of Smurf1 at S148 was abolished in Smurf1Ki/Ki osteoblasts although Smurf1 accumulation was not affected (Figure 2A).
Figure 2. Phosphorylation of Smurf1 at S148 is necessary for Smurf1 ability to regulate osteoblast differentiation in vivo.
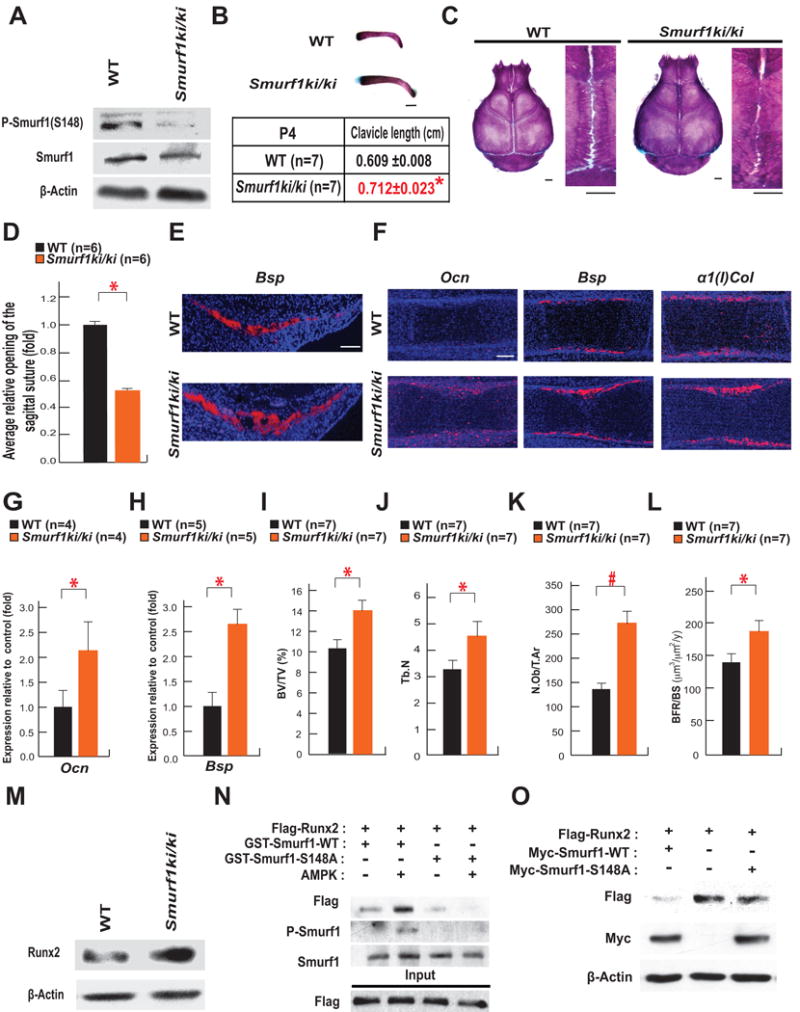
A. Western blot analysis of S148 phosphorylation in Smurf1 in WT and Smurf1ki/ki osteoblasts.
B–C. Alcian blue/Alizarin red staining of clavicles and skulls of P4 and P10 WT and Smurf1ki/ki mice respectively (scale bar, 1mm).
D. Opening of sagittal sutures in P10 Smurf1ki/ki and WT mice.
E–F. In situ hybridization analysis of Bsp in calvarial bones (E) and of Ocn, Bsp and α1(I)Col expression in femurs (F) of E14.5 WT and Smurf1ki/ki embryos (scale bar, 100 μm).
G to H. qPCR analysis of Ocn (G) and Bsp (H) expression in femurs of E14.5 WT and Smurf1ki/ki embryos (n=4–5).
I–L. Bone histomorphometric analysis of L3 and L4 vertebrae of 2 month-old WT and Smurf1ki/ki female mice (n=7). Mineralized bone volume per total volume (BV/TV) (I). Trabecular number (Tb.N) (J). Osteoblast number per tissue area (N.Ob/T.Ar) (K). The annual fractional volume of trabecular bone formed per unit trabecular surface area (BFR/BS) (L).
M. Runx2 accumulation in the skulls of P10 WT and Smurf1ki/ki mice.
N. GST-Pull down assay showing interaction of Flag-Runx2 with GST-Smurf1 WT or S148A after phosphorylation by AMPK.
O. Effect of the forced expression of WT or S148A Smurf1 on the accumulation of Runx2 in COS-7 cells.
Mean ± SEM in all panels. p values (* P ≤ 0.05, # P≤ 0.005) are from Student’s t tests.
Alcian blue/alizarin red staining of skeletal preparations showed that clavicles were significantly longer and sagittal sutures of the skulls more closed in Smurf1ki/ki than in newborn WT mice (Figure 2B–D). Accordingly, osteoblasts isolated from Smurf1−/− or Smurf1ki/ki mice formed more mineralized nodules than WT osteoblasts (Figure S2A). In vivo, Bsp expression was stronger in calvarial bones of E14.5Smurf1ki/ki embryos than in those of WT embryos (Figure 2E). The expression of Ocn started earlier and the one of Bsp was stronger in femurs of Smurf1ki/ki than in those of E14.5 WT embryos indicating that osteoblast differentiation occurred earlier in Smurf1ki/ki embryos throughout the skeleton. In contrast, no overt difference in the expression of α1(I)Col between E14.5 WT and Smurf1ki/ki embryos was noted (Figure 2F). The expression of Ocn and Bsp was also significantly higher in femurs of E14.5 Smurf1ki/ki embryos than in those of WT embryos when measured by qPCR (Figure 2G–H).
That Bsp and Ocn expression was higher in femurs of Smurf1ki/ki mice at P10 (Figure S2B–S2C) suggested that this S148A mutation in Smurf1 may affect bone biology post-natally. Indeed, two month-old Smurf1ki/ki mice had a higher bone mass and higher trabeculae number than WT littermates because of an increase in their osteoblast number and bone formation rate (Figure 2I–L). These phenotypes could be traced in part to an increase in the accumulation of Runx2 in Smurf1ki/ki skulls (Figure 2M). To ascertain that the phosphorylation of Smurf1 at S148 is needed to target Runx2 for degradation, we performed a GST-Pull-down assay and observed that the interaction between Smurf1 and Runx2 that occurred upon phosphorylation of Smurf1 by AMPK was abrogated when S148 was mutated to alanine. Accordingly, forced expression of WT but not S148A Smurf1 decreased the accumulation of Runx2 in COS-7 cells (Figure 2N–O). Taken collectively, these data establish the importance of S148 for Smurf1 ability to inhibit osteoblast differentiation and bone formation.
Smurf1 inhibits osteoblast differentiation by targeting Runx2 to degradation
If S148 is required for Smurf1 inhibition of osteoblast differentiation because it targets Runx2 for degradation, mutating S148 to alanine in Smurf1 should increase Runx2 accumulation and rescue in part, the CCD phenotype seen in Runx2+/− mice (Komori et al., 1997; Otto et al., 1997). To test this hypothesis, we analyzed Runx2+/−;Smurf1ki/ki mice.
Alcian blue/alizarin red staining of skeletal preparations showed a marked improvement of the CCD phenotype in Runx2+/−;Smurf1ki/ki compared to Runx2+/− mice (Figure 3A–C) while a western blot verified that Runx2 accumulation was increased in Runx2+/−;Smurf1ki/ki skulls (Figure 3D). This increase in Runx2 accumulation resulted in an improved osteoblast differentiation as shown by the increased Ocn expression in E15.5 Runx2+/−;Smurf1ki/ki compared to Runx2+/− embryos (Figure 3E). Ocn and Bsp expression was also significantly higher in bones of Runx2+/−;Smurf1ki/ki compared to those of Runx2+/− mice at P10 when measured by qPCR (Figure 3F–G).
Figure 3. Smurf1 phosphorylation at S148 is necessary to regulate RUNX2 in vivo.
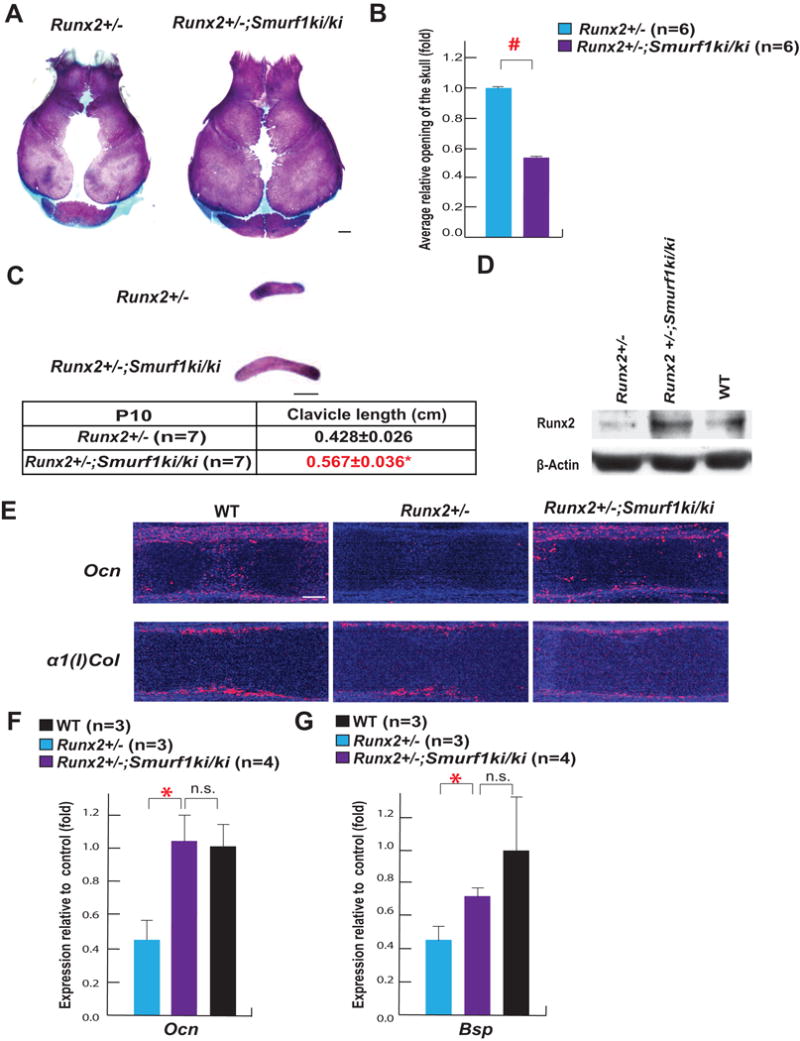
A. Alcian blue/alizarin red staining of skulls of P10 Runx2+/− and Runx2+/−;Smurf1ki/ki mice (scale bar, 1mm).
B. Area of the opening of skulls in P10 Runx2+/−;Smurf1ki/ki and Runx2+/− mice.
C. Alcian blue/alizarin red staining of clavicles of P10 Runx2+/− and Runx2+/−;Smurf1ki/ki mice (scale bar, 1mm).
D. Runx2 accumulation in skulls of P10 Runx2+/−, Runx2+/−;Smurf1ki/ki and WT skulls.
E. In situ hybridization analysis of Ocn and α1(I)Col expression in E15.5 WT, Runx2+/− and Runx2+/−;Smurf1ki/ki femurs (scale bar, 100 μm).
F–G. qPCR analysis of Ocn (F) and Bsp (G) expression in femurs of P10 WT, Runx2+/− and Runx2+/−;Smurf1ki/ki mice (n=3–4).
Mean ± SEM in all panels. p values (* P ≤ 0.05, # P≤ 0.005) are from Student’s t tests.
Hence, the ability of Smurf1 to target Runx2 for degradation explains to a large extent why this E3 ubiquitin ligase inhibits osteoblast differentiation. That the rescue of the CCD phenotype was not complete in Runx2+/−;Smurf1ki/ki mice is consistent with the notion that Smurf1 also acts through additional mechanisms to prevent osteoblast differentiation such as targeting MEKK2, Smad1 and Smad5 for degradation (Yamashita et al., 2005; Zhu et al., 1999).
S148 is needed for Smurf1 ability to regulate glucose homeostasis
To determine if S148 is necessary for the interaction of Smurf1 with its other substrates, we focused on the insulin receptor (InsR) because both Smurf1−/− mice and Smurf1ki/ki mice were hypoglycemic and hyperinsulinemic (Figure 4A–C).
Figure 4. Phosphorylation of SMURF1 at S148 is necessary for Smurf1 activity to regulate InsR degradation.
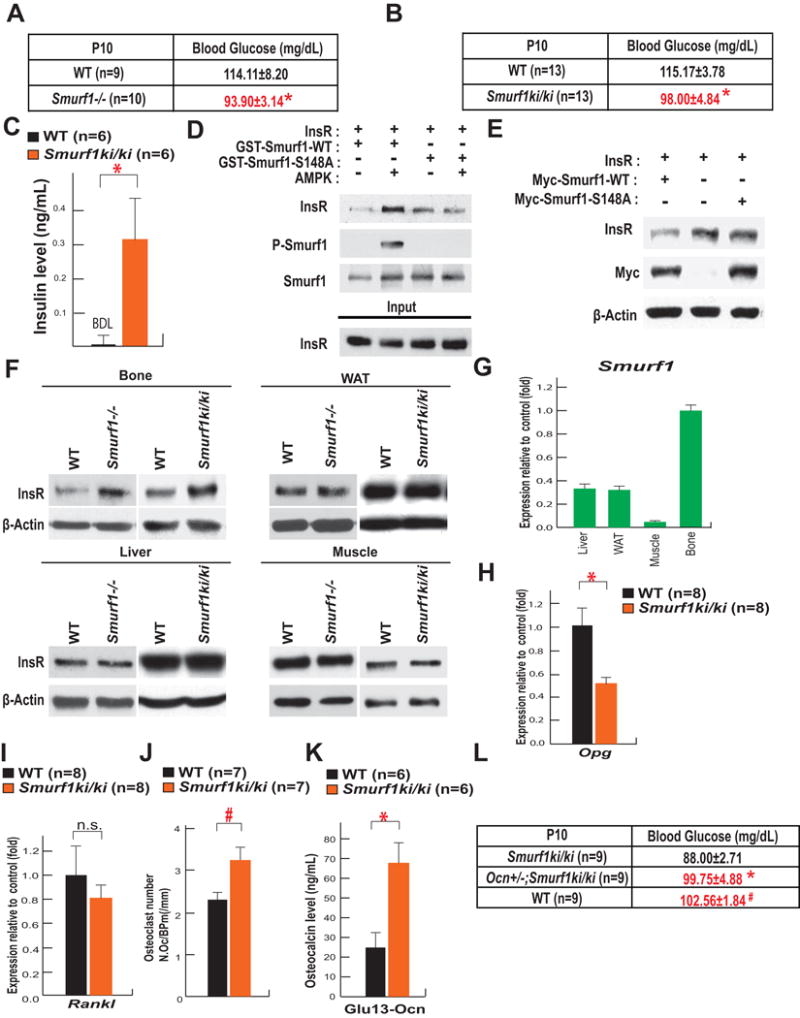
A–B. Blood glucose levels in P10 WT and Smurf1−/− mice (A), WT and Smurf1ki/ki mice (n=9–13 per group) (B).
C. Circulating insulin levels in P10 WT and Smurf1ki/ki mice (n=6). ND: not detectable.
D. GST-Pull down assay showing interaction of InsR with GST-Smurf1 WT or GST-Smurf1 S148A after AMPK phosphorylation.
E. Effect of the overexpression of WT or S148A Smurf1 on the accumulation of InsR in COS-7 cells.
F. Accumulation of the InsR in skull, liver, WAT and muscle of P10 WT, Smurf1−/− and Smurf1ki/ki mice.
G. Expression of Smurf1 in liver, WAT, muscle and skull of P10 WT mice.
H–I. Expression of Osteoprotegerin (Opg) (H) and Rankl (I) in the femur of P10 WT and Smurf1ki/ki mice (n=8).
J. Osteoclasts number (N.Oc/B.Pm (/mm)) in 2 month-old WT and Smurf1ki/ki female mice (n=7).
K. Serum levels of undercarboxylated osteocalcin in 2 month-old WT and Smurf1ki/ki mice (n=6).
L. Blood glucose levels in P10 WT, Smurf1ki/ki and Ocn+/−;Smurf1ki/ki mice (n=9).
Mean ± SEM in all panels. p values (* P ≤ 0.05, # P≤ 0.005) are from Student’s t tests.
Following its phosphorylation by AMPK, WT but not S148A Smurf1 interacts readily with the InsR (Figure 4D) and forced expression of WT but not S148A Smurf1 decreased the accumulation of InsR in COS-7 cells (Figure 4E). In vivo, the accumulation of the InsR was higher in Smurf1−/− and Smurf1ki/ki than in WT bones. That the accumulation of the InsR was not affected in other insulin target tissues, such as liver, white adipose tissue (WAT), and muscle in Smurf1−/− and Smurf1ki/ki mice (Figure 4F) is explained in part by the fact that Smurf1 expression is markedly higher in bone than in the liver, WAT and muscle (Figure 4G).
Given these observations, we tested whether the hypoglycemia seen in Smurf1ki/ki mice resulted in part, from an increase in insulin signaling in osteoblasts. Since insulin signaling in osteoblasts inhibits Osteoprotegerin (Opg) expression (Ferron et al., 2010a), we used the expression of this gene in Smurf1ki/ki femurs as a readout of insulin signaling in osteoblasts. In agreement with the increased accumulation of the InsR in bone (Figure 4F), Opg expression was decreased in Smurf1ki/ki compared to control femurs (Figure 4H) whereas Rankl expression was unchanged (Figure 4I). This decrease in Opg expression provides an explanation for the significant increase in the number of osteoclasts observed in the bones of adult Smurf1ki/ki mice (Figure 4J). This increase in bone resorption parameters was not of sufficient amplitude to compensate for the increase in bone formation parameters. As a result, Smurf1ki/ki mice had a high bone mass (Figure 2I–L).
Since bone resorption is the mechanism whereby osteocalcin, a hormone that favors insulin secretion, is activated by decarboxylation (Ferron et al., 2010a), we measured the circulating levels of undercarboxyated and bioactive osteocalcin in control and Smurf1ki/ki mice and observed that circulating undercarboxylated osteocalcin levels were approximately three-fold higher in Smurf1ki/ki than in control mice (Figure 4K). Such an increase in circulating osteocalcin levels should lead to a hyperinsulinemia and hypoglycemia as seen in Smurf1ki/ki mice (Figure 4B–C). To demonstrate that this increase in osteocalcin activity contributes to the hypoglycemia observed in Smurf1ki/ki mice, we analyzed Smurf1ki/ki mice lacking one allele of Osteocalcin (Smurf1ki/ki;Ocn+/−). As shown in Figure 4L blood glucose levels were normal in Smurf1ki/ki;Ocn+/− mice. These results reveal the existence of a Smurf1-InsR-osteoprotegerin-osteocalcin pathway taking place in osteoblasts and contributing to glucose homeostasis.
Discussion
The mechanisms regulating Runx2 accumulation in osteoblast progenitor cells have been a topic of intense investigation. This has led to the identification mostly on biochemical grounds, of several E3 ubiquitin ligases that would trigger Runx2 for degradation. Given the number of E3 ubiquitin ligases known to interact with Runx2, one could fear that disrupting the interaction of only one of them with Runx2 would not significantly hamper osteoblast differentiation in vivo. Instead, our investigation shows that deleting a single E3 ubiquitin ligase implicated in Runx2 degradation Smurf1, results in an increase in Runx2 accumulation leading to a premature osteoblast differentiation during embryonic development and increased bone formation post-natally. We further show that the majority of this function of Smurf1 requires a single amino acid, S148, be phosphorylated by AMPK (Wei et al., 2015). These results highlight the importance of Smurf1 and of this particular residue in this molecule in targeting Runx2 for degradation in vivo and regulating osteoblast differentiation.
The functions of Smurf1 in osteoblasts extend beyond osteoblast differentiation and bone formation since by favoring the degradation of the InsR in osteoblasts, Smurf1 is a regulator of circulating osteocalcin levels. This explains the existence of hyperinsulinemia and hypoglycemia in Smurf1ki/ki mice. Indeed, Smurf1 regulates bone resorption and the production of the active form of osteocalcin, a hormone that favors insulin secretion (Lee et al., 2007). As it is the case for its ability to regulate osteoblast differentiation, this function of Smurf1 requires the presence of S148. Altogether, our results provide a deeper understanding of the molecular regulation of Runx2 accumulation and of the endocrine functions of bone.
Materials and Methods
Mice generation
To generate Smurf1Ki/+ mice, a mutation was introduced into a BAC by recombineering using galK selection system (Warming et al., 2005). In the first step, the galK cassette was inserted into BAC by homologous recombination and clones were obtained through positive selection on minimal media plates in which galactose was the only carbon source. The successful recombination was validated by PCR analysis. In the second step, the galK cassette was substituted by double stranded oligonucleotides with the modified base-pair in the middle of the homology arms flanking the galK casette. This step was achieved by negatively selecting against the galK cassette by resistance to 2-deoxy-galactose (DOG) on plates with glycerol as the carbon source. Clones in which galK cassette is replaced by a mutation of interest were identified by PCR and sequencing. A frt-neo-frt (FNF) cassette was inserted upstream of the mutation and retrieved into pMCS-DTA by homologous recombination using standard protocol. The targeting vector that was verified by PCR, diagnostic digestions and sequencing, was electroporated into ES cells, positive ES cells were identified by PCR screening and sequencing. Selected ES cells were injected in 129Sv/EV blastocysts to generate chimeric mice. Chimeric mice were crossed to Gt(ROSA)26Sortm1(FLP1)Dym to remove the neomycin resistance cassette (Figure S1A–S1C).
Runx2+/− mice were generated previously (Otto et al., 1997). Smurf1+/− mice were a generous gift of Dr. Jeff Wrana (U of Toronto, Canada) (Narimatsu et al., 2009). All mice strains were maintained on a C57/129 mixed background except for Runx2+/− mice, which are maintained on C57 background. Littermates were used as controls in all experiments.
Cell culture
Primary mouse calvaria osteoblasts were cultured as described previously (Ducy and Karsenty, 1995). To determine formation of mineralized nodule, von Kossa staining was performed on osteoblasts cultured in differentiation medium supplemented with 5mM β-glycerophosphate and 100ug/mL ascorbic acid for 7 days. To detect the S148 phosphorylation in Smurf1, osteoblasts isolated from Smurf1ki/ki and WT pups were incubated overnight in glucose free KRH buffer (50mM HEPES pH7.4, 136mM NaCl, 4.7mM KCl, 1.25mM MgSO4, 1.25mM CaCl2 and 0.1% BSA) and followed by western blot analysis.
DNA transfection experiments
Myc-Smurf1 WT and S148A plasmids were constructed by inserting a full-length cDNA of mouse Smurf1 WT or S148A into pcDNA™ 3.1/myc-His B vector (Invitrogen). Myc-Smurf1 WT or S148A was co-transfected with Flag-Runx2 or the human InsR cDNA expression constructs (Wei et al., 2014; Wei et al., 2015) in COS-7 cells. DNA transfection experiments were performed using Lipofectamin 2000 (Invitrogen) according to manufacturer’s protocol.
Western blot analysis
Anti-Runx2 antibody (Santa Cruz Biotech Inc.), anti-β-ACTIN (Sigma), Anti-Smurf1 and Anti-Phospho-Ser148 Smurf1 (GenScript Inc.) were used. Others antibodies were obtained from Cell Signaling Technology. All western blot analyses were repeated at least three times, with different samples.
Gene expression analysis
RNA samples were extracted using TRIZOL reagent (Invitrogen). Using two micrograms of total RNA, cDNA preparation was carried out following standard protocols. The cDNAs were used as templates for quantitative PCR analyses using CFX-Connect realtime PCR system (Bio-Rad). Expression levels of each gene analyzed by qPCR were normalized using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression levels as an internal control. The sequences of specific primers used in this study were previously described (Obri et al., 2014).
Skeletal preparation and analysis
Alcian blue/alizarin red staining of the skeletal preparation was conducted according to standard protocols (McLeod, 1980). Littermate controls were analyzed in all experiments. Quantifications of the length of clavicles as well as the area of the opening of the skull were made using ImageJ.
Bone histomorphometry
This analysis was performed on L3 and L4 vertebrae of 2-month-old female mice as described previously (Chappard et al., 1987; Parfitt et al., 1987). Mineralized bone volume over the total tissue volume (BV/TV), osteoblast number per tissue area (N.Ob/T.Ar) as well as trabecular number (Tb.N), and bone formation rate per bone surface (BFR/BS) were measured by conducting Von Kossa/van Gieson staining, toluidine blue staining and calcein double-labeling, respectively. Osteoclasts parameters were measured by TRAP staining followed by counterstain with hematoxylin. Osteoclasts were defined as multinucleated dark red cells along the bone surface. Histomorphometric analysis was performed using the Osteomeasure System (OsteoMetrics).
Biochemistry
ELISAs were performed according to manufacturer’s instructions to measure mouse insulin (EZRMI-13K, Millipore) and undercarboxylated osteocalcin (Ferron et al., 2010b). For AMPK phosphorylation assay, GST-Smurf1 and GST-Runx2 constructs were generated and purified as previously described (Ferron et al., 2013; Wei, 2015). AMPK phosphorylation assay of GST-Smurf1 WT and S148A was performed using previously described method (Wei, 2015).
GST-Pulldown assay
GST-Smurf1 WT and GST-Smurf1S148A were bound to glutathione agarose beads (GE Healthcare) and blocked overnight at 4°C in 5%BSA with end-over-end mixing. Beads were washed with AMPK assay buffer 5 times and AMPK phosphorylation assay was followed. Beads were washed with binding buffer (50mM Tris-Hcl pH 7.5, 100mM NaCl, 50mM β-glycerophosphate, 10% glycerol, 1% Tween-20, 1mM EDTA, 25mM NaF) 5 times. Cell lysates of transfected COS-7 cells were incubated with 25μg of GST for 2 hours at 4°C with end-over-end mixing. This mixture was centrifuged and supernatants were mixed with GST-Smurf1 WT or S148A in 500μL of binding buffer for 2 hours at 4°C with end-over-end mixing. The mixtures were washed 5 times and analyzed by SDS-PAGE followed by immunoblotting.
In situ hybridization
Tissues were fixed in 4% paraformaldehyde/PBS overnight at 4°C and then embedded in paraffin after serial of dehydrations. Tissues were sectioned at 5μm. In situ hybridization was performed using 35S-labeled riboprobe as described (Ducy et al., 1997). The α1(I)Col, Bsp, and Osteocalcin probes were prepared as previously described (Takeda et al., 2001). Hybridizations were performed overnight at 55°C, and washes were performed at 63°C. Autoradiography were performed as previously described (Sundin et al., 1990) and nuclei were counterstained with DAPI.
Statistics
All data are presented as mean ± standard error of mean. In this paper, statistical analysis was performed by unpaired Student’s t test. * denotes P ≤ 0.05, and # denotes P≤ 0.005 compared to control.
Acknowledgments
We thank Dr. C.-S. Lin and F. Lee for generation of Smurf1 S148A knock-in mice, Drs. P. Ducy and J. Wrana for critical reading of the manuscript and for providing the Smurf1+/− mice, respectively. This work is supported by the RO1DK104727-A1 and PO1AG032959-06A1 (G.K.), Honjo International Scholarship (J.S.) and Mandl fellowship (J.W.). The training program in Genetics and Development (J.S.) is supported by an NIH T32 training grant from the National Institute of General Medical Science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, G.K., J.S., and J.W.; Methodology, J.S.; Investigation, J.S. and J.W.; Writing-Original Draft, G.K and J.S.; Writing-Review & Editing, G.K., J.S. and J.W.; Funding Acquisition, G.K. and J.S.; Supervision, G.K.
References
- Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–435. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- Chappard D, Palle S, Alexandre C, Vico L, Riffat G. Bone embedding in pure methyl methacrylate at low temperature preserves enzyme activities. Acta Histochem. 1987;81:183–190. doi: 10.1016/S0065-1281(87)80012-0. [DOI] [PubMed] [Google Scholar]
- Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–1869. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S, Rawlings DJ, Ballabio A, Karsenty G. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27:955–969. doi: 10.1101/gad.213827.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A, Ducy P, Karsenty G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010a;142:296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron M, Wei J, Yoshizawa T, Ducy P, Karsenty G. An ELISA-based method to quantify osteocalcin carboxylation in mice. Biochem Biophys Res Commun. 2010b;397:691–696. doi: 10.1016/j.bbrc.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DC, Wein MN, Oukka M, Hofstaetter JG, Glimcher MJ, Glimcher LH. Regulation of adult bone mass by the zinc finger adapter protein Schnurri-3. Science. 2006;312:1223–1227. doi: 10.1126/science.1126313. [DOI] [PubMed] [Google Scholar]
- Kaneki H, Guo R, Chen D, Yao Z, Schwarz EM, Zhang YE, Boyce BF, Xing L. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. The Journal of biological chemistry. 2006;281:4326–4333. doi: 10.1074/jbc.M509430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- Narimatsu M, Bose R, Pye M, Zhang L, Miller B, Ching P, Sakuma R, Luga V, Roncari L, Attisano L, et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137:295–307. doi: 10.1016/j.cell.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Obri A, Makinistoglu MP, Zhang H, Karsenty G. HDAC4 integrates PTH and sympathetic signaling in osteoblasts. J Cell Biol. 2014;205:771–780. doi: 10.1083/jcb.201403138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone Histomorphometry - Standardization of Nomenclature, Symbols, and Units. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- Sundin OH, Busse HG, Rogers MB, Gudas LJ, Eichele G. Region-specific expression in early chick and mouse embryos of Ghox-lab and Hox 1.6, vertebrate homeobox-containing genes related to Drosophila labial. Development. 1990;108:47–58. doi: 10.1242/dev.108.Supplement.47. [DOI] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, Karsenty G. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest. 2014;124:1–13. doi: 10.1172/JCI72323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, Takarada T, Lezaki T, Pessin JE, Hinoi E, et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell. 2015;161:1576–1591. doi: 10.1016/j.cell.2015.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Shimazu J, Makinistoglu PM, Maurizi A, Kajimura D, Zong H, Takarada T, Iezaki T, Pessin EJ, Hinoi E, Karsenty G. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell. 2015 doi: 10.1016/j.cell.2015.05.029. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, Zhang YE. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121:101–113. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Qiao M, Harris SE, Oyajobi BO, Mundy GR, Chen D. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. The Journal of biological chemistry. 2004;279:12854–12859. doi: 10.1074/jbc.M313294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. The Journal of biological chemistry. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]