

Abstract
In this review we discuss the mechanisms for the pleotropic effects of leptin replacement therapy to reverse liver and muscle insulin resistance in lipodystrophic individuals, as well as insulin-independent effects of leptin replacement therapy to suppress white adipose tissue lipolysis, hepatic gluconeogenesis and fasting hyperglycaemia in rodent models of poorly controlled diabetes. On the basis of these studies we conclude with a view of the potential therapeutic applications of leptin replacement therapy in humans. This review summarises a presentation given at the ‘Is leptin coming back?’ symposium at the 2015 annual meeting of the EASD. It is accompanied by two other reviews on topics from this symposium (by Thomas Meek and Gregory Morton, DOI: 10.1007/s00125-016-3898-3, and by Christoffer Clemmensen and colleagues, DOI: XXX) and an overview by the Session Chair, Ulf Smith (DOI: 10.1007/s00125-016-3894-7).
Keywords: Diabetic ketoacidosis, Leptin, Lipodystrophy, Review, Type 1 diabetes
Effects of leptin to reverse insulin resistance in patients with lipodystrophy
Patients with severe lipodystrophy manifest severe liver and muscle insulin resistance associated with hypoleptinaemia, hyperphagia and hepatic steatosis [1]. Chronic leptin replacement therapy was shown to reverse liver and muscle insulin resistance and fasting hyperglycaemia in patients with severe lipodystrophy, and this could be attributed at least in part to large reductions in ectopic lipid deposition in these tissues [1]. Previous studies have demonstrated a causal role for ectopic lipid accumulation in mediating liver and skeletal muscle insulin resistance through diacylglycerol activation of protein kinase C (PKC)-ɛ and PKC–θ in liver and muscle, respectively, which in turn leads to inhibition of insulin signalling [2, 3]. The lower ectopic lipid content in liver and skeletal muscle following leptin replacement therapy could in turn be attributed to reversal of hyperphagia since there were no detectable changes in basal rates of energy expenditure as assessed by indirect calorimetry (Fig. 1).
Fig. 1.
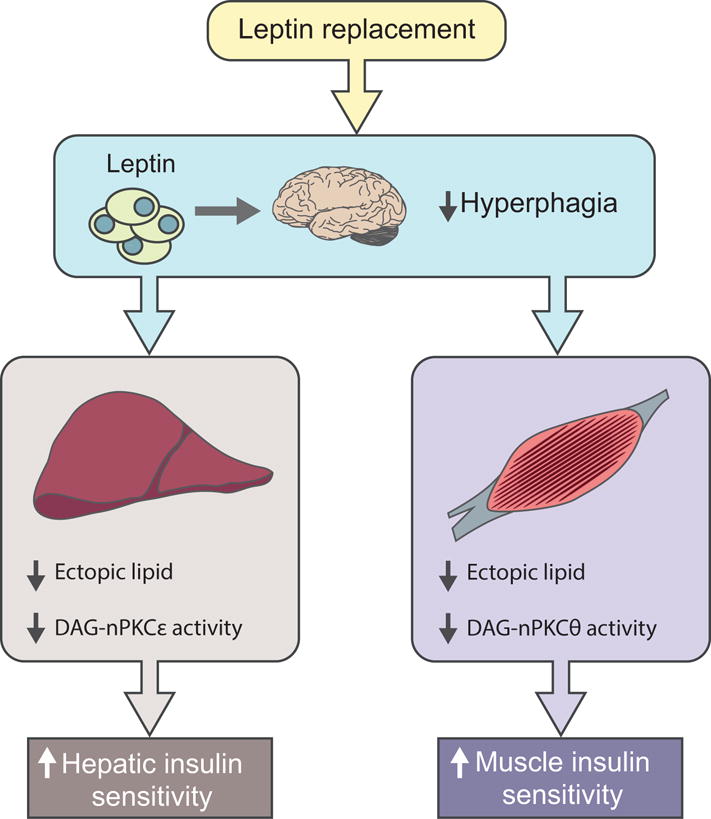
Insulin-dependent effects of leptin on liver and muscle insulin responsiveness. Leptin replacement therapy reverses liver and muscle insulin resistance in patients with severe lipodystrophy by reversing hyperphagia, which in turn leads to reduction of ectopic lipid content and improvement in insulin sensitivity in liver and skeletal muscle [1, 4]. DAG, diacylglycerol; nPKC, novel protein kinase C
Insulin-independent effects of leptin to reverse fasting hyperglycaemia and ketoacidosis in diabetic rodents
In addition to these effects of leptin to reduce hyperphagia, ectopic lipid and insulin resistance in liver and muscle in patients with severe lipodystrophy, recent studies have raised the intriguing possibility that leptin may also have insulin-independent effects to reverse hyperglycaemia and ketoacidosis in insulinopenic animal models of poorly controlled type 1 diabetes, spurring great interest in the possibility of treating type 1 diabetic patients with this hormone [5–10]. This insulin-independent glucose-lowering effect of leptin has been postulated to occur through suppression of glucagon production, suppression of glucagon responsiveness or both [5–10]. However, recent studies have called into question the existence of a direct effect of leptin on the pancreatic alpha cell [11] and have decoupled the putative glucagon-lowering effect of leptin from reversal of hyperglycaemia in rodents [12–14].
To understand the insulin-independent mechanisms by which leptin replacement therapy reverses hyperglycaemia and diabetic ketoacidosis (DKA), we studied the effects of leptin infusion in rats in DKA. Leptin therapy lowered fasting plasma glucose concentrations by more than 50% and reversed DKA [15]. In this study, we linked the effect of leptin to correct hyperglycaemia and normalise hepatic glucose production to suppression of white adipose tissue (WAT) lipolysis and reduction of hepatic acetyl-CoA content and pyruvate carboxylase activity by suppression of hypercorticosteronaemia [15] in two rat models of poorly controlled type 1 diabetes. Intra-arterial infusions of corticosterone to match plasma corticosterone concentrations or a lipid emulsion and heparin to activate lipoprotein lipase activity in normal rats to match hepatic acetyl-CoA content in untreated type 1 diabetic rats replicated the increase in hepatic glucose production seen in type 1 diabetic animals, whereas hepatic lipolysis, hepatic acetyl-CoA content and hepatic glucose production were almost fully normalised in type 1 diabetic rats treated with the potent glucocorticoid receptor antagonist mifepristone. Taken together, these data suggest a critical role for increased HPA axis activity in promoting fasting hyperglycemia and diabetic ketoacidosis in rat models of poorly controlled type 1 diabetes as a result of markedly higher rates of lipolysis and hepatic gluconeogenesis.
It is possible that acute leptin replacement therapy may also have a direct effect on muscle glucose uptake, peripheral insulin sensitivity or both. Effects of leptin on peripheral insulin sensitivity and/or glucose uptake, consistent with our previous studies of chronic leptin replacement therapy in lipodystrophic patients in which ectopic lipid content and liver and muscle insulin resistance were reversed, have been reported in some [5, 16, 17] but not all [6, 18] studies. However, our data suggest that the primary insulin-independent mechanism by which acute leptin replacement therapy reverses DKA in diabetic rodents is through reduction of hypothalamic–pituitary–adrenal (HPA)-mediated rates of whole-body lipolysis, which in turn results in reduced rates of hepatic gluconeogenesis and ketogenesis.
Taken together, these data demonstrate that excess fatty acid and glycerol flux to the liver, resulting from excess WAT lipolysis, drives the higher fasting rates of hepatic gluconeogenesis and ketogenesis by increasing hepatic acetyl-CoA concentrations, thereby allosterically activating pyruvate carboxylase and increasing the conversion of glycerol to glucose by a substrate push mechanism. Thus, these studies demonstrate that increased fatty acid and glycerol delivery to the liver and allosteric regulation of pyruvate carboxylase activity are the key factors responsible for causing the higher rates of gluconeogenesis in DKA under fasting conditions. Furthermore, it should be noted that these rodent models of poorly controlled type 1 diabetes exhibited marked elevations in hepatic gluconeogenesis despite unchanged hepatic gluconeogenic protein expression, which also challenges the canonical role for alterations in hepatic gluconeogenic gene and protein expression as the primary cause of increased hepatic gluconeogenesis in poorly controlled diabetes.
These data identify a key role for hypoleptinaemia-induced increases in HPA activity in producing the higher rates of fasting hepatic gluconeogenesis, hyperglycaemia and ketoacidosis in rodent models of type 1 diabetes as a result of glucocorticoid-mediated increases in lipolysis, and provide the mechanism for the acute correction of fasting hyperglycaemia in these animals after administration of leptin (Fig. 2):
Fig. 2.
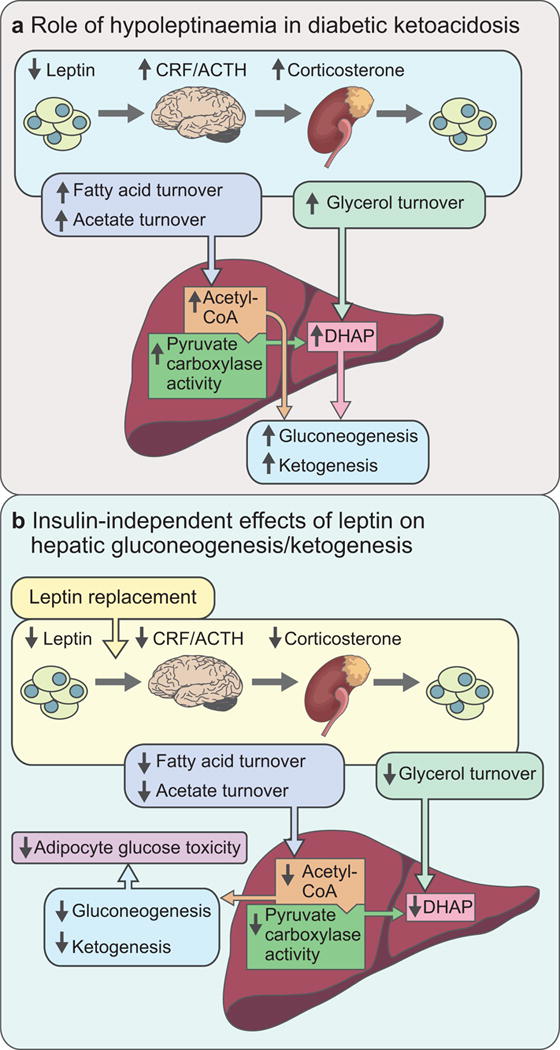
Mechanism of the effects of hypoleptinaemia (a) and its reversal by leptin replacement (b) in rat models of diabetic ketoacidosis [15]. CRF, corticotropin releasing factor; ACTH, adrenocorticotropic hormone; DHAP, dihydroxyacetone phosphate
The key findings of our study—suppression of hepatic acetyl-CoA content and hepatic glucose production by leptin via suppression of corticosterone-driven lipolysis—may appear to be inconsistent with previous studies in which the beneficial effect of leptin treatment was associated with reduced plasma glucagon concentrations in diabetic rodents [5–7, 9, 10]. We observed changes in plasma glucagon concentrations similar to those reported in prior studies: plasma glucagon was increased in both rat models of type 1 diabetes and was normalised by 24 h of leptin treatment [15]. However, the normalisation of plasma glucagon concentrations was observed more than 18 h following normalisation of plasma corticosterone concentrations, and fasting rates of lipolysis, hepatic acetyl-CoA content and hepatic glucose production, consistent with the findings of Kim et al [12], discussed earlier. Thus, it appears that, while a glucagon-lowering effect may contribute to the long-term effect of leptin to suppress hyperglycaemia, it is neither necessary nor sufficient to explain the acute glucose-normalising effect of leptin in poorly controlled diabetes.
Translation to patients
As discussed earlier, leptin replacement therapy has been shown to be efficacious at reversing hepatic and muscle insulin resistance, hyperglycaemia, hepatic steatosis and hyperphagia in patients with severe lipodystrophy [1, 19]. In this regard, metreleptin (Myalept) has recently been approved by the US Food and Drug Administration (FDA) as replacement therapy to treat the complications of leptin deficiency in patients with congenital or acquired generalised lipodystrophy. In addition, leptin replacement therapy has proved to be very effective at treating hyperphagia and severe obesity in the very rare obese individual suffering from congenital leptin deficiency [20]. With regard to the potential use of leptin treatment in patients with poorly controlled type 1 diabetes, our studies and others have raised the intriguing prospect that leptin may also have insulin-independent effects to reverse hyperglycaemia in animal models of poorly controlled type 1 diabetes, raising the possibility of treating type 1 diabetic patients with this hormone. However, given the exquisite sensitivity of the adipocyte to insulin, even low levels (70 pmol/l) of insulin would abrogate the effect of leptin to further reduce fasting rates of lipolysis, gluconeogenesis and ketogenesis by suppression of the HPA axis [15]. Therefore, based on our proposed insulin-independent mechanism for the glucose-lowering effect of leptin, it would be predicted that adjuvant leptin therapy would have minimal additional benefits in further improving glycaemic control as an adjuvant to insulin replacement therapy in most patients with type 1 diabetes who are being treated with long-acting insulin preparations or an insulin pump and therefore have sustained basal insulin concentrations (>10 μU/ml) even during fasting. Indeed, this explanation may also explain the failure of the recent study by Morton et al to find a role for leptin-induced reductions in hypercorticosteronaemia to reverse hyperglycaemia in an STZ-induced rat model of diabetes [21]. The differences in these results can most likely be attributed to the differences in the diabetic rat models [21]. Consistent with our observations, Morton et al observed that leptin treatment resulted in reductions in plasma corticosterone concentrations; however, while our study [15] focused on examining the insulin-independent effects of leptin replacement therapy on fasting rates of hepatic gluconeogenesis and fasting hyperglycemia in a type 1 diabetic rat model of DKA, in contrast, Morton et al examined the effects of leptin treatment on ‘daily-blood levels in the fed state’ in most of their studies and with sucrose in the drinking water for their adrenalectomised diabetic rats [21]. Under these fed conditions, changes in plasma glucose concentrations are mostly dependent on insulin-mediated increases in liver and muscle glucose uptake. Furthermore, in contrast to our diabetic rats, which had plasma insulin concentrations <14 pmol/l and were accordingly in DKA, as would be expected by their severe insulinopenic state, the rats studied by Morton et al had fasting plasma insulin concentrations that were >70 pmol/ml and were not in DKA, as reflected by their the ability of the diabetic control group to survive for several days without insulin or leptin treatment [21]. While leptin replacement therapy might be predicted to result in similar insulin-independent reductions in rates of fasting hepatic gluconeogenesis, ketogenesis and fasting hyperglycaemia by reducing lipolysis in patients suffering from DKA, leptin replacement therapy offers no theoretical additional advantages compared with the standard insulin treatment regimen for treating DKA, which also results in acute suppression of WAT lipolysis, gluconeogenesis and ketogenesis. Furthermore, in contrast to insulin therapy, there is no evidence that leptin treatment alone promotes glucose uptake by liver and skeletal muscle.
Summary
Taken together, these data demonstrate that leptin replacement therapy has pleotropic effects to reverse insulin resistance in humans with severe lipodystrophy, as well as an insulin-independent effect to acutely reduce fasting rates of hepatic gluconeogenesis, ketogenesis and fasting hyperglycaemia in rodent models of DKA. Leptin treatment also reduced plasma glucagon concentrations, which will also contribute to reductions in hepatic insulin resistance, ketogenesis and hepatic glucose production; however, these effects occur more than 18 h after the acute reductions of leptin to reduce fasting hyperglycaemia, hepatic gluconeogenesis and ketogenesis, and may at least in part be secondary to the reversal of glucose toxicity [22]. Leptin replacement therapy-induced reversal of liver and muscle insulin resistance and hyperglycaemia in patients with severe lipodystrophy can be attributed at least in part to reductions in ectopic lipid in liver and skeletal muscle, which in humans can be attributed mostly to reductions in hyperphagia and energy intake. The insulin-independent effects of leptin to reduce fasting rates of hepatic gluconeogenesis, ketogenesis and fasting hyperglycaemia can be attributed at least in part to suppression of HPA activity resulting in reductions in rates of lipolysis. These effects in turn lead to reductions in hepatic acetyl-CoA content and pyruvate carboxylase flux, as well as reductions in the rate of glycerol conversion to glucose.
These data highlight the role of increased lipolysis leading to increased hepatic acetyl-CoA and pyruvate carboxylase activity/flux, as well as increased conversion of glycerol to glucose as keys drivers of increased hepatic gluconeogenesis and fasting hyperglycaemia in poorly controlled diabetes. Subsequent studies have confirmed these same factors as being responsible for mediating the acute effects of fibroblast growth factor (FGF)-1 and FGF-19 to reverse DKA [14], and as the critical mediators of fasting hyperglycaemia and hepatic insulin resistance in rat models of diet-induced obesity and type 2 diabetes [23]. Thus, these data identify hepatic acetyl-CoA as a potential therapeutic target for patients with poorly controlled type 1 or type 2 diabetes and suggest that approaches to suppress increased rates of WAT lipolysis may be effective in ameliorating fasting hyperglycaemia in patients with poorly controlled diabetes. Whether leptin replacement therapy will be an effective adjuvant to insulin replacement therapy in patients with type 1 diabetes awaits the results of clinical trials. However based on our findings and proposed mechanism of action for the insulin-independent effects of leptin to reverse fasting hyperglycaemia by reducing HPA activity and rates of whole body lipolysis in rat models of poorly controlled diabetes, it would be predicted that leptin therapy would have minimal additional benefits in further improving fasting hyperglycaemia as an adjuvant to insulin replacement therapy in most patients with type 1 diabetes.
Acknowledgments
Funding
Work in the authors’ laboratories is supported by grants from the United States Public Health Service: R01 DK-40936, R01 DK-49230, R01 AG-23686, P30 DK-45735, U24 DK-059635, T32 DK-101019, The Novo Nordisk Foundation Center for Basic Metabolic Research and a Distinguished Clinical Investigator Award from the American Diabetes Association (KFP).
Abbreviations
- DKA
Diabetic ketoacidosis
- HPA
Hypothalamic–pituitary–adrenal
- PKC
Protein kinase C
- STZ
Streptozotocin
Footnotes
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
References
- 1.Petersen KF, Oral EA, Dufour S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. The Journal of clinical investigation. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shulman GI. Cellular mechanisms of insulin resistance. The Journal of clinical investigation. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. The New England journal of medicine. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- 4.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. The Journal of biological chemistry. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 5.Fujikawa T, Berglund ED, Patel VR, et al. Leptin engages a hypothalamic neurocircuitry to permit survival in the absence of insulin. Cell metabolism. 2013;18:431–444. doi: 10.1016/j.cmet.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meek TH, Matsen ME, Dorfman MD, et al. Leptin action in the ventromedial hypothalamic nucleus is sufficient, but not necessary, to normalize diabetic hyperglycemia. Endocrinology. 2013;154:3067–3076. doi: 10.1210/en.2013-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17391–17396. doi: 10.1073/pnas.1008025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinookoswong N, Wang JL, Shi ZQ. Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes. 1999;48:1487–1492. doi: 10.2337/diabetes.48.7.1487. [DOI] [PubMed] [Google Scholar]
- 9.Wang MY, Chen L, Clark GO, et al. Leptin therapy in insulin-deficient type I diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, Park BH, Wang MY, Wang ZV, Unger RH. Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14070–14075. doi: 10.1073/pnas.0806993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L, Philippe J, Unger RH. Glucagon responses of isolated alpha cells to glucose, insulin, somatostatin, and leptin. Endocrine practice: official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2011;17:819–825. doi: 10.4158/EP11101.OR. [DOI] [PubMed] [Google Scholar]
- 12.Kim GH, Szabo A, King EM, Ayala J, Ayala JE, Altarejos JY. Leptin recruits Creb-regulated transcriptional coactivator 1 to improve hyperglycemia in insulin-deficient diabetes. Molecular metabolism. 2015;4:227–236. doi: 10.1016/j.molmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meek TH, Matsen ME, Damian V, Cubelo A, Chua SC, Jr, Morton GJ. Role of melanocortin signaling in neuroendocrine and metabolic actions of leptin in male rats with uncontrolled diabetes. Endocrinology. 2014;155:4157–4167. doi: 10.1210/en.2014-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J, Shulman GI. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic–pituitary–adrenal axis. Nature communications. 2015;6:6980. doi: 10.1038/ncomms7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry RJ, Zhang XM, Zhang D, et al. Leptin reverses diabetes by suppression of the hypothalamic–pituitary–adrenal axis. Nature medicine. 2014;20:759–763. doi: 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minokoshi Y, Toda C, Okamoto S. Regulatory role of leptin in glucose and lipid metabolism in skeletal muscle. Indian journal of endocrinology and metabolism. 2012;16:S562–S568. doi: 10.4103/2230-8210.105573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toda C, Shiuchi T, Kageyama H, et al. Extracellular signal-regulated kinase in the ventromedial hypothalamus mediates leptin-induced glucose uptake in red-type skeletal muscle. Diabetes. 2013;62:2295–2307. doi: 10.2337/db12-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laker RC, Henry BA, Wadley GD, Clarke IJ, Canny BJ, McConell GK. Central infusion of leptin does not increase AMPK signaling in skeletal muscle of sheep. American journal of physiology Regulatory, integrative and comparative physiology. 2011;300:R511–R518. doi: 10.1152/ajpregu.00079.2010. [DOI] [PubMed] [Google Scholar]
- 19.Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. The New England journal of medicine. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 20.Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. The Journal of clinical investigation. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morton GJ, Meek TH, Matsen ME, Schwartz MW. Evidence against hypothalamic-pituitary-adrenal axis suppression in the antidiabetic action of leptin. The Journal of clinical investigation. 2015;125:4587–4591. doi: 10.1172/JCI82723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. The Journal of clinical investigation. 1987;79:1510–1515. doi: 10.1172/JCI112981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry RJ, Camporez JP, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]