

Abstract
Glioblastoma (GBM) is the most common malignant brain tumor in adults. We designed an adeno‐associated virus (AAV) vector for intracranial delivery of secreted, soluble tumor necrosis factor‐related apoptosis‐inducing ligand (sTRAIL) to GBM tumors in mice and combined it with the TRAIL‐sensitizing cardiac glycoside, lanatoside C (lan C). We applied this combined therapy to two different GBM models using human U87 glioma cells and primary patient‐derived GBM neural spheres in culture and in orthotopic GBM xenograft models in mice. In U87 cells, conditioned medium from AAV2‐sTRAIL expressing cells combined with lan C induced 80% cell death. Similarly, lan C sensitized primary GBM spheres to sTRAIL causing over 90% cell death. In mice bearing intracranial U87 tumors treated with AAVrh.8‐sTRAIL, administration of lan C caused a decrease in tumor‐associated Fluc signal, while tumor size increased within days of stopping the treatment. Another round of lan C treatment re‐sensitized GBM tumor to sTRAIL‐induced cell death. AAVrh.8‐sTRAIL treatment alone and combined with lanatoside C resulted in a significant decrease in tumor growth and longer survival of mice bearing orthotopic invasive GBM brain tumors. In summary, AAV‐sTRAIL combined with lanatoside C induced cell death in U87 glioma cells and patient‐derived GBM neural spheres in culture and in vivo leading to an increased in overall mice survival.
Keywords: Glioblastoma, TRAIL, Adeno‐associated virus, Gene therapy, Cardiac glycoside, Lanatoside C
Highlights
We developed an AAVrh.8 vector for local delivery of sTRAIL to the brain parenchyma surrounding GBM.
Lanatoside C can overcome acquired or intrinsic TRAIL resistance in GBM.
Intracranial injection of AAV‐sTRAIL and lanatoside C improved survival in patient‐derived xenograft orthotopic model.
Multiple injections of AAV‐sTRAIL vector around the brain tumor site is a feasible strategy.
Invasive growth of primary GBM remains a problem when combating this disease.
1. Introduction
Glioblastoma (GBM) is the most common malignant primary brain tumor in adults. Despite current therapies, median survival of patients is only 15 months (Stupp et al., 2005). New strategies for the treatment of GBM are much needed. Injection of viral vectors such as adeno‐associated virus (AAV) encoding anti‐tumor proteins is a promising strategy for GBM therapy, however, direct tumor injection with viral vectors leads to focal gene delivery and generally only a small percentage of tumor cells express the transgene (Bowers et al., 2011; Ma et al., 2014). Several AAV serotypes are capable of efficiently transducing cells in the brain after direct injection, with neurons being the primary target (Cearley et al., 2008). We have previously reported in mice that genetically modifying neurons surrounding a brain tumor with secreted anti‐tumor proteins is an effective strategy to kill tumors (Maguire et al., 2008). The transduced neurons effectively serve as a local factory for production of the anti‐tumor protein.
Tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) binds to death receptors (DRs) found specifically on tumor cell (and not normal cell) membranes, which activate a series of signaling events leading to apoptosis (Bouralexis et al., 2005; Song et al., 2003). Unfortunately, some forms of cancer, particularly adult GBMs, are notoriously resistant to treatment with TRAIL (Hao et al., 2001; Panner et al., 2005; Seol, 2011). Previously, through drug screening, we identified the cardiac glycoside lanatoside C (lan C) to sensitize GBM cells to TRAIL in culture and in a subcutaneous GBM mouse model (Badr et al., 2011). A low dose of lan C combined with TRAIL killed over 90% of U87 GBM cells, while it had no significant effect on primary fibroblasts. Cardiac glycosides such as lan C have shown to act as neuroprotective agents, which shows that these compounds can efficiently penetrate the brain (Johansson et al., 2001; Wang et al., 2006). Major limitations of using TRAIL for brain tumor therapy are the inability of TRAIL protein to cross the blood–brain barrier as well as its short half‐life in vivo and potential liver toxicity (Duiker et al., 2012; Xiang et al., 2004).
In the present study, we developed an AAVrh.8 serotype vector for the delivery of a secreted, soluble form of TRAIL (sTRAIL) to GBM tumors in the mouse brain via intracranial (i.c.) injection. In this case, the normal brain surrounding the tumor secretes active TRAIL, which in turn finds and binds to death receptors on GBM cells inducing apoptosis upon systemic administration of lanatoside C. We showed that this combined strategy has a therapeutic benefit in two different intracranial GBM models, U87 cells as well as patient‐derived invasive GBM neural spheres.
2. Methods
2.1. Cell culture and lentiviral vectors
293T cells and U87 human GBM cells obtained from American Type Culture Collection (ATCC; Manassas, VA) were cultured in high‐glucose Dulbecco's Modified Eagle Medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO) and 100 U/mL penicillin/100 μg/mL streptomycin (Invitrogen). Primary human GBM8 stem‐like cells have been previously described (Wakimoto et al., 2009) and were cultured as neural spheres in serum‐free NeuroCult™ NS‐A Basal Medium with Proliferation Supplement (StemCell Technologies, Vancouver, BC), supplemented with 20 ng/mL recombinant epidermal growth factor (EGF; R&D Systems, Minneapolis, MN), 10 ng/mL basic fibroblast growth factor (bFGF; Peprotech, Rocky Hill, NJ) and 2 μg/mL heparin (Sigma). All cells were cultured at 37 °C under 5% CO2 humidified atmosphere.
For in culture use, AAV vectors were packaged as AAV2 serotype. U87 cells were transduced with the AAV2‐CBA‐GFP or AAV2‐CBA‐sTRAIL vector at a ratio of 1000 genome copy (g.c.)/cell. Three days post‐infection, the medium from these cells containing sTRAIL or control was harvested. U87 cells were cultured in 24‐well plates and one day after seeding, medium was removed and conditioned medium from sTRAIL or GFP‐expressing cells was added for the following groups: (1) control medium, (2) control medium + DMSO, (3) control medium + 0.25 μM lan C, (4) sTRAIL medium, (5) sTRAIL medium + DMSO, (6) sTRAIL medium + 0.25 μM lan C. Twenty‐four hours after treatment, quantification of cell viability was performed by measuring ATP in metabolically active cells using the commercially available CellTiter‐Glo® assay per manufacturer's protocol (Promega, Madison, WI). The same experiment was repeated on GBM8 cells cultured as described above after allowing them to form neural spheres for at least two days before treatment was initiated.
To generate U87 cells and GBM8 neural spheres stably expressing firefly luciferase (Fluc) and mCherry fluorescent protein, these cells were transduced with a lentivirus vector based on CSCW2‐Fluc‐IRES‐mCherry at a multiplicity of infection of 10 transducing units/cell as previously described (Maguire et al., 2008) generating U87‐Fluc/mCherry (U87‐FM) or GBM8‐Fluc/mCherry (GBM8‐FM).
2.2. Adeno‐associated virus (AAV) vectors
The pAAV‐CBA‐sTRAIL vector consists of a transgene cassette for soluble, secreted TNF‐related apoptosis‐inducing ligand (sTRAIL) carrying amino acid (a.a.) 1–150 from human Flt3L, an isoleucine zipper domain, and the extracellular domain (a.a. 114–281) of the human TRAIL, based on the previously reported h‐Flex‐zipper‐TRAIL (Maguire et al., 2013a; Wu et al., 2001). The transgene is controlled by a hybrid cytomegalovirus immediate early enhancer/chicken beta‐actin promoter (CMV IE/CBA), while the woodchuck hepatitis virus post‐transcriptional regulatory element (WPRE) is downstream of the transgene. As a control, a similar vector expressing GFP was used.
AAV vectors were produced using triple transfection of 293T cells as previously described (Broekman et al., 2006). Briefly, AAV helper plasmid Fd6, AAV2 rep/cap plasmid (used to generate AAV2 for in vitro culture use) or AAVrh.8 rep/cap plasmid (to generate AAVrh.8 for in vivo use) and the AAV2 ITR‐containing transgene expression plasmid (sTRAIL or GFP, both single‐stranded genomes) were used for triple transfection of 293T cells by the calcium phosphate method. Three days after transfection, the cells were harvested, lysed and purified using iodixanol gradient ultracentrifugation followed by anion exchange chromatography. Vectors were titered by quantitative polymerase chain reaction (qPCR) for AAV genomes using primers against the poly A signal sequence. A typical titer of 1013 genome copies (g.c.) per mL was obtained using this strategy.
2.3. Lanatoside C
Lanatoside C was purchased from Sigma, resuspended in DMSO at 20 mg/mL concentration, and stored in aliquots at −80 °C. Aliquots were thawed one time and any remaining solution was discarded.
2.4. Animal studies
All animal experiments were approved by the Massachusetts General Hospital (MGH) Subcommittee on Research Animal Care and performed in accordance with their guidelines and regulations as set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Female six‐to eight‐week old athymic nude mice were anesthetized with a mixture of 100 mg/kg ketamine and 5 mg/kg xylazine in 0.9% sterile saline. GBM cells were intracranially injected at a rate of 0.4 μL/min using a Micro 4 Microsyringe Pump Controller (World Precision Instruments, Sarasota, FL) attached to a Hamilton syringe with a 33‐gauge needle (Hamilton, Reno, NV) into the mid‐left striatum at the following coordinates in mm from bregma: +0.5 anterior‐posterior, +2.0 medio‐lateral, −2.5 dorso‐ventral. In the U87 glioma model, each mouse was injected with approximately 105 U87‐FM cells in 2 μL of Opti‐MEM. In the primary GBM8 stem‐like cell model, neural spheres were dissociated using Accutase (Innovative Cell Technologies, San Diego, CA), two days before implantation and allowed to form small spheres. Approximately 105 GBM8‐FM cells (3000 intact spheres) resuspended in 2 μL Opti‐MEM were injected in each mouse into the left mid‐striatum using the same coordinates as above.
AAVrh.8‐CBA‐sTRAIL or AAVrh.8‐CBA‐GFP vectors were infused using the Microsyringe Pump Controller attached to a Hamilton syringe with a 33‐gauge needle at a rate of 0.2 μL/min. Coordinates were either the same as for tumor cell injections, or adapted for the multiple injections experiment to the following coordinates from bregma: +0.7 anterior‐posterior, +2.0 medio‐lateral; +0.3 anterior‐posterior, +2.2 medio‐lateral; +0.3 anterior‐posterior, +1.8 medio‐lateral; and −2.5 dorso‐ventral.
2.5. Bioluminescence imaging
d‐luciferin was purchased from Gold Biotechnology® (St. Louis, MO) and resuspended at 25 mg/mL in PBS. For Fluc imaging, mice were injected i.p. with 200 mg/kg body weight of d‐luciferin, and imaged 10 min later using the IVIS® Spectrum optical imaging system fitted with an XGI‐8 Gas Anesthesia System (Caliper Life Sciences, Hopkinton, MA). Bioluminescent images were acquired using the auto‐exposure function. Data analysis for signal intensities and image comparisons were performed using Living Image® software (Caliper Life Sciences).
2.6. Statistical analysis
GraphPad Prism v6.01 software (LaJolla, CA) was used for statistical analysis of all data. A p‐value less than 0.05 was considered to be statistically significant. For analysis between multiple groups, a one‐way analysis of variance (ANOVA) was performed followed by Sidak's multiple comparison test to compare differences between two groups. An unpaired two‐tailed t‐test was used for the comparison of two samples. Survival was analyzed using Kaplan–Meier curves and log‐rank (Mantel–Cox) tests.
3. Results
3.1. Secreted TRAIL in producer cell conditioned medium kills recipient glioma cells when combined with lanatoside C
Previously, we have shown that lanatoside C (lan C) can sensitize glioma cells to TRAIL‐induced cell death, thus overcoming intrinsic resistance both in cell culture and in a subcutaneous glioma mouse model (Badr et al., 2011). We have designed an AAV vector to express a secreted soluble form of TRAIL (sTRAIL) (Maguire et al., 2013a; Wu et al., 2001). To confirm that the AAV vector produces secreted active TRAIL, U87 cells were transduced with AAV2‐CBA‐sTRAIL or AAV2‐CBA‐GFP control (referred to as AAV‐sTRAIL or AAV‐GFP) and conditioned medium was collected. This method of gene transfer yields a TRAIL concentration of approximately 125 ng/mL in the conditioned medium from AAV‐sTRAIL transduced cells (Maguire et al., 2013a). We treated U87 cells with this conditioned medium or recombinant TRAIL protein alone or in combination with lanatoside C. Treating with sTRAIL‐containing conditioned medium did not cause any cell death as compared to the control showing that these cells are resistant to TRAIL (Figure 1B). On the other hand, a significantly lower number of viable cells (around 50%) were observed after treatment with 0.25 μM lan C alone (p < 0.001), showing that lan C has some toxicity on U87 cells in culture at this dose (Figure 1B). Combining sTRAIL‐containing medium and lan C resulted in a significant decrease in cell viability between control‐treated cells (around 80%; p < 0.001), cells treated with AAV‐sTRAIL alone (p < 0.001) or cells treated with lan C alone (p = 0.0107; Figure 1B). A similar effect was observed with recombinant TRAIL protein, with significantly less viable cells in the TRAIL + lan C treated wells compared to TRAIL treatment alone (p < 0.001). While U87 cells are resistant to TRAIL‐induced cell death, GBM8 neural spheres were more sensitive to sTRAIL and had higher response to lan C (Figure 1C). However, combined treatment with lan C resulted in a significantly lower number of viable cells compared to control medium alone (around 90%; p < 0.001), sTRAIL‐containing medium alone (p = 0.0014), and control medium + lan C (p = 0.0137; Figure 1C). These results show that upon AAV transduction, sTRAIL is secreted in an active form and can kill GBM cells in combination with lanatoside C.
Figure 1.
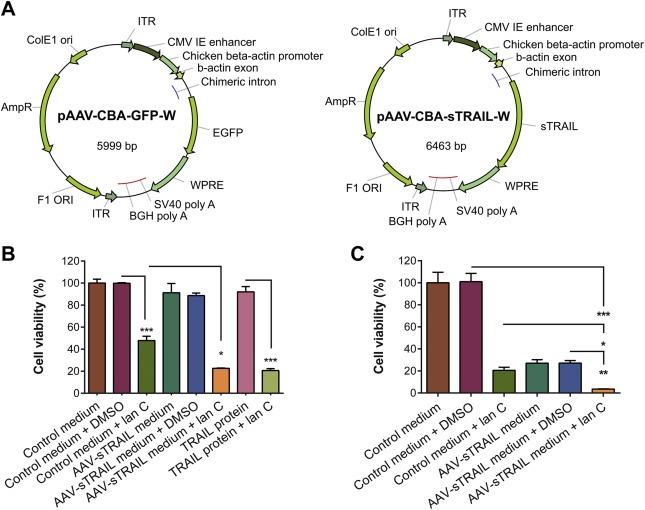
Conditioned media from cells transduced with AAV2‐sTRAIL kills recipient U87 glioma cells when used in combination with lanatoside C. U87 cells were transduced with AAV‐GFP (control) or AAV‐sTRAIL, of which vector maps are shown in (A). Three days later, conditioned medium was transferred onto non‐transduced U87 cells. (B) U87 cells show insensitivity to the treatment with TRAIL protein or AAV‐sTRAIL medium alone, while a significant decrease in cell viability is observed when TRAIL protein or AAV‐sTRAIL is combined with lanatoside C, compared to AAV‐GFP control. (C) GBM8 cells neural spheres were found to be sensitive to the treatment with lanatoside C or AAV‐sTRAIL alone, but AAV‐sTRAIL combined with lanatoside C induced a significant decrease in cell viability, compared to AAV‐GFP control medium. Data presented as % cell viability in which the control group is set at 100% (*p < 0.05, **p < 0.01, ***p < 0.001).
3.2. Lanatoside C is a potent sensitizer of TRAIL‐mediated cell death in a GBM mouse model
We sought to test the effect of lan C on its ability to sensitize human GBM tumors to sTRAIL delivered by AAVrh.8 vector (hereafter referred to as AAV‐sTRAIL), in an intracranial mouse model. The AAVrh.8 capsid was chosen for its excellent ability to transduce normal murine brain cells (Maguire et al., 2008). In this model, 105 U87‐FM cells were stereotactically injected into the striatum of nude mice. Tumors were allowed to form, as monitored by Fluc bioluminescence imaging. Ten days post‐tumor implantation, convection‐enhanced delivery was used to inject the AAV‐sTRAIL vector into the mouse brain at the same coordinates that were used for tumor cell implantation, which primarily results in the transduction of neurons and minimal tumor cells (Maguire et al., 2008). Five days post‐injection of AAV‐sTRAIL, mice were randomized into two groups (n = 6 per group) each receiving alternating treatment of lan C. Group A received daily i.p. injections of lan C (7.5 mg/kg) for five days while group B received vehicle treatment. Next, Group B received daily i.p. injection of lan C for five days, while group A was taken off the drug. Finally, lan C treatment was stopped in group B, while group A was kept on lan C for the remainder of the study (15 days; Figure 2A, B). In group A, all mice bearing intracranial U87 tumors responded to lan C, as observed by a rapid 14‐fold decrease in tumor‐associated Fluc signal after 5 days of treatment (Figure 2A). When taken off lan C, tumors started to regrow very quickly and a significant 37‐fold increase (p = 0.0190) in tumor Fluc signal was observed. When lan C was re‐administered to this group, tumor‐associated Fluc signal decreased 37‐fold over time (p = 0.0365), and remained stable until the end of the experiment (Figure 2A). On the other hand, tumor‐associated Fluc signal continued to increase in Group B upon vehicle treatment, reaching a 15‐fold increase in ten days post AAV‐sTRAIL injection. When receiving lan C, a response to treatment was observed as a 17‐fold decrease in the total Fluc signal from the tumor. Once lan C treatment in group B was stopped, tumors started to regrow, resulting in a significant 380‐fold increase in tumor‐associated signal (p < 0.001) over the course of ten days (Figure 2B). All remaining mice from both groups were sacrificed 40 days after tumor inoculation, with all mice in group A showing no increase in tumor‐associated signal, indicating signs of stable disease (Figure 2A). Histology on brain sections using hematoxylin and eosin (H&E) staining confirmed a visual difference in tumor size between the two groups (Figure 2C). These results show that lan C could re‐sensitize tumors with acquired TRAIL resistance. As a control and to show that the decrease in Fluc signal is not a lan C artifact on bioluminescence signal detection, we repeated the same experiment as above but infused mice with a control AAVrh.8‐CBA‐GFP vector (referred to as AAV‐GFP). Five days post‐vector implantation, lanatoside C was administered for 7 days, however, the tumor size continued to increase over time significantly (p = 0.0246) showing that the AAV‐GFP control vector combined with lan C does not have any effect on tumor‐associated Fluc signal (Figure 2D).
Figure 2.
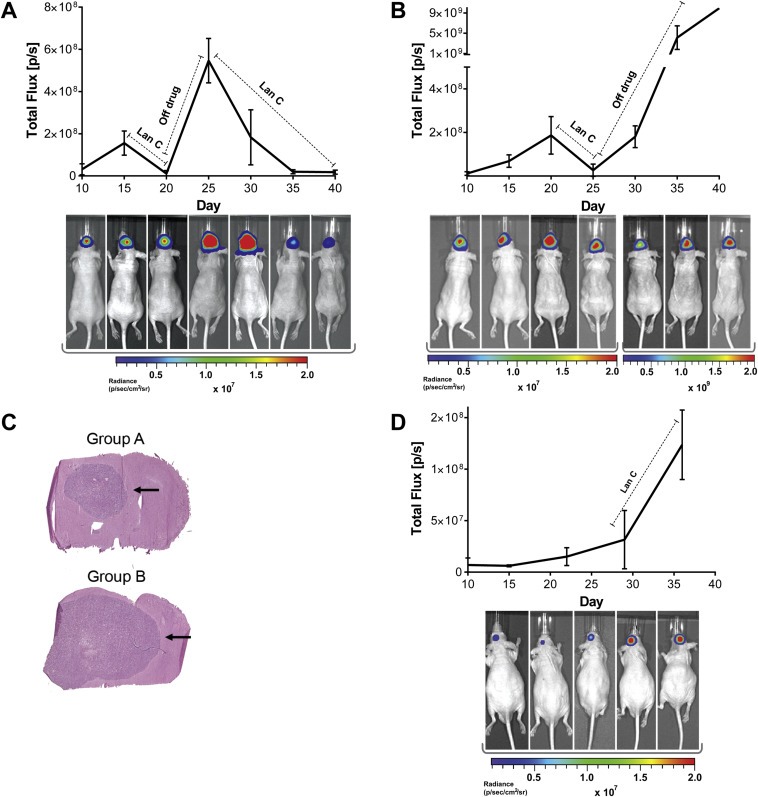
Lanatoside C cycling resensitizes U87 glioma cells to TRAIL‐induced apoptosis. (A) Mice bearing U87‐FM brain tumors were intracranially infused with AAV‐sTRAIL vector. Five days post‐AAV‐sTRAIL injection, mice were treated with lanatoside C for five days, then were taken off the drug for five days, and then kept on lan C until the end of the study. (B) Same as (A), except that ten days after AAV‐sTRAIL injection, mice were treated with lan C for five days, and then taken off the drug until the end of the study. (C) A representative H&E stained brain section at the end of the study, of a mouse from each of the groups. (D) Similar to (A) and (B), except that U87‐FM tumors were intracranially infused with AAV‐GFP control vector. Bioluminescence imaging was used to measure tumor volume. A representative mouse from each group is shown at each time point. Please note that all the time points in (A) and (C) and the first four time points in (B) are in the range of 1 × 106–2 × 107 photons/sec, while the last three time points in (B) are in the range of 1 × 108–2 × 109 photons/sec to prevent saturation of the signal.
3.3. AAVrh.8‐sTRAIL combined with lanatoside C decreases invasive patient‐derived GBM tumor growth leading to an increased survival
Having demonstrated the sTRAIL‐sensitizing effect of lan C on intracranial human GBM tumors in vivo, we then evaluated this combined therapy in an invasive patient‐derived GBM orthotopic xenograft model using GBM8 stem‐like cells cultured as neural spheres (Wakimoto et al., 2009). As before, GBM8‐FM neural spheres were stereotactically implanted in the striatum of nude mice. Fourteen days later, mice were imaged for tumor‐associated Fluc signal and divided into four groups such that the mean signal in each group is similar (n = 8 per group; Figure 3 and Supplementary Figure 1A). At day 22 post‐tumor inoculation, where no significant difference in mean Fluc signal was present between the groups (p = 0.9955; Supplementary Figure 1A), convection‐enhanced delivery was used to infuse mice brain with 1010 g.c. of vector at the site of tumor implantation. Daily i.p. injections of 7.5 mg/kg lan C were given to two of the groups, starting the day after AAV infusion, with the treatment groups being as follows: (1) AAV‐GFP + vehicle, (2) AAV‐GFP + lan C, (3) AAV‐sTRAIL + vehicle, (4) AAV‐sTRAIL + lan C, (n = 8 per group). Tumor volume was monitored once a week by Fluc imaging. After the first week, the Fluc signal in the AAV‐GFP + vehicle and AAV‐GFP + lan C groups had increased by 8.5‐fold and 17‐fold, respectively. In contrast, at the same time point, the Fluc signal in mice treated with AAV‐sTRAIL + vehicle and AAV‐sTRAIL + lan C groups showed a 4‐fold and 7‐fold decrease in Fluc signal, and therefore tumor volume, respectively (Figure 3A,B). The two control groups reached a maximum Fluc signal between two and three weeks post‐treatment, after which they were sacrificed due to reaching predetermined humane endpoint criteria. At day 30, tumors treated with AAV‐sTRAIL + vehicle started to re‐grow, probably due to acquired resistance of the tumor to sTRAIL, reaching a 20‐fold increase in Fluc signal over 4 weeks. Conversely, mice in the combination treatment group (AAV‐sTRAIL + lan C) showed only a 1.5‐fold increase in Fluc signal over the same period (Figure 3A, B). At day 38 post‐tumor implantation, an overall significant difference in tumor‐associated Fluc signal between all four groups was observed (p = 0.0305). However, post‐hoc analysis showed that individual differences approached but did not achieve significance between the AAV‐GFP + vehicle vs. AAV‐sTRAIL group (p = 0.0556) and the AAV‐GFP + vehicle vs. AAV‐sTRAIL + lan C group (p = 0.0540).
Figure 3.
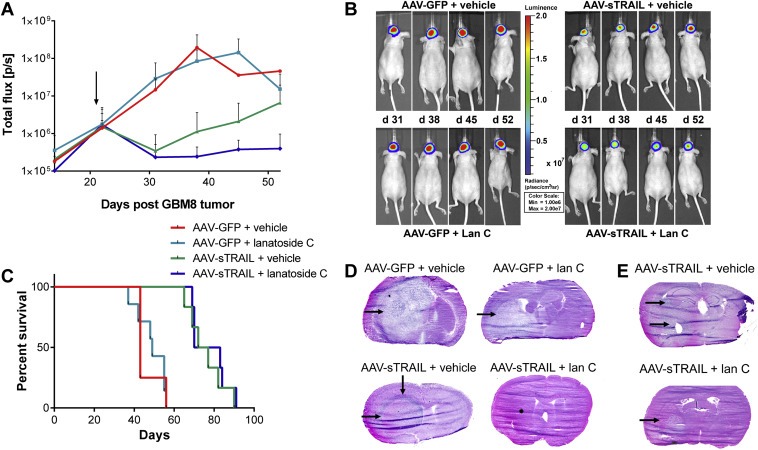
AAVrh.8‐sTRAIL combined with lanatoside C slows invasive primary glioma growth. (A) Mice were intracranially injected with primary GBM8‐FM stem‐like glioma neural spheres. After 21 days, mice were divided into four groups which were treated with AAV‐GFP + vehicle, AAV‐GFP + lan C, AAV‐sTRAIL + vehicle or AAV‐sTRAIL + lan C. The arrow indicates the start of treatment. (B) Weekly bioluminescence imaging was used to assess tumor response to treatment. A representative mouse at significant time points is shown. (C) Kaplan–Meier survival curves for the different groups. (D, E) H&E staining of brain slices for the different groups. A representative image is shown.
An overall significant difference in survival between all groups was observed, although the tumor signal eventually increased in the AAV‐sTRAIL and AAV‐sTRAIL + lan C groups, regardless of therapy (p < 0.0001). Between‐group analysis showed that there was no significant difference in survival between the AAV‐GFP + vehicle vs. AAV‐GFP + lan C group which shows that lan C on its own did not provide a therapeutic benefit. On the other hand, a significant difference was found in the AAV‐GFP + vehicle vs. AAV‐sTRAIL + vehicle group (p = 0.0013), the AAV‐GFP + vehicle vs. AAV‐sTRAIL + lan C group (p = 0.0006), the AAV‐GFP + lan C vs. AAV‐sTRAIL + vehicle group (p = 0.0004), and the AAV‐GFP + lan C vs. AAV‐sTRAIL + lan C group (p = 0.0002) showing that sTRAIL provided therapeutic benefit (Figure 3C). Surprisingly, there was no significant difference between AAV‐sTRAIL + vehicle vs. AAV‐sTRAIL + lan C group which shows that despite the decrease in tumor volume (as observed by Fluc imaging), the addition of lan C did not provide survival benefit. Median survival was 43 days for the AAV‐GFP + vehicle group, 49 days for the AAV‐GFP + lan C group, 74.5 days for the AAV‐sTRAIL + vehicle and 76.5 days for the AAV‐sTRAIL + lan C group (Figure 3C). To corroborate these findings ex vivo, histological analysis using H&E staining on brains of mice in the different treatment groups was performed. Mice treated with the AAV‐GFP + vehicle control showed large multi‐lobed tumors (Figure 3D). Large tumors at the injected side were also apparent in the AAV‐GFP + lan C treated mice (Figure 3D). In contrast, mice treated with AAV‐sTRAIL + vehicle had more diffuse tumors with a “ring‐like” phenotype around the site of vector injection in the striatum. Secondary tumor masses were observed away from the injection site, and tumor cell infiltration was found along the corpus callosum (Figure 3D, E). In mice treated with both AAV‐sTRAIL and lan C, no visible tumor masses were observed at the site of vector injection showing efficient therapy against the primary tumor mass, which confirms Fluc imaging, however, tumor cells appeared to have migrated away from the therapeutic zone with highest sTRAIL expression and secondary tumors were found more caudally, e.g. in the hippocampus (Figure 3E).
3.4. Multi‐site injection of AAVrh.8‐sTRAIL combined with lanatoside C combats invasiveness
The observation that GBM8 stem‐like cells were able to escape the “zone of resistance” around the tumor led us to hypothesize that multi‐site injections of AAV‐sTRAIL around the tumor would prevent this by creating a larger barrier to tumor growth. The same experiment as above was repeated and mice were divided into four groups so that the average signal between groups was similar (p > 0.9999; Supplementary Figure 1B). At 21 days post‐tumor implantation, three injections were performed around the site of tumor implantation using convection‐enhanced delivery, to slowly infuse 1010 g.c. of AAV‐GFP or AAV‐sTRAIL vector at each site. The next day, daily i.p. injection of lan C was initiated resulting in the following treatment groups: (1) AAV‐GFP + vehicle (n = 4), (2) AAV‐GFP + lan C (n = 4), AAV‐sTRAIL + vehicle (n = 8), AAV‐sTRAIL + lan C (n = 8). After the first week, the tumor‐associated Fluc signal had increased 23‐fold and 16‐fold in the AAV‐GFP + vehicle and AAV‐GFP + lan C groups, respectively. A peak Fluc signal was reached in these groups two weeks post‐treatment and these mice were sacrificed due to humane endpoint (Figure 4A, B). At this time there was a significant difference between all treatment groups (p = 0.0002; Supplementary Figure 2A). The AAV‐sTRAIL + vehicle group showed a significant decrease in tumor growth as compared to the two control groups, and the addition of lan C further enhanced this therapy. AAV‐sTRAIL + vehicle and AAV‐sTRAIL + lan C groups increased only 2‐fold and 1‐fold, respectively, one week post‐treatment and the signal remained stable two‐to‐four weeks post‐treatment. Finally, in the AAV‐sTRAIL + vehicle group, a 17‐fold increase in tumor‐associate Fluc signal was observed four‐to‐seven weeks post‐treatment, and all remaining animals in this group were sacrificed at this time after reaching the humane endpoint (Figure 4A). At 7 weeks post‐treatment, there was a significant difference in tumor‐associated Fluc signal between the AAV‐sTRAIL + vehicle vs. AAV‐sTRAIL + lan C groups (p < 0.0001; Supplementary Figure 2B). In the AAV‐sTRAIL + lan C group, the signal had decreased 5‐fold at four weeks compared to two weeks post‐treatment. After week eight, however, a significant 14‐fold increase in Fluc signal was observed (p = 0.0067) and one week later (9 weeks post treatment), all remaining animals were sacrificed (Figure 4A) due to tumor burden.
Figure 4.
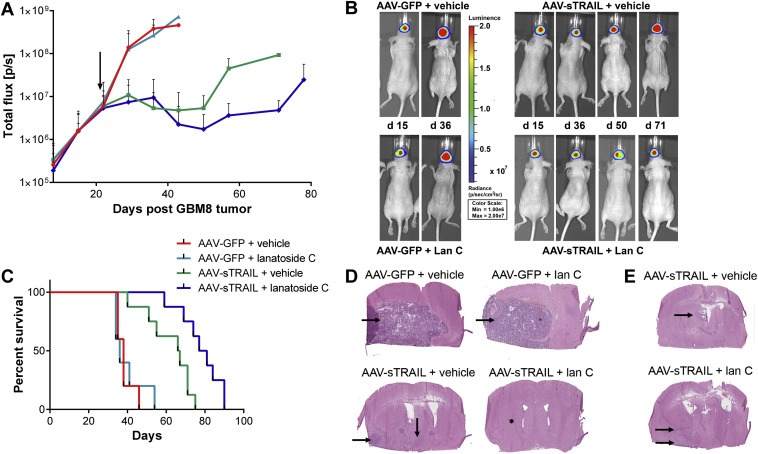
Multi‐site injection of AAVrh.8‐sTRAIL combined with lanatoside C extends survival over AAV‐sTRAIL alone. Mice bearing intracranial primary GBM8‐FM stem‐like cells glioma tumor were infused with AAV‐sTRAIL or AAV‐GFP at three different sites around the tumor. Each group of mice was then divided into 2 subgroups which either received vehicle or lan C. (A, B) Weekly bioluminescence imaging was used to assess tumor response to treatment. A representative mouse at significant time points is shown in (B). The arrow indicates the start of treatment. (C) Kaplan–Meier survival curves for the different groups are shown. (D, E) H&E staining of brain slices for the different groups. A representative image is shown.
Overall there was a significant difference in survival between all treatment groups (p < 0.0001). Analysis between groups showed that there was no significant difference in the AAV‐GFP + vehicle vs. AAV‐GFP + lan C groups, with a median survival of 38 and 36 days, respectively. Differences in survival were significant in the AAV‐GFP + vehicle vs. AAV‐sTRAIL + vehicle (p = 0.0006) and AAV‐GFP + vehicle vs. AAV‐sTRAIL + lan C (p = 0.0002) groups. Similarly, there was a significant difference in the AAV‐GFP + lan C vs. AAV‐sTRAIL + vehicle (p = 0.0028) and AAV‐GFP + lan C vs. AAV‐sTRAIL + lan C (p = 0.0001) groups. Finally, in contrast to the experiment with a single vector injection, there was a significant difference in survival in the AAV‐sTRAIL + vehicle vs. AAV‐sTRAIL + lan C group (p = 0.0062; Figure 4C) with a median survival of 66.5 and 79 days, respectively. Histological analysis again showed large lobular tumors in the AAV‐GFP + vehicle and the AAV‐GFP + lan C treated groups on H&E stained brain sections (Figure 4D). In mice treated with sTRAIL + vehicle, few visible tumor cells were found at the site of tumor injection, with tumor cells migrating ventrally and caudally (Figure 4D, E). No tumor cells were found at the injection site in mice treated with AAV‐sTRAIL + lan C, however, cells appeared to have migrated away from the injection site towards the hippocampus (Figure 4D, E). All of these data suggest that multiple injection of AAV‐sTRAIL vector is required to achieve widespread gene delivery to treat GBM tumors in combination with lan C.
4. Discussion
The lack of successful novel treatments for glioblastoma (GBM) underscores the complexity of this disease. TRAIL has been regarded as an anti‐cancer agent, however, some types of cancer, including gliomas, are resistant to TRAIL‐induced cell death (Hao et al., 2001; Panner et al., 2005; Seol, 2011). This can be caused by downregulation of death receptors (Ding et al., 2011), expression of decoy receptors on cells (Pan et al., 1997, 1997, 1997), deregulation of the mTOR signaling pathway, which can result in overexpression of c‐FLIP (Panner et al., 2005) and induction of NF‐κB by TRAIL (Ibrahim et al., 2001; Khanbolooki et al., 2006). Through drug screening, we have previously shown that the family of cardiac glycosides, including lanatoside C (lan C), sensitizes glioma cells to TRAIL‐induced cell death, partially through upregulation of death receptors (Badr et al., 2011). Cardiac glycosides have been recently shown to provide neuroprotection against ischemic stroke (Johansson et al., 2001; Wang et al., 2006), suggesting efficient penetration of these drugs to the brain. The major disadvantages of using TRAIL for brain tumor therapy are its inability to cross the blood–brain barrier and its short half‐life in vivo with potential liver toxicity (Duiker et al., 2012; Xiang et al., 2004). To address these problems, we explored AAV‐mediated gene delivery of sTRAIL directly to the brain tumor milieu. Using this approach, the brain parenchyma is engineered to synthesize and secrete soluble TRAIL (sTRAIL), which can form a “zone of resistance” around the tumor. Direct injection into the brain bypasses the blood–brain barrier, and as long as AAV‐sTRAIL is present, new sTRAIL will be synthesized, which can bind to death receptors found only on GBM cells and not the normal brain. Co‐administration of lan C can then sensitize the resistant GBM cells to TRAIL‐induced apoptosis. Clinical trials have already evaluated the use of TRAIL, AAV vectors and lan C, separately, and therefore this approach has the potential to be translated for use in the clinic.
Another disadvantage of using TRAIL for cancer therapy is the ability of the tumor to acquire resistance to this treatment. Here, we showed that lanatoside C can sensitize GBM tumor cells to AAV‐sTRAIL therapy. However, once mice were taken off lan C therapy, the GBM tumor treated with AAV‐sTRAIL started to regrow, potentially due to acquired resistance. Importantly, lan C re‐administration could reverse this growth and re‐sensitize the resistant GBM tumor cells to sTRAIL. These results suggest that lan C could not only overcome intrinsic sTRAIL resistance, but could restore TRAIL‐acquired resistance in GBM. The exact mechanism of GBM sensitization to sTRAIL by lan C is still unknown. In previous work, we showed that lanatoside C upregulates death receptor 5 (DR5) on GBM cells, which could be partially responsible for TRAIL sensitization (Badr et al., 2011). Different TRAIL sensitizers could affect different pathways leading to a more‐ or less‐pronounced effect on tumor regression. Combining lan C with other sensitizers could therefore be beneficial to achieve an even greater effect of TRAIL‐induced apoptosis. For example, the proteasome inhibitor bortezomib has been shown as an effective TRAIL sensitizer on different glioma cell lines (Jane et al., 2011), through downregulation of c‐FLIP and increased expression of DRs (de Wilt et al., 2013) as well as activation of the NF‐κB pathway. Furthermore, the high basal level of NF‐κB was an indication of resistance against TRAIL‐induced apoptosis, while no correlation was found between sensitivity of the cells and expression of other pro‐ or anti‐apoptotic proteins. This is in line with the hypothesis that NF‐κB inhibition is important in TRAIL sensitization, which is also observed in our studies with lan C (Badr et al., 2011; Jane et al., 2011).
One of the current pitfalls in testing novel GBM therapeutics is the use of in vivo animal models that do not recapitulate a phenocopy of the human tumor. Typical cell lines such as U87 form local tumors that do not invade the brain and therefore do not mimic the behavior of human GBM. Thus, we tested the AAV‐sTRAIL and lan C therapy using primary cells dissociated from GBM patient specimen and grown as stem‐like cell neural spheres which invade the mouse brain upon intracranial injection, similar to the original human tumor (Wakimoto et al., 2009). A single injection of AAV‐sTRAIL showed an initial tumor response, which eventually progressed probably due to acquired resistance to sTRAIL. The addition of lan C showed an enhanced GBM response to sTRAIL, but not an increase in overall survival due to GBM cells escaping the “zone of resistance” around the vector injection site, with the highest concentration of sTRAIL, as well as secondary tumor formation away from the injection site due to tumor cell migration. On the other hand, multiple vector injection prolonged mice survival even further in combination with lan C. Interestingly, GBM cells appeared to have migrated more caudally and towards the ventral side of the brain in animals injected with AAV‐sTRAIL. Due to the nature of i.c. injections, sTRAIL might have had a stronger effect in the dorsal side of the brain close to the injection site, where the highest concentration was present. While both the single‐ and multi‐injection approach of sTRAIL combined with lan C showed a modest survival benefit, animals eventually succumbed to the disease. The invasive growth of GBM is a major problem in controlling this disease. Single cells that escape the therapeutic zone of resistance can form a new tumor elsewhere in the brain, as observed here, where tumor cells were found in the hippocampus. Widespread expression of sTRAIL in the brain is probably necessary in order to combat these invasive cells. One way of achieving this would be systemic injection of an AAV vector that can bypass the blood–brain barrier (e.g. AAV9), which can lead to widespread gene delivery in the brain (Foust et al., 2009; Gray et al., 2011; Maguire et al., 2013b; Zhang et al., 2011).
In conclusion, we developed an AAV serotype rh.8 vector to engineer the brain parenchyma surrounding the tumor to synthesize and secrete sTRAIL, which in turn finds and binds DRs on GBM cells and kill them upon administration of lan C. While GBM is intrinsically resistant to TRAIL therapy, or could acquire resistance during treatment, co‐administration of lanatoside C can re‐sensitize these cells to TRAIL‐induced cell death. Treatment with AAV‐sTRAIL alone or in combination with lanatoside C resulted in a significantly improved survival in two different mouse models, however, in both cases, mice eventually succumbed to the disease due to migrating tumor cells. New methods for widespread gene delivery to the brain would enhance GBM response to the combined sTRAIL and lan C therapy.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
This study was supported by grants from the National Institutes of Health (NIH), the National Institute of Neurological Disorders and Stroke (NINDS) R01NS064983 and the National Cancer Institute (NCI) R01CA166077 (BAT), the American Brain Tumor Association Discovery Grant and NIH/NINDS R21NS081374 (CM). The authors acknowledge the support from 1S10RR025504 Shared Instrumentation Grant IVIS imaging system that was used to acquire imaging data. M.H.W.C. was supported by a scholarship from KWF Kankerbestrijding (Dutch Cancer Society). The authors would like to thank Kevin Conway for the production of lentivirus vectors at the MGH Vector Core, Charlestown, MA, USA (supported by NIH/NINDS P30NS045776; BAT).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.11.011.
Crommentuijn Matheus H.W., Maguire Casey A., Niers Johanna M., Vandertop W. Peter, Badr Christian E., Würdinger Thomas, Tannous Bakhos A., (2016), Intracranial AAV‐sTRAIL combined with lanatoside C prolongs survival in an orthotopic xenograft mouse model of invasive glioblastoma, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.11.011.
References
- Badr, C.E. , Wurdinger, T. , Nilsson, J. , Niers, J.M. , Whalen, M. , Degterev, A. , Tannous, B.A. , 2011. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor-related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro-oncology. 13, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouralexis, S. , Findlay, D.M. , Evdokiou, A. , 2005. Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis. 10, 35–51. [DOI] [PubMed] [Google Scholar]
- Bowers, W.J. , Breakefield, X.O. , Sena-Esteves, M. , 2011. Genetic therapy for the nervous system. Hum. Mol. Genet. 20, R28–R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman, M.L. , Comer, L.A. , Hyman, B.T. , Sena-Esteves, M. , 2006. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 138, 501–510. [DOI] [PubMed] [Google Scholar]
- Cearley, C.N. , Vandenberghe, L.H. , Parente, M.K. , Carnish, E.R. , Wilson, J.M. , Wolfe, J.H. , 2008. Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol. Ther. 16, 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, L. , Yuan, C. , Wei, F. , Wang, G. , Zhang, J. , Bellail, A.C. , Zhang, Z. , Olson, J.J. , Hao, C. , 2011. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Invest. 29, 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duiker, E.W. , Dijkers, E.C. , Lambers Heerspink, H. , de Jong, S. , van der Zee, A.G. , Jager, P.L. , Kosterink, J.G. , de Hooge Vries, E.G. , Lub-de, M.N. , 2012. Development of a radioiodinated apoptosis-inducing ligand, rhTRAIL, and a radiolabelled agonist TRAIL receptor antibody for clinical imaging studies. Br. J. Pharmacol. 165, 2203–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust, K.D. , Nurre, E. , Montgomery, C.L. , Hernandez, A. , Chan, C.M. , Kaspar, B.K. , 2009. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, S.J. , Matagne, V. , Bachaboina, L. , Yadav, S. , Ojeda, S.R. , Samulski, R.J. , 2011. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 19, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, C. , Beguinot, F. , Condorelli, G. , Trencia, A. , Van Meir, E.G. , Yong, V.W. , Parney, I.F. , Roa, W.H. , Petruk, K.C. , 2001. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 61, 1162–1170. [PubMed] [Google Scholar]
- Ibrahim, S.M. , Ringel, J. , Schmidt, C. , Ringel, B. , Müller, P. , Koczan, D. , Thiesen, H.J. , Löhr, M. , 2001. Pancreatic adenocarcinoma cell lines show variable susceptibility to TRAIL-mediated cell death. Pancreas. 23, 72–79. [DOI] [PubMed] [Google Scholar]
- Jane, E.P. , Premkumar, D.R. , Pollack, I.F. , 2011. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol. Cancer Ther. 10, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, S. , Lindholm, P. , Gullbo, J. , Larsson, R. , Bohlin, L. , Claeson, P. , 2001. Cytotoxicity of digitoxin and related cardiac glycosides in human tumor cells. Anticancer Drugs. 12, 475–483. [DOI] [PubMed] [Google Scholar]
- Khanbolooki, S. , Nawrocki, S.T. , Arumugam, T. , Andtbacka, R. , Pino, M.S. , Kurzrock, R. , Logsdon, C.D. , Abbruzzese, J.L. , McConkey, D.J. , 2006. Nuclear factor-kappaB maintains TRAIL resistance in human pancreatic cancer cells. Mol. Cancer Ther. 5, 2251–2260. [DOI] [PubMed] [Google Scholar]
- Ma, H.-I.I. , Hueng, D.-Y.Y. , Shui, H.-A.A. , Han, J.-M.M. , Wang, C.-H.H. , Lai, Y.-H.H. , Cheng, S.-Y.Y. , Xiao, X. , Chen, M.-T.T. , Yang, Y.-P.P. , 2014. Intratumoral decorin gene delivery by AAV vector inhibits brain glioblastomas and prolongs survival of animals by inducing cell differentiation. Int. J. Mol. Sci. 15, 4393–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, C.A. , Bovenberg, M.S. , Crommentuijn, M.H.W. , Niers, J.M. , Kerami, M. , Teng, J. , Sena-Esteves, M. , Badr, C.E. , Tannous, B.A. , 2013. Triple bioluminescence imaging for in vivo monitoring of cellular processes. Mol. Ther. Nucleic Acids. 2, e99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, C.A. , Crommentuijn, M.H.W. , Mu, D. , Hudry, E. , Serrano-Pozo, A. , Hyman, B.T. , Tannous, B.A. , 2013. Mouse gender influences brain transduction by intravascularly administered AAV9. Mol. Ther. 21, 1470–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, C.A. , Meijer, D.H. , LeRoy, S.G. , Tierney, L.A. , Broekman, M.L. , Costa, F.F. , Breakefield, X.O. , Stemmer-Rachamimov, A. , Sena-Esteves, M. , 2008. Preventing growth of brain tumors by creating a zone of resistance. Mol. Ther. 16, 1695–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, G. , Ni, J. , Wei, Y.F. , Yu, G. , Gentz, R. , Dixit, V.M. , 1997. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 277, 815–818. (New York, N.Y.) [DOI] [PubMed] [Google Scholar]
- Pan, G. , O'Rourke, K. , Chinnaiyan, A.M. , Gentz, R. , Ebner, R. , Ni, J. , Dixit, V.M. , 1997. The receptor for the cytotoxic ligand TRAIL. Science. 276, 111–113. [DOI] [PubMed] [Google Scholar]
- Panner, A. , James, C.D. , Berger, M.S. , Pieper, R.O. , 2005. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol. Cell. Biol. 25, 8809–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol, D.-W.W. , 2011. p53-Independent up-regulation of a TRAIL receptor DR5 by proteasome inhibitors: a mechanism for proteasome inhibitor-enhanced TRAIL-induced apoptosis. Biochem. Biophys. Res. Commun. 416, 222–225. [DOI] [PubMed] [Google Scholar]
- Sheridan, J.P. , Marsters, S.A. , Pitti, R.M. , Gurney, A. , Skubatch, M. , Baldwin, D. , Ramakrishnan, L. , Gray, C.L. , Baker, K. , Wood, W.I. , Goddard, A.D. , Godowski, P. , Ashkenazi, A. , 1997. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 277, 818–821. [DOI] [PubMed] [Google Scholar]
- Song, J.H. , Song, D.K. , Pyrzynska, B. , Petruk, K.C. , Van Meir, E.G. , Hao, C. , 2003. TRAIL triggers apoptosis in human malignant glioma cells through extrinsic and intrinsic pathways. Brain Pathol. 13, 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp, R. , Mason, W.P. , van den Bent, M.J. , Weller, M. , Fisher, B. , Taphoorn, M.J. , Belanger, K. , Brandes, A.A. , Marosi, C. , Bogdahn, U. , Curschmann, J. , Janzer, R.C. , Ludwin, S.K. , Gorlia, T. , Allgeier, A. , Lacombe, D. , Cairncross, J.G. , Eisenhauer, E. , Mirimanoff, R.O. , 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Wakimoto, H. , Kesari, S. , Farrell, C.J. , Curry, W.T. , Zaupa, C. , Aghi, M. , Kuroda, T. , Stemmer-Rachamimov, A. , Shah, K. , Liu, T.-C.C. , Jeyaretna, D.S. , Debasitis, J. , Pruszak, J. , Martuza, R.L. , Rabkin, S.D. , 2009. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 69, 3472–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.K. , Portbury, S. , Thomas, M.B. , Barney, S. , Ricca, D.J. , Morris, D.L. , Warner, D.S. , Lo, D.C. , 2006. Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proc. Natl. Acad. Sci. U.S.A. 103, 10461–10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilt, L.H. , Kroon, J. , Jansen, G. , de Jong, S. , Peters, G.J. , Kruyt, F.A. , 2013. Bortezomib and TRAIL: a perfect match for apoptotic elimination of tumour cells?. Crit. Rev. Oncol. Hematol. 85, 363–372. [DOI] [PubMed] [Google Scholar]
- Wu, X. , He, Y. , Falo, L.D. , Hui, K.M. , Huang, L. , 2001. Regression of human mammary adenocarcinoma by systemic administration of a recombinant gene encoding the hFlex-TRAIL fusion protein. Mol. Ther. 3, 368–374. [DOI] [PubMed] [Google Scholar]
- Xiang, H. , Nguyen, C.B. , Kelley, S.K. , Dybdal, N. , Escandón, E. , 2004. Tissue distribution, stability, and pharmacokinetics of Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in human colon carcinoma COLO205 tumor-bearing nude mice. Drug Metab. Dispos. 32, 1230–1238. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Yang, B. , Mu, X. , Ahmed, S.S. , Su, Q. , He, R. , Wang, H. , Mueller, C. , Sena-Esteves, M. , Brown, R. , Xu, Z. , Gao, G. , 2011. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 19, 1440–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data