

Abstract
VZV IE62 is an essential, immediate-early, tegument protein and consists of five domains. We generated recombinant viruses carrying mutations in the first three IE62 domains and tested their influence on VZV replication kinetics. The mutations in domain I did not affect replication kinetics while domain II mutations, disrupting the DNA binding and dimerization domain (DBD), were lethal for VZV replication. Mutations in domain III of the nuclear localization signal (NLS) and the two phosphorylation sites S686A/S722A resulted in slower growth in early and late infection respectively and were associated with IE62 accumulation in the cytoplasm and nucleus respectively. This study mapped the functional domains of IE62 in context of viral infection, indicating that DNA binding and dimerization domain is essential for VZV replication. In addition, the correct localization of IE62, whether nuclear or cytoplasmic, at different points in the viral life cycle, is important for normal progression of VZV replication.
Keywords: IE62, VZV, NLS, putative phosphorylation site, TAD, DBD, dimerization
Introduction
Varicella-zoster virus (VZV) is a human herpesvirus that causes varicella (chicken pox), and establishes a life-long latency during primary infection. VZV is lymphotropic and neurotropic and establishes latency within cells of the dorsal root ganglia. Reactivation of the virus causes herpes zoster (shingles). The VZV DNA genome, 125 kb in length, is contained in an icosahedral nucleocapsid surrounded by a proteinaceous layer called the tegument. The VZV tegument is comprised of viral proteins, including the IE4, IE62, IE63, ORF9, ORF10, ORF47 and ORF66 proteins, several of which function in regulating viral gene expression (Arvin and Gilden, 2013). Numerous cellular proteins are also associated with HSV-1 and HCMV virion and tegument (Lippe, 2012) and thus probably with VZV also.
The VZV genome encodes at least 71 open reading frames. During lytic infection, the viral genes are expressed in a temporal cascade consisting of immediate early (IE) [transcriptional regulators], early (E) [replication factors], and late (L) [structural proteins] (Reichelt et al., 2009). IE62 is considered the primary regulator of the expression of most VZV genes, in conjunction with other viral and cellular factors. Several other VZV proteins have also been shown to transactivate and/or transrepress viral promoters, including the IE4, IE63, ORF10 and ORF61 proteins (Moriuchi et al., 1993; Baudoux et al., 2000; Spengler et al., 2000; Sato et al., 2003; and Wang et al., 2013).
The lytic phase of VZV infection requires entry of the virus into the host cells following the fusion of the virus envelope with the host cell membrane. The release of the virus tegument-coated nucleocapsid occurs in the cytoplasm and, by analogy with information from other alphaherpesviruses, is followed by de-tegumentation of the nucleocapsid. The nucleocapsid is transported into the nucleus along with VZV transactivators from the inner tegument layer including IE4, IE62 and ORF10. IE62 is localized to the nucleus early in infection (Reichelt et al., 2009) and initiates immediate early gene expression (Arvin and Gilden, 2013). Later in infection, IE62 is shuttled to the cytoplasm for incorporation into the tegument of newly produced virions (Lynch et al., 2002). This translocation requires phosphorylation of IE62 by the VZV ORF66 kinase and possibly other cellular kinases (Kinchington et al., 2000; Eisfeld et al., 2006; and Cilloniz et al., 2007).
IE62 protein is a 1310 amino acid protein divided into five functional domains (Fig. 1A). The acidic activation domain (TAD) is located in the N-terminal region and is represented in domain I (aa 1-467) (Peng et al., 2003). This domain is also the site of interaction with human mediator complex, Sp family members, YY1 and USF-1 (Fig. 1B) (Rahaus et al., 2003; Peng et al., 2003; Yang et al., 2006; Yang et al., 2008; Yamamoto et al., 2009; Khalil et al., 2014; and Khalil et al., 2015b). VZV ORF9 and IE4 also interact with IE62 through this domain (Spengler et al., 2000; and Cilloniz et al., 2007). The highly conserved DNA binding and dimerization domain (DBD) follows the acidic activation domain and is included in domain II (aa 468-640) (Perera, 2000). This domain is crucial for the interaction with TATA-box binding protein (TBP) and TFIIB (Fig. 1C) (Perera, 2000). IE63 also binds to IE62 domain II (Lynch et al., 2002). The nuclear localization signal (NLS) is in domain III (aa 641-734). This region is phosphorylated during lytic infection and influences the transactivation function of the protein (Kinchington and Turse, 1998). Domain IV (aa 735-1147) has also been implicated in IE62 gene regulatory function (Baudoux et al., 1995). Domain V (aa 1148-1310) contains the SEAC domain and is thought to be involved in transcriptional activation (Baudoux et al., 1995). In this study, we focus on domains I-III, since they are predicted to have clearly defined functions important for VZV but have not yet been assessed in the context of VZV infection. Our approach is to use targeted mutagenesis of the VZV genome to introduce mutations into IE62 domains I, II and III and evaluate their influence on the replication of VZV, as well as the intracellular localization of IE62 and IE63 in melanoma cells.
FIG. 1.
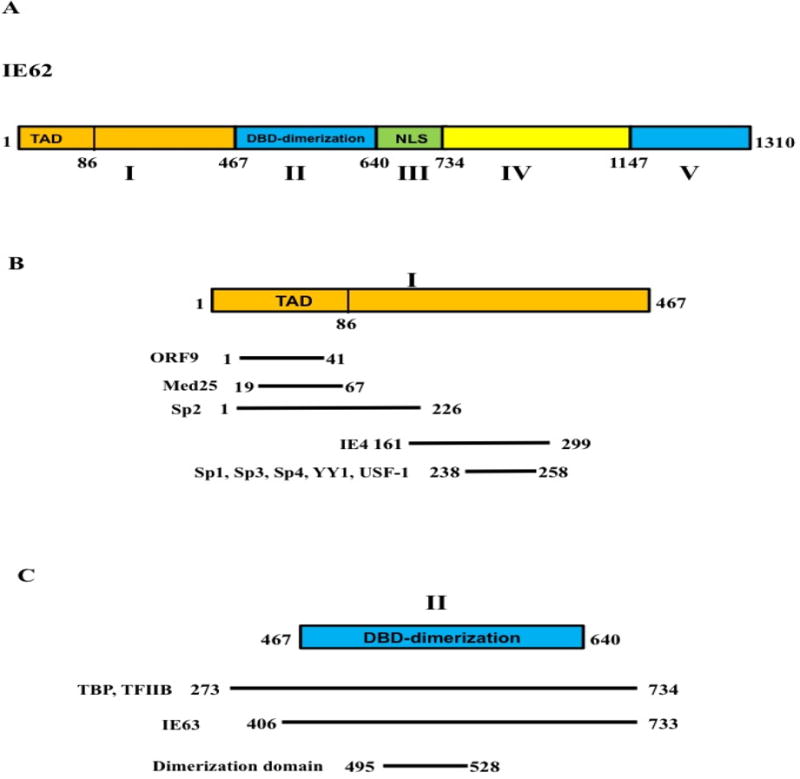
The domain structure of IE62. A) A schematic diagram showing the functional domains of IE62. B) A schematic diagram showing domain I of IE62, the interacting proteins and their site of interaction C) A schematic diagram showing the DNA binding and dimerization domain of IE62 and the site of interaction of TBP, TFIIB and IE63.
The study of the roles that different IE62 domains play in the virus replication cycle is essential to our further understanding of the intricacies of VZV growth. This is particularly important, given the central position of IE62 as the major viral transactivator. Apart from the value of these findings to the molecular virology of VZV, this approach will also allow us to select potential targets for the improvement of the current varicella and zoster vaccines.
Materials and Methods
Cell Lines
Melanoma cells were propagated in culture medium (MEM) supplemented with 10% fetal bovine serum, non-essential amino acids, penicillin-G, streptomycin, and amphotericin as described by Khalil et al., (2014). VZV strain pOka was propagated in melanoma cell monolayers as described by Khalil et al., (2015a).
Generation of recombinant mutant viruses carrying mutations in IE62 domains. Recombinant viruses were generated using cosmids derived from the VZV parent Oka virus (pOka) (Niizuma et al., 2003). The pOka genome was divided into four overlapping segments and incorporated into four SuperCos 1 cosmid vectors designated pvFsp73 (nucleotides 1-33128), pvSpe14 (21796-61868), pvPme2 (53756-96035), and pvSpe23 (94055-125123). The pvSpe23 cosmid has a substitution mutation removing one of the AvrII restriction enzyme sites, leaving a unique AvrII site beginning at base pair 18092 for use in the mutagenesis process. ORF62 and ORF63 are located in the internal repeat short region in the cosmid pvSpe23, and ORF70 and ORF71 are in the terminal repeat short region in the same cosmid. The pvSpe23 cosmid was digested with EcoRI/AvrII and the 17kb fragment was inserted into the pLitmus28 plasmid vector to make pLitmusORF59-65 containing ORFs 59-65. The pvSpe23 cosmid was digested with AscI/AvrII and the 12.2kb fragment was inserted into the pLitmus vector to make pLitmusORF66-71 containing ORFs 66-71. This strategy of using the cosmids containing the whole VZV or HSV genome with some overlap to help in the recombination events is similar to strategies for inserting mutations in diploid genes in HSV-1 genome using recombination events too (Knipe et al., 1979; and DeLuca et al., 1984). pLitmus ORF59-65 and pLitmus ORF66-71 with mutations in the IE62 nuclear localization sequence were generated by inserting point mutations in aa681-684 of IE62 as described previously (Kinchington and Taruse, 1998). Mutagenic primers were designed to create the S686A and S722A alanine substitutions and two mutant viruses were generated with point mutations in the TAD. The first mutant has an F50A mutation and the second mutant virus changes aa57-60 to alanines as described (Yamamoto et al., 2009). A K548E mutation was also introduced into the pOka genome.
The deletions within ORF62 were generated using mutagenic primers that removed sequences for aa238-258 and aa495-528. For each mutation, two complimentary primers were designed to anneal in a 20 or 30 nucleotides region containing the nucleotide targeted for mutation. All nucleotides of the primer were complimentary to the ORF62 DNA with the exception of the desired mutation so that newly generated DNA fragments would incorporate the mutations at the corresponding location. Two reactions were used in the first round of PCR: a 5′ outside primer was used with a 3′ mutagenic primer (with pLitmus ORF59-65 as the template) and a 3′ outside primer was used with a 5′ mutagenic primer. These two PCR reactions yielded two overlapping double stranded fragments of DNA, each containing the desired mutation in the overlapping sequence. For the second round of PCR, the two products from the first round were purified and combined and PCR was done using only the outside primers from the first round of reactions. Because the products from the first round contained short overlapping regions with the mutations, the second round of PCR resulted in a 1.4kb fragment of DNA that spanned the annealing sites of the outside primers and internally contained the targeted mutations. In order to generate a DNA fragment encoding both the S686A and S722A mutations, two rounds of PCR were first performed to generate one mutation, and the resulting 1.4kb DNA fragment was used as a template in subsequent two rounds of PCR to generate the second mutation.
Each 1.4kb fragment was cloned into pCR4-Topo vector to generate ORF62-Topo and sequenced to verify the mutations. The ORF62-Topo plasmids, pLitmus ORF59-65, and pLitmus ORF66-71 were digested with AgeI, which cleaves each plasmid twice to generate a ~1kb fragment. The fragments from ORF62-TOPO containing the mutations were ligated into both of the pLitmus plasmids. To insert the ORF62 mutations back into the pvSpe23 cosmid, a NheI/AvrII fragment was excised from the pLitmus ORF 59–65 plasmids and ligated back into pvSpe23. Finally, to insert the ORF71 mutations into pvSpe23, the AscI/AvrII fragment from the pLitmus ORF 66-71 plasmids was ligated into the corresponding mutant ORF62 pvSpe23 cosmid.
Generation of recombinant virus by cosmid co-transfection and measurement of virus replication kinetics
Recombinant viruses were isolated by transfection of melanoma cells with either wild type or mutated Spe23 cosmid and the other three intact cosmids, Fsp73, Spe14, and Pme2. To confirm the targeted mutations in IE62, genomic DNA was extracted from virus-infected melanoma cells with the DNAzol reagent (Invitrogen, Carlsbad, CA). A PCR fragment covering the mutated region was amplified from the genomic DNA by using Pfu polymerase (Stratagene, La Jolla, CA), gel-purified with a QIAquick gel extraction kit (Qiagen, Inc., Valencia, CA), and sequenced (Elim Biopharm, Inc., Hayward, CA). The replication kinetics of recombinant viruses was assessed by an infectious focus assay with immunostaining to detect plaques as previously described (Chaudhuri et al., 2008; and Moffat et al., 1995). Briefly, 6-well assay plates and 24-well titer plates were seeded with melanoma cells. The 6-well plates were incubated with wild type or mutant viruses for variable times, and several dilutions of the samples were taken for infectious focus assay. Titer plates were incubated for 4 days then fixed with 4% paraformaldehyde for immunohistochemical staining using anti-VZV monoclonal antibody (Meridian). A one-way ANOVA analysis of variance followed by Tukey’s post hoc test was used for the statistical significance determination.
Immunofluorescence Microscopy
For confocal microscopy, melanoma cells were seeded on sterile coverslips and infected with 2,000 pfu of pOKA, pOKA ORF62/71 S686/722A, and pOKA ORF62/71NLS mutants. Infected and uninfected control cells were fixed with 4% formaldehyde at 24 and 48 hpi. After blocking with 1% fish gelatin for 1 hr at RT, cells were incubated with a rabbit anti-ORF63 antibody and a murine monoclonal anti-ORF62 antibody. Cells were washed and incubated for 1 hr at RT with fluorescein isothiocyanate-labeled anti-rabbit and Texas Red-labeled anti-mouse secondary antibodies (Jackson ImmunoResearch, Inc.). Cell nuclei were counterstained with Hoechst 22358 (Invitrogen, Carlsbad, CA). Analysis was performed with a Leica TCSSP2 confocal laser scanning microscopy (Heidelberg, Germany).
Whole cell lysate preparation and western blot analysis
Whole cell lysates of VZV infected and uninfected melanoma cells were prepared in lysis buffer (50 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 1 mM EDTA, 0.1% Triton X-100 and protease inhibitor cocktail (Roche, Mannheim, GE, added per the manufacturer’s instructions) and analyzed for IE62 and IE63 expression by immunoblot as previously described (Khalil et al., 2015a). Antibodies against full length IE62 and IE63 were used as described previously (Spengler et al., 2000; and Zuranski et al., 2005). Rabbit polyclonal antibody for β-tubulin was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and mouse monoclonal antibody for α-tubulin was purchased from Sigma-Aldrich (St-Louis, MO). Quantification of the relative amounts of IE62 and IE63 normalized to α-tubulin or β-tubulin in loading controls was performed using a Bio-Rad GS700 imagining densitometer (Bio-Rad, Hercules, CA). Statistical significance was determined using a one-way analysis of variance, followed by Tukey’s post hoc test.
Plasmids
The wild type pCMV62 expressing plasmid was constructed to express IE62 under the control of the cytomegalovirus immediate-early (IE) promoter as described previously (Perera et al., 1992 and 1993). Two mutant forms of pCMV-ORF62 were constructed to have mutations in IE62 dimerization domain. Mutagenic primers were designed to mutate amino acids 500–501 from AR-EE and amino acids 524–525 from QY-AA. These point mutations were generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene, LaJolla, CA). The primers were synthesized by IDT (Coralville, IA). The mutations were verified by sequencing at the Roswell Park Cancer Institute sequencing facility, Buffalo NY. The TA-Luc reporter plasmid was described previously (Yang et al., 2006). The luciferase reporter plasmid 62-Luc containing ORF62 promoter was described (Narayanan et al., 2007). The reporter 29-Luc containing ORF29 promoter was constructed using pGL2 plasmid and 142 bp upstream of ORF29 translation start site.
Reporter gene assays
Luciferase reporter gene assay experiments were carried out in melanoma cells as described (Yang et al., 2004; and Khalil et al., 2015b). 2 × 105 melanoma cells were seeded in each well of 12-well plates 24 h before transfection. Cells were transfected with one microgram of each reporter vector using Lipofectamine reagent (Invitrogen, Carlsbad, CA), along with 5 ng of pEF1α-RL plasmid (Promega, Madison, WI) and increasing concentrations of wild type and mutant pCMV-ORF62 ranging from 5 to 20 ng. Dual luciferase activities were normalized to the Renilla luciferase activities. pcDNA was transfected to add equal amounts of total DNA in each set of experiments. The cells were lysed 48 h post transfection in 250 μl of lysis buffer (50 mM HEPES, pH 7.4, 250 mM NaCl, 1% NP-40, 1 mM EDTA). Control experiments without transfection of pCMV-ORF62 were performed for each reporter to determine basal expression. Dual-luciferase assays were done according to the manufacturer’s instruction. Transfection experiments were carried out at least three times.
Results
Influence of IE62 domain I mutations on VZV replication kinetics. In order to test the influence of IE62 domain I on replication kinetics, we constructed three recombinant viruses with mutations in domain I: an F50A point mutation, an aa57-60 alanine substitution and an aa238-258 deletion mutant. Based on the experiments with plasmid constructs, these changes are predicted to disrupt the interaction with human mediator complex components, Sp family members, YY1 and USF-1 (Peng et al., 2003; Yang et al., 2006; Yamamoto et al., 2009; Khalil et al., 2014; and Khalil et al., 2015b). A one-way ANOVA analysis of variance was used for the statistical significance determination. The statistical test was performed on the variance between foci number from individual wells containing samples from the same time point. No significant differences were observed when the replication kinetics of these three mutant viruses was compared with wild type pOka over 5 days post-infection (P>0.05) (Fig. 2).
FIG. 2.
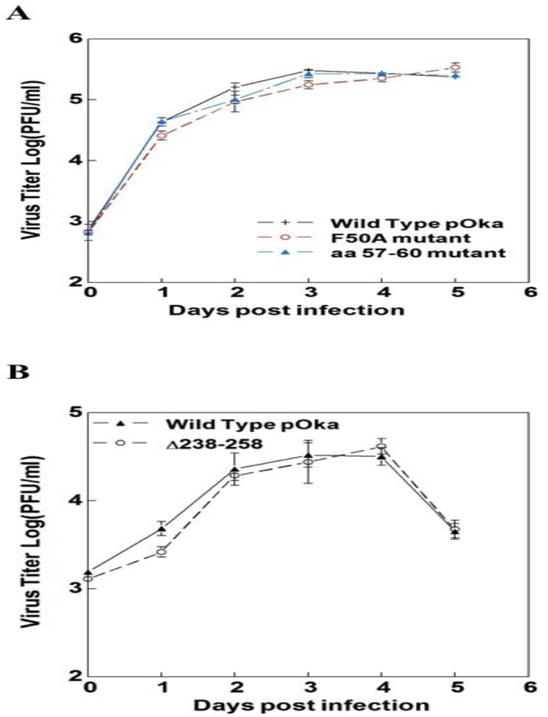
Replication kinetics of the domain I mutants. Replication kinetics of A) the pOka wild type and ORF62/ORF71 F50A and amino acids 57–60 mutant viruses and B) the pOka wild type and ORF62/ORF71 Δ238–258 mutant viruses are shown in melanoma cells. Cells were inoculated at 103 PFU/ml with wild type and mutant viruses and infectious virus yields were determined in triplicate for 5 days after inoculation. Each point is the mean of triplicate experiment. A one-way ANOVA analysis of variance followed by Tukey’s post hoc test was used for the statistical significance determination. Standard errors represented by error bars.
Influence of IE62 domain II and III mutations on VZV replication kinetics and IE62 and IE63 expression. Evaluating the effects of mutations of domain II and III showed that a K548E point mutation (DNA binding) and an aa495-528 deletion (dimerization) were both lethal for VZV replication. In experiments with plasmid constructs, these mutations attenuate the DNA binding activity and transcriptional activation of IE62 with model and VZV promoters (Tyler and Everett, 1994; and White et al., 2010). Virus was not recovered from melanoma cells transfected with either of these mutant cosmids in three experiments, while wild type virus was generated in the same assays using wild type cosmid.
Transfections with cosmids carrying the NLS and S686A/S722A mutations, however, yield infectious virus consistently. A one-way ANOVA analysis of variance was used for the statistical significance determination. The statistical test was performed on the variance between plaque numbers from three wells containing samples from the same time point. Replication kinetics was tested in duplicate, in two independent experiments. When the replication kinetics of the NLS mutant were compared to wild type pOka in melanoma cells, NLS mutant titers were significantly lower during the first two days post infection (P<0.001). After that, it grew as well as the wild type pOka from days 3–5 post-infection. On the other hand, the S686A/S722A mutant virus grew similarly to the wild type virus in the first three days post-infection. Following that, the S686A/S722A phosphorylation mutation was associated with lower titers on day 4 and 5 post infection (P<0.001) (Fig. 3A).
FIG. 3.
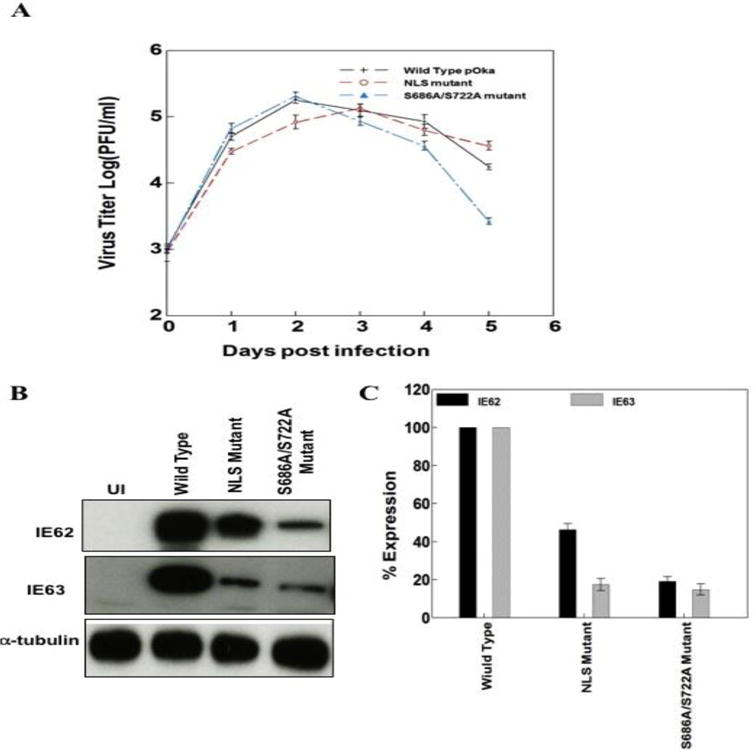
NLS and phosphorylation site mutations influence VZV replication in melanoma cells. A) Replication kinetics of the pOka wild type and ORF62/ORF71 NLS and S686A/S722A mutant viruses. Cells were inoculated using 103 PFU/ml of wild type and mutant viruses and infectious virus yields were determined in triplicate for 5 days. Each point is the mean of triplicate experiment. B) Western blot analysis showing the levels of IE62, IE63 in the wild type and mutant pOka. α-tubulin was used as a loading control. C) Histogram summarizing the accumulation of IE62 and IE63 in the wild type and mutant pOka from three independent preparations. A one-way ANOVA analysis of variance was used for the statistical significance determination. Standard errors represented by error bars.
To determine whether the influence of domain III mutations on replication kinetics correlates with the transcriptional activation efficiency of IE62, we then evaluated the effect of NLS and S686A/S722A mutations on the accumulation of two essential immediate-early proteins IE62 and IE63 in melanoma cells. We examined IE62 and IE63 levels at 36 hrs post infection. IE62 and IE63 accumulation decreased significantly by about 5 fold with the S686A/S722A mutant virus. IE62 and IE63 expression was also significantly reduced in the IE62 NLS mutant virus infected cells compared to wild type (Fig. 3B and C).
Influence of nuclear localization sequence and putative phosphorylation site mutations on IE62 and IE63 cellular localization. In wild type virus infection, IE62 is predominantly nuclear up to 24 hours post infection, presumably to activate transcription from VZV promoters; at later times, it is present mainly in the cytoplasm of the host cells, presumably, to be incorporated into the tegument layer, contributing to virion formation (Fig. 4 and 5). When examined by confocal microscopy, the NLS mutant exhibited a predominantly cytoplasmic localization of IE62 at both 24 and 48 hours post infection, in contrast to wild type pOka, indicating that the NLS mutation influences IE62 distribution during VZV replication. On the other hand, IE62 produced in melanoma cells infected with the S686A/S722A mutant had a predominantly nuclear localization at both 24 and 48 hours post infection, again differing from pOka. Thus, mutation of the putative phosphorylation site of IE62 also modulates the cytoplasmic localization of IE62 in VZV infected cells (Fig. 4 and 5) but in a quite different way from the NLS mutation.
FIG. 4.
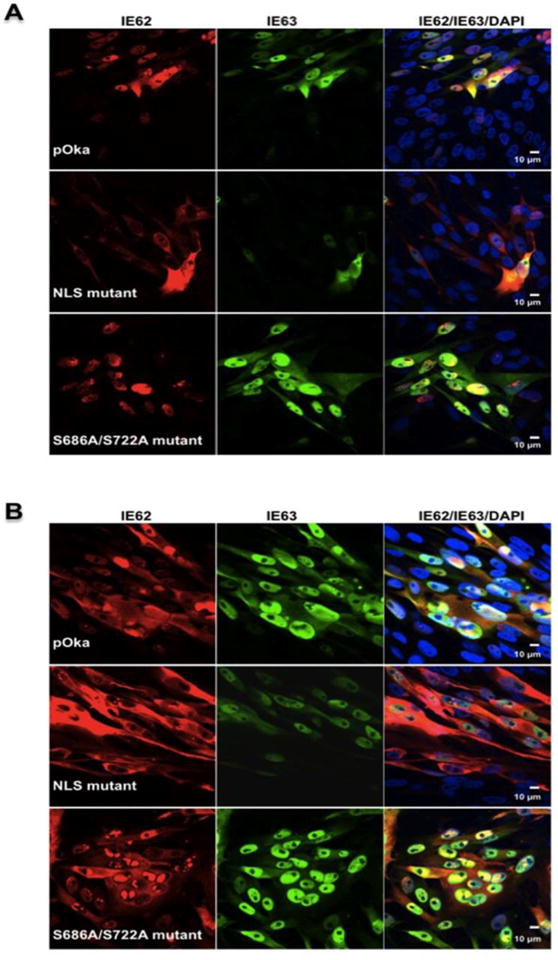
NLS and phosphorylation site mutations influence IE62 intracellular localization. The intracellular localization of the wild type, ORF62/ORF71 NLS and S686A/S722A mutant viruses at A) 24 hpi and B) 48 hpi. Infected melanoma cells were stained with antibodies to IE62, IE63 and with Texas Red (IE62, red) and fluorescein isothiocyanate (IE63, green) conjugated secondary antibodies. Cell nuclei were counterstained with Hoechst 22358 (blue); size bars: 10 μm.
FIG. 5.
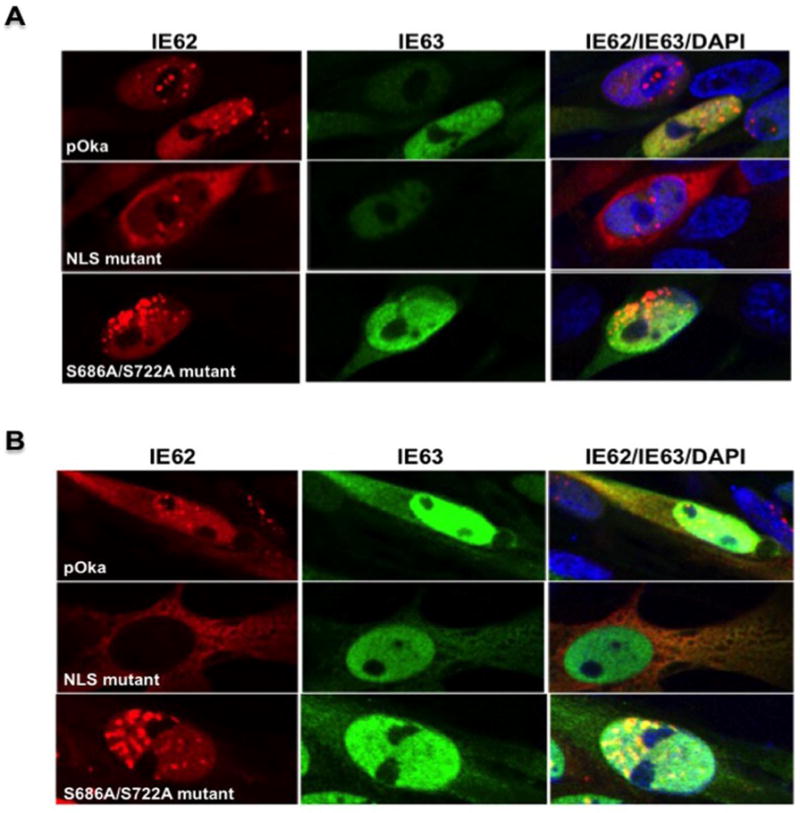
NLS and phosphorylation site mutations influence IE62 intracellular localization in melanoma cells. Enlarged images of melanoma cells infected with the wild type, ORF62/ORF71 NLS and S686A/S722A mutant viruses at A) 24 hpi and B) 48 hpi. Infected melanoma cells were stained with antibodies to IE62 and IE63 and with Texas Red (IE62, red) and fluorescein isothiocyanate (IE63, green) conjugated secondary antibodies. Cell nuclei were counterstained with Hoechst 22358 (blue).
In contrast, there is no detectable influence on IE63 localization in cells infected with NLS and S686A/S722A mutant viruses compared to the wild type pOka (Fig. 4A and B). IE63 is distributed in the nucleus and the cytoplasm of the VZV infected cells both at 24 and 48 hours post infection. However, there is a clear decrease in the level of expression of IE63 protein in cells infected with NLS mutant virus compared to the S686A/S722A mutant and the wild type viruses.
Analysis of the IE62 dimerization domain using reporter gene assays. Since the domain II dimerization mutant (Δ495–528) was lethal for virus formation, and this region had not been explored in the context of reporter gene assays, we compared the behavior of wild type and dimerization point mutations in pCMV-ORF62 using three reporter constructs. The first is a model promoter containing only a TATA element, and the others contain the promoter regions of the ORF29 and ORF62 genes respectively. We tested two IE62 dimerization domain point mutants. One has point mutations in amino acids 500–501 from AR to EE and the second has alanine substitutions of the QY residues at aa524-525. These four amino acids are conserved in the dimerization domain of HSV ICP4. Luciferase activities obtained from each reporter plasmid in the absence of ORF62 represented basal activity levels and were normalized to 1. Reporter gene activities in the presence of ORF62 transfection were displayed as induction (n-fold) of luciferase activities relative to the basal activity.
As shown in Fig. 6A, B and C, these mutations in the IE62 dimerization domain inhibited significantly the activation of both the model promoter and the two VZV promoters (P<0.001). Also, there was no response to increasing concentrations of IE62 with the dimerization domain mutations on any of the three reporters. The wild type IE62 expression level was similar to IE62 expression in these two dimerization domain mutants as detected by western blot analysis (Fig. 6D).
FIG. 6.
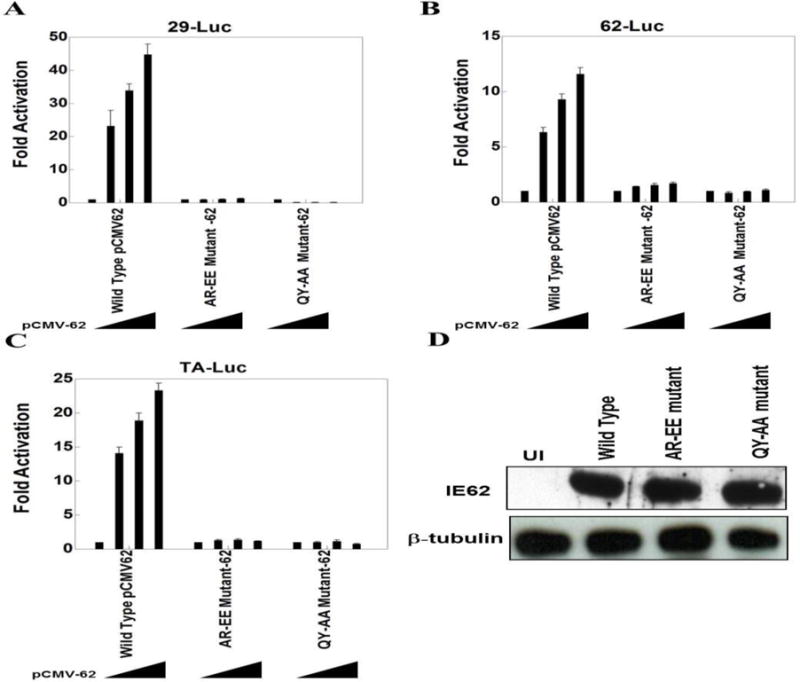
The IE62 dimerization domain influences transactivation in vitro. Results of triplicate transient transfection assays determining the effect of IE62 dimerization domain mutations on IE62 mediated transactivation are shown for A) the ORF29 promoter, B) the ORF62 promoter, and C) the TA-Luc model promoter. Luciferase activities in the absence of IE62 were normalized to one. Promoter activities in the presence of IE62 are reported as induction (n-fold) of the luciferase activity over the basal activity. Statistical significance was determined by a one-way ANOVA analysis of variance followed by Tukey’s post hoc test. D) Western blot analyses show expression levels of IE62, and β-tubulin. β-tubulin was used as loading control in experiments.
Discussion
In the current investigation, we examined the role of IE62 domains on virus replication in melanoma cells. We generated recombinant mutant viruses containing mutations in domains I, II and III, using mutations that had been previously tested in several studies in vitro.
For the domain I investigation we focused on three mutations. Two of these were in the TAD, while the third was in a region previously shown to act as a platform for interaction with cellular transcription factors (Sp family members, YY1 and USF-1). The F50A and aa57-60 mutations (in the TAD) have been shown to be required for interaction with the human mediator complex and for the transcriptional activation of IE62 (Yamamoto et al., 2009). The deletion of amino acids 238–258 ablated the interaction of IE62 with Sp1, Sp3, Sp4, YY1 and USF-1 (Peng et al., 2003; Yang et al., 2006; Khalil et al., 2014; and Khalil et al., 2015b). The binding of these cellular transcription factors to VZV promoters (e.g. ORF3, gI, gE, ORF28/29, ORF10) and the downstream region of oriS were reported previously (Ito et al., 2003; Peng et al., 2003, Yang et al., 2004; Berarducci et al., 2007; Che et al., 2007; Khalil et al., 2008, and Khalil et al., 2013). Mutation of their DNA binding sites on VZV promoters inhibited the activation of these VZV promoters, suggesting that their interaction with promoters (and IE62) is important for efficient VZV replication (Khalil et al., 2012; and Khalil et al., 2013).
These three domain I mutant viruses grew as well as the wild type pOka suggesting that the absence of the interaction of these cellular transcription factors and human mediator complex with IE62 may be compensated for in an indirect way in a cultured cell line. For example, Sp family members, YY1 and USF-1 contain more than one zinc finger domain and have the ability to bind to DNA and promoters, perhaps in several different ways. Also, all of them bind to TBP as well as IE62 and the human mediator complex (Chiang and Roeder, 1995; Torigoe et al., 2003; and Wu et al., 2003). IE62 binds to DNA and TBP through domain II of IE62 (DBD) that may act as an alternative mediator for the interaction with these cellular factors in situations in which their primary binding site is no longer functional. Still another possibility is that the mutations in domain I that lacked a phenotype in melanoma cells could have effects on virus replication in other cell types.
In domain II, on the other hand, the K548E mutation in the DNA binding domain, as well as the complete removal of the dimerization domain by deletion of aa495-528, was lethal for VZV replication. These results demonstrate that the DNA binding and dimerization domains are essential for VZV replication in infected cells. Tyler et al., (1994) showed that mutations that diminished site-specific DNA binding also decreased transactivation. Specifically, they identified the critical lysine residue (K548) which, when mutated to glutamic acid, resulted in an almost complete loss of transactivation and DNA-binding activity under their experimental conditions in vitro. K548 also lies within the region homologous to the AntP homeodomain. Many alphaherpesvirus transactivators contain a region with homology to the DNA recognition helix of Drosophila AntP homeodomain (Tyler et al., 1994).
Interestingly, the vOka vaccine strain of VZV contains a mutation of interest within domain II of IE62. This mutation resulted in the substitution of proline for leucine at position 446 (L446P), proximal to the minimal IE62 DNA-binding domain. Reversion of the proline mutation to wild type leucine has been linked to occurrence of varicella rash formation in vaccinated individuals (Quinlivan et al., 2007). For that reason, we also tested the influence of the L446P mutation on virus replication in melanoma cells but it had no significant effect (data not shown).
The role played by the IE62 dimerization domain in VZV replication or in IE62 transactivation has not been studied. In this study, we were able to determine a role for the putative dimerization domain in IE62 transcriptional activation. The two IE62 mutants with point mutations in the dimerization domain substantially inhibit the transcriptional activation of IE62 both in model and VZV promoters. Our results show that the dimerization domain in the DBD is involved in the IE62 transactivation process, and that deleting it is lethal for viral growth.
IE62 has a nuclear localization sequence (NLS), present in domain III of IE62 (a.a 674-685) adjacent to the DBD. Previous studies using plasmid constructs showed that mutation of IE62 NLS influences the cellular localization of the protein and results in the concentration of the protein in the cytoplasm of the host cells (Kinchington and Turse, 1998).
Domain III of IE62 contains not only the NLS but also the phosphorylation site. The two serine residues identified by Kinchington et al., (2000) to be important for ORF66 phosphorylation of IE62 are serine 686 and serine 722. Mutation of either of these two serine residues not only inhibited the phosphorylation of IE62 by ORF66 but also the shuttling of the protein from the nucleus to the cytoplasm.
The influence of the IE62 NLS and putative phosphorylation site on VZV replication in melanoma cells was tested using two recombinant mutant viruses. The two IE62 mutations inhibited the replication kinetics of recombinant mutant viruses. While the ORF62/ORF71 NLS mutant virus grew more slowly than pOka virus at day 1 and 2 postinfection, the ORF62/ORF71 S686A/S722A mutant virus was less vigorous at day 4 and 5 postinfection. These two mutations also altered the localization of IE62 from the nucleus to the cytoplasm and from the cytoplasm to the nucleus respectively. The NLS mutation that decreased the amount of nuclear IE62 early in infection may inhibit the efficiency of virus gene expression at this stage, since IE62 is activating most, if not all of the VZV genes. Hence, this NLS mutation likely inhibits virus replication at early times of infection (day 1 and 2). The S686A/S722A mutation that inhibited ORF66 phosphorylation of IE62 (Kinchington et al., 2001) and the shuttling of IE62 to the cytoplasm may inhibit the incorporation of IE62 into virions. Thus, it inhibits the replication late during infection (day 4 and 5) although VZV spreads by cell fusion rather than by infectious virions in melanoma cells. It is quite likely that this mutant virus would be impaired in other cell types that do not fuse when infected with VZV, such as T cells, or where fusion is less important, such as ARPE-19 and HFFs. The behavior of the VZV IE62 domain I–III mutants described in this paper is summarized in Table 1.
Table 1.
Summary of the effects of mutations in IE62 domains I–III.
| IE62 domain | Mutation | Effect on virus replication kinetics | Effect on IE62 localization | Effect on IE62 and IE63 expression |
|---|---|---|---|---|
| Domain I | -F50A | No effect | NA | NA |
| -aa57-60 mutant | No effect | NA | NA | |
| -Δ238–258 | No effect | NA | NA | |
| Domain II | -K548E | Lethal | NA | NA |
| -Δ495–528 | Lethal | NA | NA | |
| Domain III | -NLS mutant | Inhibition at days 1–2 post infection | IE62 predominant in cytoplasm | Low accumulation of IE62 and IE63 |
| -S686A/S722A | Inhibition at days 4–5 post infection | IE62 predominant in nucleus | Low accumulation of IE62 and IE63 |
The levels of IE62 and IE63 decreased in melanoma cells infected with domain III mutant viruses (NLS and S686A/S722A mutant). The switch in IE62 localization due to these mutations may be a reason for this phenomenon because it may cause a decrease in IE62 transcriptional activation efficiency. Domain III is the main site of IE62 phosphorylation. Possibly the change of IE62 modification status due to these mutations alters the stability of IE62 and could also cause a decrease in the level of IE62. This in turn, would likely lead to a decrease in the level of IE63.
The presence of small residual amounts of IE62 in the target compartment for each mutation (Fig. 4 and 5) might suggest that the NLS and the phosphorylation of IE62 are not the only factors controlling the localization of IE62 inside the host cell. For example, the protein-protein interaction network might be another player affecting localization. We know that IE62 interacts with numerous proteins distributed in both cellular compartments, including USF-1 and Sp1 (Peng et al., 2003; and Rahaus et al., 2003) as well as IE63 (Lynch et al., 2002). These cellular and viral proteins may act as additional mediators for the division of IE62 between the nucleus and the cytoplasm. Still another possibility is that leakage in the nuclear membrane of infected cells during infection leads, at late times, to syncytium formation, and might be a cause of the presence of IE62 in both nucleus and cytoplasm in the mutant viruses.
Final IE62 domains paper research highlights.
-
-
Mutation of IE62 domain I did not affect VZV replication in melanoma cells.
-
-
IE62 domain II and III are important for VZV replication in melanoma cells.
-
-
Mutations of IE62 domain II (DBD) were lethal for virus replication.
-
-
Mutations of IE62 NLS and phosphorylation sites inhibited VZV replication.
-
-
NLS and S686A/S722A mutations altered localization of IE62 during early and late infection.
Acknowledgments
This work was supported by grants AI018449, AI053846 and AI020459 from the National Institutes of Health. We thank Lee Wang, MD. and Shinobu Yamamoto, Ph.D. for their assistance with this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arvin AM, Gilden D. Varicella-Zoster Virus. In: Howley PM, Knipe DM, editors. Fields Virology. Vol. 2. Philadelphia: Lippincott Williams and Wilkins Press; 2013. pp. 2015–2184. [Google Scholar]
- Baudoux L, Defechereux P, Schoonbroodt S, Merville MP, Rentier B, Piette J. Mutational analysis of varicella-zoster virus major immediate-early protein IE62. Nucleic Acids Res. 1995;23:1341–1349. doi: 10.1093/nar/23.8.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoux L, Defechereux P, Rentier B, Piette J. Gene activation by varicella-zoster virus IE4 protein requires its dimerization and involves both the arginine-rich sequence, the central part, and the carboxyl-terminal cysteine-rich region. J Biol Chem. 2000;275:32822–32831. doi: 10.1074/jbc.M001444200. [DOI] [PubMed] [Google Scholar]
- Berarducci B, Sommer M, Zerboni L, Rajamani J, Arvin A. Cellular and viral factors regulate the varicella-zoster virus gE promoter during viral replication. J Virol. 2007;81:10258–10267. doi: 10.1128/JVI.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri V, Sommer M, Rajamani J, Zerboni L, Arvin AM. Functions of Varicella-zoster virus ORF23 capsid protein in viral replication and the pathogenesis of skin infection. J Virol. 2008;82:10231–10246. doi: 10.1128/JVI.01890-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che X, Berarducci B, Sommer M, Ruyechan WT, Arvin AM. The ubiquitous cellular transcriptional factor USF targets the varicella-zoster virus open reading frame 10 promoter and determines virulence in human skin xenografts in SCIDhu mice in vivo. J Virol. 2007;81:3229–3239. doi: 10.1128/JVI.02537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CM, Roeder RG. Cloning of an intrinsic human TFIID subunit that interacts with multiple transcriptional activators. Science. 1995;267:531–536. doi: 10.1126/science.7824954. [DOI] [PubMed] [Google Scholar]
- Cilloniz C, Jackson W, Grose C, Czechowski D, Hay J, Ruyechan WT. The varicella-zoster virus (VZV) ORF9 protein interacts with the IE62 major VZV transactivator. J Virol. 2007;81:761–774. doi: 10.1128/JVI.01274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca NA, Courtney MA, Schaffer PA. Temperature-sensitive mutants in herpes simplex virus type 1 ICP4 permissive for early gene expression. J Virol. 1984;52:767–776. doi: 10.1128/jvi.52.3.767-776.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Turse SE, Jackson SA, Lerner EC, Kinchington PR. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J Virol. 2006;80:1710–1723. doi: 10.1128/JVI.80.4.1710-1723.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Sommer MH, Zerboni L, He H, Boucaud D, Hay J, Ruyechan W, Arvin AM. Promoter sequences of varicella-zoster virus glycoprotein I targeted by cellular transactivating factors Sp1 and USF determine virulence in skin and T cells in SCIDhu mice in vivo. J Virol. 2003;77:489–498. doi: 10.1128/JVI.77.1.489-498.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Hay J, Ruyechan WT. The cellular transcription factors Sp1 and Sp3 suppress varicella zoster virus origin-dependent DNA replication. J Virol. 2008;82:11723–11733. doi: 10.1128/JVI.01322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Robinson M, Sommer M, Arvin A, Hay J, Ruyechan WT. An Sp1/Sp3 site in the downstream region of varicella zoster virus oriS influences origin-dependent DNA replication and flanking gene transcription and is important for VZV replication in vitro and in human skin. J Virol. 2012;86:13070–13080. doi: 10.1128/JVI.01538-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Sommer M, Arvin A, Hay J, Ruyechan WT. Regulation of the Varicella-zoster virus ORF3 promoter by cellular and viral factors. Virology. 2013;440:171–181. doi: 10.1016/j.virol.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Sommer M, Arvin A, Hay J, Ruyechan WT. Cellular transcription factor YY1 mediates the varicella-zoster virus (VZV) IE62 transcriptional activation. Virology. 2014;449:244–253. doi: 10.1016/j.virol.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Sommer M, Hay J, Ruyechan WT, Arvin AM. Varicella-zoster virus (VZV) origin of DNA replication oriS influences origin-dependent DNA replication and flanking gene transcription. Virology. 2015a;481:179–186. doi: 10.1016/j.virol.2015.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil MI, Ruyechan WT, Hay J, Arvin A. Differential effects of Sp cellular transcription factors on viral promoter activation by varicella-zoster virus (VZV) IE62 protein. Virology. 2015b;485:47–57. doi: 10.1016/j.virol.2015.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchington PR, Turse SE. Regulated nuclear localization of the varicella-zoster virus major regulatory protein, IE62. J Infect Dis. 1998;178:S16–21. doi: 10.1086/514263. [DOI] [PubMed] [Google Scholar]
- Kinchington P, Fite K, Turse SE. Nuclear accumulation of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, is inhibited by phosphorylation mediated by the VZV open reading frame 66 protein kinase. J Virol. 2000;74:2265–2277. doi: 10.1128/jvi.74.5.2265-2277.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchington PR, Fite K, Seman A, Turse SE. Virion association of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, requires expression of the VZV open reading frame 66 protein kinase. J Virol. 2001;75:9106–9113. doi: 10.1128/JVI.75.19.9106-9113.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Ruyechan WT, Honess RW, Roizman B. Molecular genetics of herpes simplex virus: the terminal a sequence of L and S components are obligatorily identical and constitute a part of a structural gene mapping predominantly in the S component. Proc Natl Acad Sci USA. 1979;76:4534–4538. doi: 10.1073/pnas.76.9.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippe R. Deciphering novel host-herpesvirus interactions by virion proteomics. Front Microbiol. 2012;3:181–82. doi: 10.3389/fmicb.2012.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JM, Kenyon TK, Grose C, Hay J, Ruyechan WT. Physical and functional interaction between the varicella zoster virus IE63 and IE62 proteins. Virology. 2002;302:71–82. doi: 10.1006/viro.2002.1555. [DOI] [PubMed] [Google Scholar]
- Moffat JF, Stein MD, Kaneshima H, Arvin AM. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol. 1995;69:5236–5242. doi: 10.1128/jvi.69.9.5236-5242.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriuchi H, Moriuchi M, Straus SE, Cohen JI. Varicella-zoster virus open reading frame 10 protein, the herpes simplex virus VP16 homolog, transactivates herpesvirus immediate-early gene promoters. J Virol. 1993;67:2739–2746. doi: 10.1128/jvi.67.5.2739-2746.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A, Ruyechan WT, Kristie TM. The coactivator host cell factor-1 mediates Set1 and MLL H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc Natl Acad Sci USA. 2007;104:10835–10840. doi: 10.1073/pnas.0704351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niizuma T, Zerboni L, Sommer MH, Ito H, Hinchliffe S, Arvin AM. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J Virol. 2003;77:6062–6065. doi: 10.1128/JVI.77.10.6062-6065.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, He H, Hay J, Ruyechan WT. Interaction between the varicella zoster virus IE62 major transactivator and cellular transcription factor Sp1. J Biol Chem. 2003;278:38068–38075. doi: 10.1074/jbc.M302259200. [DOI] [PubMed] [Google Scholar]
- Perera LP, Mosca JD, Sadeghi-Zadeh M, Ruyechan WT, Hay J. The varicella-zoster virus immediate early protein, IE62, can positively regulate its cognate promoter. Virology. 1992;191:346–354. doi: 10.1016/0042-6822(92)90197-w. [DOI] [PubMed] [Google Scholar]
- Perera LP, Mosca JD, Ruyechan WT, Hayward GS, Straus SE, Hay J. A major transactivator of varicella-zoster virus, the immediate-early protein IE62, contains a potent N-terminal activation domain. J Virol. 1993;67:4474–4483. doi: 10.1128/jvi.67.8.4474-4483.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera LP. The TATA motif specifies the differential activation of minimal promoters by varicella zoster virus immediate-early regulatory protein IE62. J Biol Chem. 2000;275:487–496. doi: 10.1074/jbc.275.1.487. [DOI] [PubMed] [Google Scholar]
- Quinlivan ML, Gershon AA, Al Bassam BB, Steinberg SP, LaRussa P, Nicholas RA, Breuer J. Natural selection for rashforming genotypes of the varicella-zoster vaccine virus detected within immunized human hosts. Proc Natl Acad Sci USA. 2007;104:208–212. doi: 10.1073/pnas.0605688104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaus M, Desloges N, Yang M, Ruyechan WT, Wolff MH. Transcription factor USF, expressed during the entire phase of varicella-zoster virus infection, interacts physically with the major viral transactivator IE62 and plays a significant role in virus replication. J Gen Virol. 2003;84:2957–2967. doi: 10.1099/vir.0.19335-0. [DOI] [PubMed] [Google Scholar]
- Reichelt M, Brady J, Arvin AM. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J Virol. 2009;83:3904–3918. doi: 10.1128/JVI.02137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato B, Ito H, Hinchliffe S, Sommer MH, Zerboni L, Arvin AM. Mutational analysis of open reading frames 62 and 71, encoding the varicella-zoster virus immediate-early transactivating protein, IE62, and effects on replication in vitro and in skin xenografts in the SCID-hu mouse in vivo. J Virol. 2003;77:5607–5620. doi: 10.1128/JVI.77.10.5607-5620.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler ML, Ruyechan WT, Hay J. Physical interaction between two varicella zoster virus gene regulatory proteins, IE4 and IE62. Virology. 2000;272:375–381. doi: 10.1006/viro.2000.0389. [DOI] [PubMed] [Google Scholar]
- Torigoe T, Izumi H, Yoshida Y, Ishiguchi H, Okamoto T, Itoh H, Kohno K. Low pH enhances Sp1 DNA binding activity and interaction with TBP. Nucleic Acids Res. 2003;31:4523–4530. doi: 10.1093/nar/gkg487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler JK, Allen KE, Everett RD. Mutation of a single lysine residue severely impairs the DNA recognition and regulatory functions of the VZV gene 62 transactivator protein. Nucleic Acids Res. 1994;22:270–278. doi: 10.1093/nar/22.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler JK, Everett RD. The DNA binding domains of the varicella-zoster virus gene 62 and herpes simplex virus type 1 ICP4 transactivator proteins heterodimerize and bind to DNA. Nucleic Acids Res. 1994;22:711–721. doi: 10.1093/nar/22.5.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Rajamani J, Sommer M, Zerboni L, Arvin AM. Identification of a hydrophobic domain in varicella-zoster virus ORF61 necessary for ORF61 self-interaction, viral replication, and skin pathogenesis. J Virol. 2013;87:4075–4079. doi: 10.1128/JVI.02963-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Peng H, Hay J, Ruyechan WT. Role of the IE62 consensus binding site in transactivation by the varicella-zoster virus IE62 protein. J Virol. 2010;84:3767–3779. doi: 10.1128/JVI.02522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Zhou T, Chiang CM. Human mediator enhances activator-facilitated recruitment of RNA polymerase II and promoter recognition by TATA-binding protein (TBP) independently of TBP-associated factors. Mol Cell Biol. 2003;23:6229–6242. doi: 10.1128/MCB.23.17.6229-6242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Eletsky A, Szyperski T, Hay J, Ruyechan WT. Analysis of the varicella-zoster virus IE62 N-terminal acidic transactivating domain and its interaction with the human mediator complex. J Virol. 2009;83:6300–6305. doi: 10.1128/JVI.00054-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Hay J, Ruyechan WT. The DNA element controlling expression of the varicella-zoster virus open reading frame 28 and 29 genes consists of two divergent unidirectional promoters which have a common USF site. J Virol. 2004;78:10939–10952. doi: 10.1128/JVI.78.20.10939-10952.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Peng H, Hay J, Ruyechan WT. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J Virol. 2006;80:7339–7353. doi: 10.1128/JVI.00309-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Hay J, Ruyechan WT. Varicella-zoster virus IE62 protein utilizes the human mediator complex in promoter activation. J Virol. 2008;82:12154–12163. doi: 10.1128/JVI.01693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuranski T, Nawar H, Czechowski D, Lynch JM, Arvin A, Hay J, Ruyechan WT. Cell-type-dependent activation of the cellular EF-1alpha promoter by the varicella-zoster virus IE63 protein. Virology. 2005;338:35–42. doi: 10.1016/j.virol.2005.05.005. [DOI] [PubMed] [Google Scholar]