

Abstract
This Review discusses the various types of non-coding oligonucleotides, which have garnered extensive interest as new alternatives for targeted cancer therapies over small molecule inhibitors and monoclonal antibodies. These oligonucleotides can target any hallmark of cancer, no longer limited to so-called “druggable” targets. Thus, any identified gene that plays a key role in cancer progression or drug resistance can be exploited with oligonucleotides. Among them, small-interfering RNAs (siRNAs) are frequently utilized for gene silencing due to the robust and well established mechanism of RNA interference. Despite promising advantages, clinical translation of siRNAs is hindered by the lack of effective delivery platforms. This Review provides general criteria and consideration of nanoparticle development for systemic siRNA delivery. Different classes of nanoparticle candidates for siRNA delivery are discussed, and the progress in clinical trials for systemic cancer treatment is reviewed. Lastly, this Review presents HER2 (human epidermal growth factor receptor type 2)-positive breast cancer as one example that could benefit significantly from siRNA technology. How siRNA-based therapeutics can overcome cancer resistance to such therapies is discussed.
Keywords: breast cancer, oligonucleotide, siRNA, nanoparticle, targeted therapy, targeted delivery
Graphical abstract
1. Introduction
The recent launching of the visionary Precision Medicine Initiative by President Barack Obama seeks to integrate individual variability in genes, environment, and lifestyle for personalized disease treatment and prevention. As the second most common cause of death in the US after heart disease, cancer remains one of the most fatal diseases. Oncology drug discovery is therefore at the forefront of the initiative. The initiative recognizes the issue of drug resistance and seeks to develop solutions. Progress in this regard will largely rely on programs such as the Cancer Genome Atlas (TCGA) project. TCGA researchers have begun to identify genomic aberrations and affected regulatory networks that enable aspects of cancer progression including proliferation, angiogenesis, invasion, drug resistance, and metastasis.1, 2 Unfortunately, many of the identified attractive therapeutic targets are considered ‘undruggable’ by conventional means (e.g., small molecule inhibitors, antibodies).
Advances in developing non-coding RNA molecules have provided a potential alternative strategy. RNA interference (RNAi) can easily be designed to modulate virtually any gene with a known mRNA sequence.3 RNAi could also negate the dedicated costs and efforts associated with traditional chemical compound screening strategies in drug discovery. Knocking down expression of oncoproteins at the mRNA level is potentially a more effective approach because this process inhibits the synthesis of the active proteins, whereas monoclonal antibodies and small molecule inhibitors merely block their activity and do not halt the synthesis of new active oncoproteins. Despite their promise, few oligonucleotide-based therapies have reached the clinic due to inherent issues with bioavailability. The development of a versatile nanoparticle-based delivery platform is ever pressing for translating RNAi into the clinic.
This Review provides an overview on the various oligonucleotide technologies and nanoparticle delivery platforms that have reached clinical trials. Insight into the shortcomings and limitations of the first-generation delivery platforms and rationale for current state-of-the-art nanoparticle development will be discussed. Lastly, specific examples utilizing oligonucleotide-based strategies for treating HER2-positive (HER2+) breast cancer will be provided along with perspectives regarding their potential translational impact.
2. Non-coding oligonucleotides as therapeutics in cancer
The functional roles of oligonucleotides (nucleic acids), beyond their use in encoding genes and proteins, were discovered in the 1990s. The identified non-coding oligonucleotides were shown to have a role in regulating gene expression and cell function in all organisms.4 Figure 1 shows the research trend for each class of oligonucleotides and reveals that siRNAs and miRNAs have garnered the most interest since 2005. Although these oligonucleotides have promises in many disease applications, this Review will be limited to applications in cancer.
Figure 1.
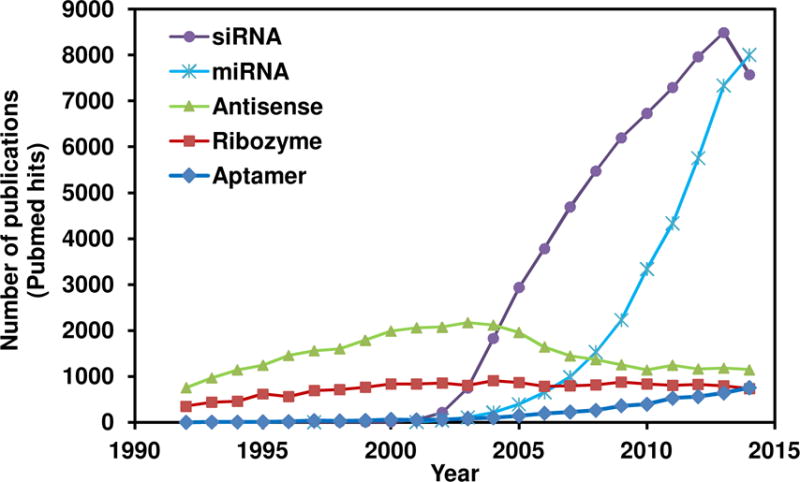
Trend of research in oligonucleotides. The number of publications each year (1992–2014) based on Pubmed queries with specified keywords.
Small interfering RNAs (siRNAs)
Small interfering RNAs (siRNAs) are small (19–25 nt) double-stranded RNAs, which are incorporated into a protein complex called RNA-induced silencing complex (RISC) upon cellular internalization.5 Each siRNA has two strands, a sense strand and an antisense strand. The sense strand will be degraded by an endonuclease of RISC, argonaute 2. The antisense strand will guide RISC towards complementary target mRNA and induce mRNA cleavage. The siRNA strand containing the thermodynamically less stable 5′-end is preferentially incorporated as the antisense strand of RISC. Hence, chemical modifications are sometimes performed on siRNA strands to favor the incorporation of the intended antisense strand with RISC by modulating their thermodynamic asymmetry.6 siRNA machinery (RISC) can also be recycled upon degrading each mRNA. siRNAs have only one mechanism of gene ablation, which is mRNA cleavage, unlike antisense oligos, which have many mechanisms of action (see next section). Although siRNAs and miRNAs share the same RISC-mediated RNA cleavage, siRNAs are optimized and designed to target certain genes with high specificity, while miRNA can have multiple or unknown targets (see next section). Thus siRNAs are deemed more effective and controllable than antisense oligos and miRNAs, and their investigations pre-clinically and in clinics will be reviewed in the next sections.
miRNAs
miRNAs (mature microRNAs) are involved in regulating post-transcriptional gene expression and thus serve as one of the mechanisms that regulate cellular events and homeostasis.7 miRNA mimics (small, chemically modified double-stranded RNAs that mimic endogenous miRNAs) follow the sequence of existing miRNAs presiding in various cell functions. Due to the ability to target multiple genes, the role of miRNAs in non-targeted cells can be uncertain. Further, miRNA expression can be upregulated or downregulated in cancer to promote cancer’s survival advantages. As miRNAs can behave as oncogenes or tumor suppressors,8 one can strategize with miRNAs therapeutically by either suppressing oncogenic miRNAs or introducing tumor suppressor miRNAs (e.g., miRNA mimics). The first miRNA mimic that entered clinical trial in 2013 utilizes a liposome-based technology to deliver miR-34a in cancer patients (primary or metastatic with liver involvement), exploiting their natural tendency to accumulate in the liver. The miR-34a was found to downregulate mRNA expression of several genes such as ERC1, RRAS, PHF19, WTAP, CTNNB1, SIPA1, DNAJB1, MYCN, and TRA2A.9 This broad targeting ability can theoretically enhance therapeutic potential, but it also increases propensity for negative unwanted side effects.
Antisense oligonucleotides
Antisense oligonucleotides modulate gene expression by altering mRNA splicing pattern, blocking mRNA translation (by providing steric hindrance), and inducing degradation of targeted mRNA by the endogenous enzyme RNase H.4 RNAs are inherently unstable in biological system due to the presence of nucleases and have poor pharmacokinetic profiles due to rapid kidney clearance. To overcome these issues, several chemical modifications of the antisense oligos have been performed. Phosphorothioate (PS) backbone modification is one of the earliest and widely used for oligos currently in clinics. PS modification increases the antisense’s stability to nuclease degradation10 and promotes binding to plasma proteins, which prevents rapid renal clearance and promotes uptake by certain cell types with scavenger receptors (e.g., kidney and liver cells).11 In newer generations of antisense therapeutics, additional modifications (e.g., sugar modification, base modification, direct conjugation to targeting ligands) are also performed in addition to PS to further improve their performance, as reviewed elsewhere.12 One of the most advanced antisense oligos for cancer in clinical trials (i.e., reaching the NDA filing stage) is Genasense (Genta Inc.). Genasense was developed to block the production of the Bcl-2 protein, a key anti-apoptotic oncoprotein in cancer.13 It was later rejected by the FDA for treating melanoma and chronic lymphocytic leukemia because the primary endpoint of improving overall survival was not met.14 ISIS Pharmaceuticals is another leading company in antisense development. The most advanced antisense in their pipeline for cancer is OGX-011, which targets clusterin in castration-resistant prostate cancer. However, the phase III SYNERGY trial did not show significant improvement in overall survival.15 Other next-generation antisense drugs for cancer developed by ISIS Pharmaceuticals include ISIS-STAT3-2.5Rx for targeting STAT316 in hepatocellular carcinoma and lymphoma and ISIS-AR-2.5Rx for targeting AR17 in prostate cancer. ISIS-STAT3-2.5Rx showed some clinical response in lymphoma patients (Phase I, 2014) and has currently progressed to Phase II studies.18 ISIS-AR-2.5Rx is currently in the phase I/II stage, but there are no published results to date.
Ribozymes
Ribozymes are considered self-processing RNAs in that they do not require proteins for catalysis. Angiozyme (Ribozyme Pharmaceuticals) is the first ribozyme that reached clinical trials for cancer treatment by targeting the vascular endothelial growth factor receptor-1 (VEGFR-1) in patients with renal cancer. Phase I results (2005) in patients with refractory solid tumors showed a favorable safety profile, and 25% of patients had stable diseases for more than 6 months.19 Angiozyme was recently evaluated with metastatic breast cancer patients (Phase II, 2012) but did not show clinical efficacy.20
Aptamers
Aptamers unlike other non-coding RNAs, rely on their tertiary and quaternary structure for interacting and binding with target proteins.21 Aptamers can bind proteins in a similar manner to antibodies but with less immunogenicity. Therefore, aptamers may serve as improved alternatives to current therapeutic antibodies. Like antibodies, most of the aptamer’s targets are still confined to only extracellular or membrane proteins. AS1411 (Antisoma PLC) was designed to inhibit nucleolin activity and was the first aptamer to reach clinical trials for cancer treatment. Extended phase I (2006) and phase II (2014) studies have shown promising outcomes in patients with metastatic renal cell carcinoma.22
Challenges and Limitations
Challenges and Limitations most applications of oligonucleotides without delivery platforms are confined to blood or clearance organs (e.g., liver and kidney). Thus, lymphoma, kidney cancer, and liver cancer are the main candidates amenable to such technology. However, delivering sufficient therapeutic levels of oligonucleotides to other solid tumors (e.g., breast, prostate, pancreatic cancer) upon systemic administration remains a challenge. Molecular complexes and nanoparticle platforms have been introduced and widely studied in order to address these unmet needs.
3. Clinical translation of siRNA-based cancer therapeutics
Systemic (intravenous) administration of siRNAs is considered more feasible and applicable than local treatment to target a wider spectrum of cancers, including advanced cancer or metastasis. Systemic delivery of siRNAs must overcome several barriers before reaching their intended site, which is the cytosol of cancer cells. When introduced in blood circulation, naked siRNAs exhibit potential for stimulating an innate immune response and are susceptible to blood enzyme degradation. Several siRNA modification strategies have been studied to address these issues: (1) backbone modifications such as phosphorothioate or boranophosphate linkages, (2) modifications of 2′-OH group on the pentose sugar such as 2′-fluoro, 2′-O-methyl, 2′-O-(2-methoxylethyl), 2′-O-(2,4-dinitrophenyl), and locked nucleic acids, and (3) modifications of the termini such as 5′-phosphate, 5′-O-methyl, and 3′-deoxythymidine.5, 23 However, extensive modifications may hamper siRNA silencing efficacy by negatively affecting RISC incorporation and target afffinity.24, 25 Due to their small size, siRNAs also suffer a short circulation half-life due to rapid kidney clearance. In addition, these modified siRNAs still lack the ability to home specifically to cancer cells.
Nanoparticles are considered the most promising carriers for siRNA delivery over such alternatives as viral-based carriers, which have concerns regarding immunogenic response and insertional mutagenesis.26 Nanoparticles can deliver hundreds to thousands of siRNA molecules per an uptake event,27 while there are only 1–10 siRNA molecules in the conjugates (with antibodies or aptamers).28 As a result, this review will only focus on non-viral vectors/platforms for siRNA delivery.
4. Nanoparticles for siRNA delivery: common rationale and concepts
Prolonging siRNAs’ half-life
Prolonging siRNAs’ half-life cannot be achieved by modifying siRNAs alone. Due to their small size, siRNAs will be cleared rapidly by kidney filtration. Similarly, nanoparticles larger than 200 nm can be trapped and cleared by the liver and spleen. Thus, nanoparticles loaded with siRNAs at a size range of 30–200 nm can potentially prolong systemic clearance.29 Besides size considerations, surface characteristics also dictate their fates in vivo.30 Charged nanoparticles can bind with opsonins like immunoglobulin and complement proteins.29 This binding promotes phagocytosis by means of Fcγ and complement receptors, respectively, in the reticuloendothelial system (RES). This also leads to faster blood clearance by the liver and spleen. To minimize the nanoparticle uptake by RES, a hydrophilic neutral polymer is often used to shield the surface charge of the nanoparticles. Polyethyleneglycol (PEG)31 is one of the most often used stealth polymers. Others are dextran and sialic acid.32 This prolonged circulation of siRNA-nanoparticles will increase the likelihood of their accumulation in tumors.
The toxicity of cationic nanoparticles
The toxicity of cationic nanoparticles must be taken into account when used as a siRNA delivery platform. Although cationic compounds (polymer or lipid) are commonly used for siRNA delivery due to their ability to load negatively charged siRNAs via electrostatic interaction, they are considered toxic to cells and notorious for poor blood compatibility. Cellular damage can be caused by direct interactions between the cationic groups and cellular components or indirectly by reactive oxidative species formed in the presence of cationic compounds.33 Further, cationic nanoparticles can interact with red blood cells, causing hemolysis.34 Since PEGylation of cationic compounds can serve to shield the surface charge of nanoparticles, the stealth effects of PEG also enhance the safety profile and blood compatibility of cationic materials.35, 36 In addition to PEG, charge neutralization upon binding with anionic-charged siRNAs may be accomplished to reduce the toxicity of cationic charged particles.37
Targeting tumors by nanoparticles
Targeting tumors by nanoparticles can be achieved by two simultaneous strategies. First, passive targeting of nanoparticles to the tumor area is made possible by the enhanced permeability and retention (EPR) effect.38 This effect describes tumors with abnormal molecular and fluid transport dynamics due to leaky vasculature and poor lymphatic drainage, and is contributed by rapidly growing tumors requiring extra nutrient, oxygen, and blood supply.39 This pathological characteristic allows nanoparticles of size 30–200 nm to escape blood vessels and accumulate in tumor tissue. The longer the blood circulation half-life, the more nanoparticles will be able to accumulate in the tumors. In clinical setting, this phenomenon is diverse among different cancer types and affected by several complex biological characteristics, such as angiogenesis, morphology of tumor vasculature, tumor genetic profiles, and components of tumor microenvironment (ECM, immune cells).39 While passive delivery to human tumors has been evidenced by enhanced tumor accumulation of drugs40 and siRNAs41, 42 when delivered by nanoparticle platforms, the field still requires significant further exploration. One of the challenges is the lack of controlled studies that tease out how different components of the tumor microenvironment or tumor types affect the EPR effect. Additional reviews on the EPR effect and nanomedicine can be found elsewhere.39, 43 Once accumulated in tumors by the EPR effect, active targeting can be achieved by decorating outer surface of nanoparticles with targeting agents (homing targets),44 such as monoclonal antibodies, single-chain variable fragments (scFv), targeting peptides, transferrin, folic acid, and aptamers. This strategy exploits the binding between targeting agents and receptors (or other membrane proteins) on the tumors, which is followed by cellular uptake of the particles termed receptor-mediated endocytosis.45 The efficiency of active targeting is contributed by several factors, such as receptor expression levels, binding affinity between receptors and targeting agents, number and accessibility of targeting agents to the receptors, and the uptake kinetics of the cells.39 Alternatively, cationic nanoparticles may enter cells upon electrostatic interaction with negatively charged cell membranes, a process called adsorptive endocytosis,46 which is less specific than the aforementioned receptor-mediated endocytosis. Other physiological properties of nanoparticles (size, shape, surface chemistry, surface roughness) also impact the cellular uptake profile.29 Active targeting should be deemed as a complementary strategy to the EPR effect rather than a distinct strategy because nanoparticles need to be in the vicinity of tumor area by passive targeting first. Upon tumor accumulation, active targeting then enhances the internalization of nanoparticles by target cells, improving its efficacy. In addition to utilizing overexpressed markers on cancer cells, active targeting may also potentiate the EPR effect/tumor accumulation by targeting tumor vasculature with overexpressed endothelial markers,47 such as tumor-penetrating iRGD peptide targeting αv integrins.48
Endosomal escape
Endosomal escape is considered a major cellular barrier for siRNA delivery. The primary route of nanoparticle uptake to cells is endocytosis. Upon endocytosis, early endosomes containing siRNA-nanoparticles will later fuse with sorting endosomes, late endosomes, and eventually lysosomes in which various nucleases and acidity will degrade siRNAs.49 To avoid lysosomal degradation, nanoparticles have to be capable of compromising endosomal membranes so that siRNAs can escape from the endosome into the cytosol where they can function. For example, liposomal-based platforms must reorganize and bind the anionic phospholipids on the endosomal membrane. This binding destabilizes the endosome membrane, allowing endosomal escape of siRNAs (flip-flop mechanism).50 Polymers with protonatable amines (e.g., Polyethylenimine (PEI)) can also promote endosomal escape by proton sponge effects.49
To summarize, in order for siRNA delivery to be feasible as illustrated in Figure 2, the nanoconstructs need to (1) be intravenously injectable and thus dispersible in saline, (2) have prolonged blood circulation (avoiding rapid clearance by the kidney, liver, and spleen) so they can seek and accumulate in the tumor, (3) protect siRNAs from blood enzyme degradation, (4) be taken up effectively into cells, (5) escape the endosome and release siRNAs in the cytoplasm, and (6) have low toxicity.
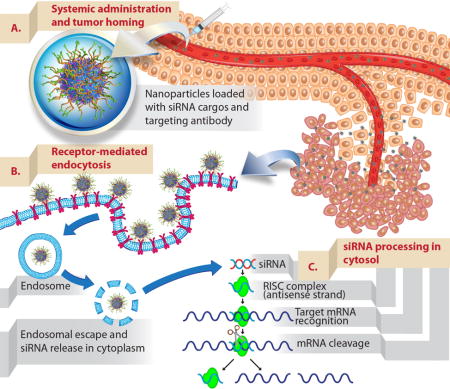
Figure 2. Schematic illustration of delivering siRNAs to tumors upon intravenous administration.
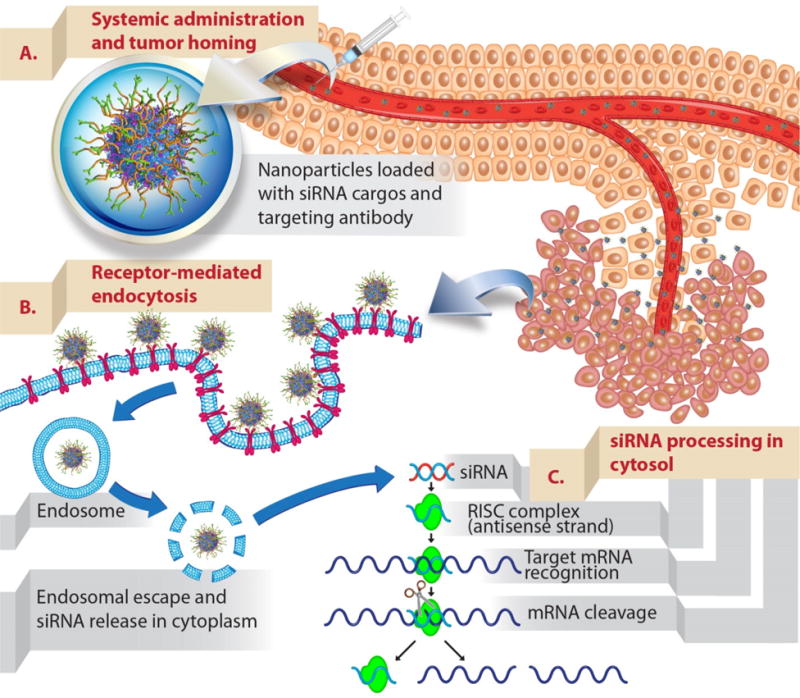
(A) Nanoparticles (mesoporous silica nanoparticles surface functionalized with polymer and targeting antibody used as an example) that were developed as siRNA carriers need to be able to protect siRNAs from blood degradation and prolong their blood residence time. This will enhance the propensity of siRNA-nanoparticles to accumulate in tumors via the EPR effect (see text). (B) Nanoparticles are typically taken up to cancer cells by endocytosis. The uptake can be further promoted by conjugating targeting ligands onto nanoparticles (active targeting). Upon endocytosis, siRNAs need to escape endosome to their site of action, cytosol. (C) Delivered siRNAs are then processed by intracellular machinery (RNA-induced silencing complex) and degrade their target complementary mRNA.
5. Different classes of nanoparticles for systemic delivery of siRNAs
This section provides a general overview of nanoparticle platforms (Figure 3) investigated for siRNA delivery. Emphasis is placed on those that show efficacy in animal models upon systemic administration. The three main classes of materials under investigation are lipid-based, polymer-based, and inorganic nanoparticles.
Figure 3.
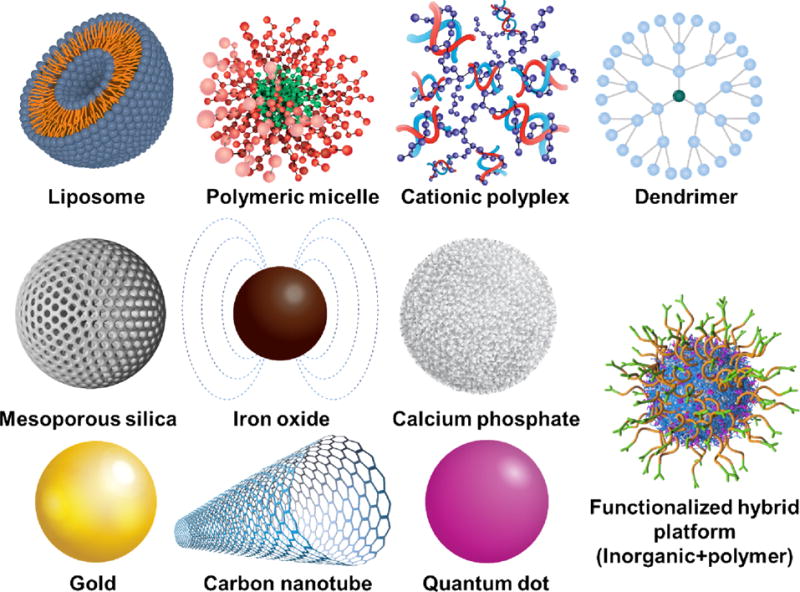
Different classes of nanoparticle platforms being developed for siRNA delivery. Three main classes of nanoparticle platforms include lipid-based, polymer-based and inorganic-based nanoparticles. Adapted from refs.36, 100
5.1 Lipid-based nanoparticles
Lipid-based nanoparticles or liposomes are among the earliest class of materials for systemic siRNA delivery that advanced to clinical trials. They are known for their excellent biocompatibility and biodegradability. When put in solution with siRNAs, siRNA-liposomes can self-assemble and encapsulate the siRNA. Endosomal escape of siRNA relies on flip-flop mechanism as discussed in the previous section. A thorough catalog of lipids utilized for encapsulation is summarized elsewhere.51 A majority of lipid-based nanoparticles exploit their natural tendency to accumulate in the liver (major clearance organ).52 Specific examples of lipid-based platforms for siRNA delivery in clinical trials will be discussed in the clinical trial section.
5.2 Polymer-based nanoparticles
A cyclodextrin-based nanoparticle was the first polymeric nanoparticle to enter clinical trials for systemic siRNA delivery to cancer.53 Since then, there have been several attempts to develop polymeric-based platforms for siRNA delivery. Attractive attributes include ease of chemical modification to introduce functionalities (e.g., endosomal escape components, targeting components), controlled release properties, and a large selection of functional polymers, while challenges for polymeric carriers are toxicity concerns and some issues of scalability and reproducibility. Traditionally, polymers used for siRNA delivery are cationic in order to complex with negatively charged siRNAs (electrostatic interaction). Also, siRNAs can be conjugated directly on the polymer. More detailed reviews on polymers (polyplex, conjugate, micelle, and dendrimer) for siRNA delivery can be found elsewhere.54
5.3 Inorganic nanoparticles
Inorganic nanoparticles exhibit unique tunable properties including the rigid surface amenable to chemical modification and their unique intrinsic properties (e.g., superparamagnetic property of iron oxide NPs,55 size-dependent optical features of quantum dots,56 photothermal property of gold and silver NPs,57 antioxidant property of mesoporous silica nanoparticle (MSNP),58 drug-loading capacity in MSNP pores). They are also typically easier to synthesize with high reproducibility and scalability. Inorganic materials do not destabilize as easily as polyplexes or liposomes when challenged with charged compounds in blood or on cell membranes. In most cases, to enable siRNA delivery, inorganic nanoparticles are typically surface-modified with other polymers or lipids, generating a hybrid compound. One hurdle for clinical translation of inorganic nanoparticles is potential toxicity due to their physicochemical properties (size, shape, charge), rigidity, and non-degradability. Nevertheless, some materials within this class are considered biologically inert (e.g., gold NPs),59 and some are degradable60 to safe components in the body (e.g., calcium phosphate,61 iron oxide,60 and mesoporous silica).62 The most widely studied inorganic nanoparticles are described as follows.
Mesoporous silica nanoparticles (MSNPs)
Mesoporous silica nanoparticles (MSNPs) have many favorable properties as drug delivery carriers such as tailorable mesoporous structures, high specific surface areas, large pore volumes, ease of controlling size, and high synthesis scalability.63 Thus, they have been studied extensively and evaluated for their biomedical applications. Mesoporous silica is biodegradable to non-toxic components (e.g., silicic acid) that can be cleared by kidneys.62 Silica nanoparticles (C dots) were approved for clinical trials as injectable PET tracers, and the Phase I safety profile was favorable.64 Therefore, the translation of other silica-based nanoparticles to clinics should be feasible.
MSNPs have been widely researched for siRNA delivery in vitro and in vivo. Efficacy in animals upon systemic administration has been attained when MSNPs were coated with cationic polymers including PEI-cyclodextrin65, PDMAEMA66, and PEI-PEG36, 67 to promote siRNA and/or cellular uptake by adsorptive endocytosis due to lack of targeting agent. Recently, our group reported an antibody-conjugated PEG-PEI-MSNP for targeted siRNA delivery, which results in growth inhibition of drug-resistant HER2-positive breast tumors.36
Iron oxide nanoparticles
Iron oxide nanoparticles have been widely evaluated in biomedical and clinical applications. Examples of FDA-approved injectable iron oxide nanoparticles include ferumoxtran-10 as an MRI imaging agent and ferumoxytol as an iron replacement product for anemia in patients with chronic kidney disease. Iron oxide nanoparticles are deemed safe because iron exists in and is essential for the body. In addition, iron oxide nanoparticles can be guided to the target site by magnetic drug delivery.68 Lastly, superparamagnetic iron oxide nanoparticles (SPIONs) can generate heat under alternating magnetic fields and have been explored for magnetic hyperthermia treatment.55 Since iron oxide nanoparticles have been approved by the FDA, the translation of this class of materials to clinics is feasible. Wu et al.69 modified SPIONs with PEI-PEG and conjugated them with targeting agents (RGD peptides) for delivery of survivin siRNA to hepatocellular carcinoma xenografts. Given every other day, the targeted nanoparticles were more effective than their non-targeted counterpart, exemplified by gene silencing activity and increased apoptotic activity in tumors.
Calcium phosphate nanoparticles
Calcium phosphate nanoparticles unlike other inorganic nanoparticles, possess the unique property of being able to self-induce endosomal escape. Calcium phosphate can dissolve in acidic conditions61 thereby increasing endosomal osmotic pressure and, in turn, causes endosomes to swell and eventually rupture. Yang et al. utilized calcium phosphate nanoparticles coated with a lipid bilayer, PEG, and anisamide (targeting sigma receptor) for siRNA delivery to NSCLC xenograft.70 Promising in vivo efficacy (tumor growth inhibition) upon systemic administration was also reported.
Gold nanoparticles
Gold nanoparticles are of interest for biomedical applications due to their property of surface plasmon resonance.59 They can be utilized for bioimaging diagnosis and photothermal therapy. SiRNA delivery with gold nanoparticles has also been widely studied in vitro. Studies that have evaluated response in animal models upon systemic administration are much less prevalent. For example, relying on passive delivery, Jensen et al. successfully delivered siBcl2L12 to glioblastoma by conjugating siRNAs and PEG directly on gold nanoparticles.71
Carbon nanotubes
Carbon nanotubes have also been evaluated as siRNA carriers. However, toxicity is of major concern for this class of material. The mechanisms of carbon nanotube toxicity include oxidative stress, inflammatory responses, and DNA damage.72 Most studies that employ carbon nanotubes to deliver siRNAs in vivo employ local injection (intratumoral injection).73 While carbon nanotubes have been widely studied for drug delivery, in vivo efficacy as siRNA carriers upon systemic administration have yet to be reported for cancer treatment.
Quantum dots
Quantum dots have unique electronic and optical properties that are tunable to different sizes and shapes.56 While they may be useful tools for molecular biology as imaging agents, the toxicity of their components (i.e., cadmium and tellurium) is an issue.74 This will likely limit the translation of quantum dots to in-human clinical applications in the near future. Non-toxic quantum dots are currently under research in a developmental stage.75 Quantum dots have not yet been evaluated in vivo for siRNA delivery.
6. Translation of systemic siRNA delivery for cancer treatment in clinics
The systemic delivery of siRNAs in their infancy relied on modified siRNAs to enhance their stability and allow cellular uptake in lieu of a carrier. However, due to their small size, a majority of them will be cleared by the kidneys with a filtration size cutoff of 10 nm. Therefore, the application of these modified siRNAs are primarily restricted to renal cancer.76
Nanoparticle platforms have been developed to overcome this barrier for siRNA delivery. As mentioned previously, the first nanoparticle system that reached clinical trial (in 2008) for siRNA delivery to solid tumors was the cyclodextrin nanoparticle system (CALAA-01) developed at Calando Pharmaceuticals. It was designed to deliver siRNA against the M2 subunit of ribonuclease reductase to melanoma. Although clinical data were only reported for three patients, it was the first successful systemic siRNA delivery by targeted nanoparticles to human tumors, as evidenced by target gene and protein knockdown, and mRNA cleavage (RACE analysis).41 Since then, there have been several developments in siRNA delivery. Table 1 summarizes the nanoparticle systems that have reached clinical trials for cancer treatment upon systemic administration, along with the references that describe the technology and/or the clinical studies. A majority of these technologies are lipid-based, exploiting their natural tendency to accumulate in the liver (major clearance organ).52
Table 1. Clinical Trials of nanoparticle-mediated siRNA delivery upon systemic administration for cancer treatment.
The drug names, companies, material descriptions, targeted diseases, and their clinical status are shown.
| Drug | Company | Gene target | Description | Diseases/Indications | Clinical status (Trial number) | Outcomes/Limitations |
|---|---|---|---|---|---|---|
| CALAA-0141, 77 | Calando Pharma | RRM2 | Cyclodextrin-based nanoparticles with transferrin as a targeting agent | Solid tumor/advanced melanoma | Phase I, terminated (NCT00689065) | Stable disease observed in one (out of three) patient; biopsies show successful gene knockdown. However, construct is rapidly destabilized in kidneys78 and could explain adverse events in patients (e.g., liver/kidney toxicity, platelet drop, prolonged APPT), which were also found in animals receiving the polymer delivery components alone. |
| ALN-VSP0242 | Alnylam Pharma | KSP + VEGF | Stable nucleic acid lipid particles (SNALPs) | Advanced solid tumors with liver involvement | Phase I, completed (NCT00882180) | 4 out of 24 patients receiving >0.7 mg/kg achieved stable disease, 1 of which achieved complete response. One patient died from liver failure possibly from disease progression and/or drug-induced injury to the small amount of remaining normal liver. Phase II will establish pharmacodynamics changes and clinical outcome. |
| Phase I, completed (NCT01158079) | ||||||
| Atu02779, 80 | Silence Therapeutics | PKN3 | Cationic lipoplex containing siRNA | Advanced solid tumors | Phase I, completed (NCT00938574) | In phase I, 14 out of 34 patients had stable disease. Given with gemcitabine, Phase IIa data indicated that higher and more frequent doses needed to be investigated prior to full Phase II study. |
| Pancreatic ductal carcinoma | Phase I/II, ongoing (NCT01808638) | |||||
| TKM-08030181 | Tekmira Pharma (now called Arbutus Biopharma) | PLK1 | Stable nucleic acid lipid particles (SNALPs) | Solid tumors with liver involvement | Phase I, completed (NCT01437007) | Stable disease (4/9) if >0.6 mg/kg, but max tolerated dose = 0.75 mg/kg. |
| Neuroendocrine tumors and adrenocortical carcinoma | Phase I/II, completed (NCT01262235) | Outcomes are not available yet. | ||||
| Hepatocellular carcinoma (HCC) | Phase I/II, ongoing (NCT02191878) | Phase IIa clinical trial has also been modified to treat chronic HBV patients enrolled in the HCC trial | ||||
| DCR-MYC82 | Dicerna Pharma | MYC | Lipid nanoparticles | Solid tumors, myeloma and lymphoma | Phase I, recruiting (NCT02110563) | Interim data report mild adverse events, and some metabolic response and tumor shrinkage.83 |
| Hepatocellular carcinoma | Phase I/II, recruiting (NCT02314052) | Recruiting, no published result yet | ||||
| siRNA-EphA2-DOPC84 | MD Anderson Cancer Center | EphA2 | Neutral liposomes | Advanced cancer | Phase I, recruiting (NCT01591356) | Liposome without targeting agent (Recruiting) |
Delivering siRNAs successfully to other solid tumors in humans remains a challenge. For example, Atu027 was designed to modulate PKN3 expression in the vascular endothelium (not cancer cells).80 By reducing vascular leakage/permeability with siPKN3, Atu027 may limit the metastatic potential of the cancer. In a completed Phase I study, Atu027 was found to stabilize diseases in 41% of the patients with advanced solid tumors at the end of treatment (i.e., 8 weeks).80 Accordingly, it will be evaluated in combination with gemcitabine in patients with pancreatic cancer in the Phase I/II trial. While the development of Atu027 signifies important progress in the field of siRNA delivery, it does not contain a targeting agent and hence has not yet addressed the need to deliver siRNAs specifically to cancer cells. Other particles are lipid-based nanoparticles, which face the same limitations of liver homing. Clinical trials evaluating these compounds in solid tumors beyond the liver are ongoing (Table 1), and their efficacy remains to be seen. More detailed review on siRNA platform in cancer clinical trials can be found in a recent review.85
As an example of how siRNA based therapeutics can overcome shortcomings of current standard therapies, we will present a case study with HER2+ breast cancer.
7. Overview of HER2+ breast cancer and HER2 protein
Breast cancer is the most frequently diagnosed cancer in women, and, after lung cancer, the second leading cause of cancer death in women in the US since 1950.86 The current risk of American women developing breast cancer in their lifetime is one in eight. It is estimated that 234,190 new cases will be diagnosed in the US in 2015.86 For metastatic breast cancer, the five-year survival rate is 25%. HER2+ breast cancer is a clinical subtype that presents HER2 overexpression on the tumor cell surface, caused by the amplification of HER2 oncogene and related genetic elements in the amplicon on chromosome 17.87 This clinical subtype accounts for approximately 15–25% of invasive breast cancer.88
HER2 (ERBB2) belongs to a family of transmembrane receptor tyrosine kinases (RTKs), which also include HER1 (EGFR; epidermal growth factor receptor), HER3 (ERBB3), and HER4 (ERBB4). RTKs have key roles in regulating several cellular processes such as proliferation, migration, metabolism, differentiation, and survival, particularly during embryogenesis.89, 90 In normal cells, this signaling network is tightly regulated. However, when these genes mutate, amplify, or overexpress, they become oncogenes that are responsible for the onset, progression, and aggressiveness of many types of cancer.
8. Targeted RNAi-based Therapeutic Strategies for HER2+ Breast Cancer
The standard treatment for HER2+ breast cancer (and first-line in metastatic HER2+ breast cancer) is a combination of HER2-targeted therapies (trastuzumab + pertuzumab) and a taxane agent (docetaxel or paclitaxel). However, it cannot manage advanced cancer beyond prolonging survival and, even still, most HER2+ patients progress. Thus, non-coding RNA molecules such as HER2 siRNA (siHER2) serve as potential alternatives to antibodies and small molecule inhibitors. Knocking down HER2 at the mRNA level is potentially a more effective approach than merely blocking HER2 activities by conventional monoclonal antibodies or small molecule inhibitors because it circumvents resistance mediated by incomplete receptor blockage, alternative splicing and post-translational modification, mutations, and sustained phosphorylation mediated by other proteins.91 Further, while monoclonal antibodies and small molecule inhibitors can target only certain accessible proteins (so-called “druggable targets”), RNAi can be designed to modulate virtually any gene with known mRNA sequences.
To date, no siRNA therapeutics for HER2+ breast cancer have reached clinical trials. In preclinical studies, a handful of studies have explored RNAi-based strategies targeting HER2+ breast cancer. Inoue et al.92 utilized polymalic acid conjugated with HER2 antisense and trastuzumab (HER2-antibody) for targeted delivery. In addition to serving as a targeting agent, trastuzumab can elicit therapeutic effects presenting a potential synergy between HER2 antisense and trastuzumab by which HER2 antisense inhibits new HER2 receptor synthesis, and trastuzumab blocks the activity of existing HER2 receptors. In addition to targeting HER2, two studies investigated siPLK1 (siRNA against polo-like kinase 1 (PLK1)) as an anticancer strategy. PLK1 is an attractive therapeutic target in oncology due to its integral role in promoting cell division, and its overexpression is correlated with aggressive behavior in multiple tumor types.93 Yao et al.94 complexed siPLK1 with a peptide fusion protein containing HER2 scFv for targeted siRNA delivery. Later, Dou et al.95 employed a similar strategy but instead used PEG-PLA based nanoparticles conjugated with HER2 scFv. Both studies showed successful targeted delivery, accumulation, and silencing of PLK1 in breast tumor models. Despite the promising results, these studies all utilized the BT474 xenograft model, a trastuzumab-sensitive cell line.
Recently, our group has reported that treatment with siHER2 could induce growth arrest with effective GI50’s in the low nanomolar range (<10 nM) in a panel of 15 trastuzumab-resistant (8 of which are also lapatinib-resistant) HER2+ breast cancer cell lines (Figure 4). The effect (e.g., cell death) was also specific to HER2+ cancer cells and not HER2-cells.
Figure 4. Trastuzumab and siHER2 response in 21 HER2-positive cell lines and 2 HER2-negative cell lines.
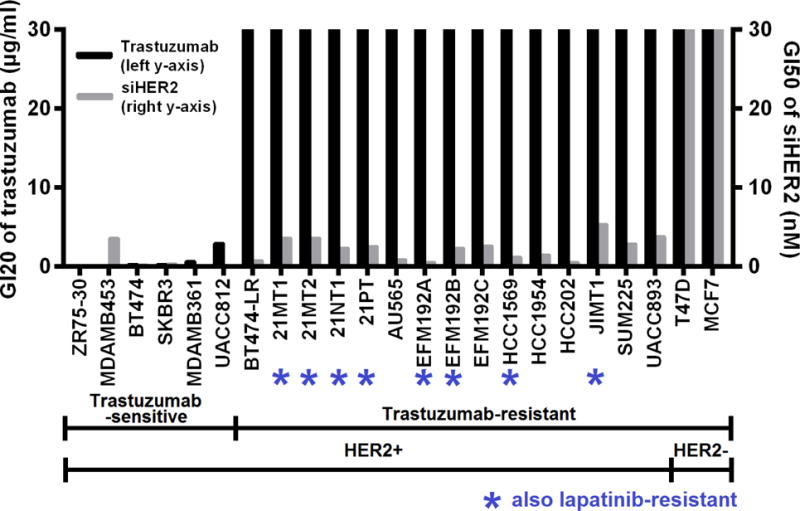
Cells were treated with trastuzumab in a dose range of 0–30 μg/ml or siHER2 in a dose range of 0–30 nM. Cell viability was measured 5 days after treatment. GI20 values (dose required to achieve 20% inhibition of cell growth) of trastuzumab and GI50 values (dose required to achieve 50% inhibition of cell growth) of siHER2 across the cell lines are reported. If required growth inhibition was not achieved, 30 μg/ml and 30 nM were reported for trastuzumab and siHER2, respectively. Modified with Permission from John Wiley and Sons36
For systemic delivery of the siHER2, our group has developed an MSNP-based delivery platform conjugated with trastuzumab as the targeting agent.36 The nanoconstruct consisted of a MSNP core (~50 nm) with surface modifications of cross-linked PEI and PEG. PEI promoted the endosomal escape of the siRNA, while PEG provided steric effects to prevent aggregation, protected siRNA against enzyme degradation, and enhanced overall blood compatibility. Further conjugation with trastuzumab provided homing capabilities and showed enhanced specificity (see Figure 5).36 The nanoconstructs effectively stimulated apoptotic death in HER2+ breast cancer cells grown in vitro, while sparing HER2 non-amplified cells. As in the Inoue et al. study, we also observed a synergistic effect of trastuzumab and siHER2 upon evaluation in trastuzumab-sensitive cells such as BT474. More importantly, the siHER2-nanoconstruct displayed efficacy in refractory HER2+ xenograft tumors. As shown in Figures 5A and B, HER2 protein levels could be reduced by 60% in orthotopic HCC1954 xenografts following a single intravenous injection of the siHER2-nanoconstructs. While the tumor was shown to be resistant to regimens of trastuzumab with or without paclitaxel (Figure 5C), siHER2-nanoconstructs could achieve significant tumor growth inhibition after 2 doses (Figure 5D). This ability to overcome resistance with siHER2 despite targeting the same pathway as trastuzumab emphasizes a key advantage of RNAi. This suggests great promise in RNAi technology as alternative and efficient agents to conventional small molecule inhibitors and monoclonal antibodies.
Figure 5. In vivo HER2 reduction and growth inhibition of orthotopic HCC1954 tumors.
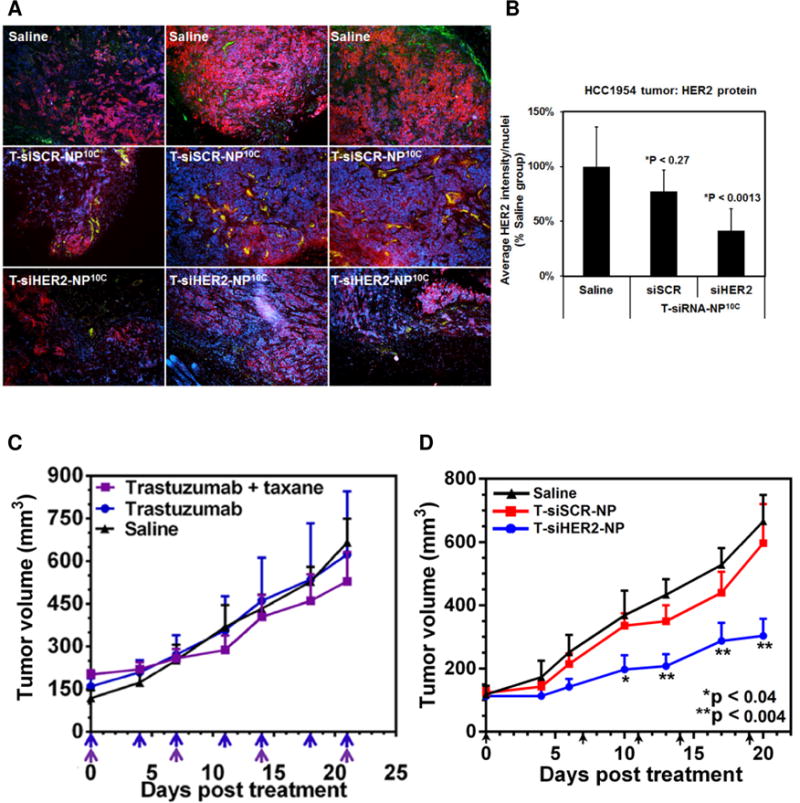
(A) Representative immunofluorescent images of tumor tissues collected from mice (n = 4/group) at four days post i.v. injection with one dose of MSNP-based nanoconstructs loaded with siHER2 or siSCR (1.25 mg siRNA/kg) or PBS control. (B) Quantitative HER2 levels of the tissues (means ± SD). Images were analyzed by CellProfiler (red = HER2 protein; green = CD31 endothelial marker; blue = DAPI staining cell nuclei). (C) Tumor growth in mice bearing orthotopic HCC1954 tumor xenografts showed resistance to 5 mg/kg trastuzumab with or without 3 mg/kg paclitaxel but (D) responded to T-siHER2-NP, given via IV injection – receiving the same treatments as (A) but multiple doses (days of injection are indicated by arrows). Tumor volumes are presented as means ± SEM. Specified p-values are against the saline control. Reproduced with Permission from John Wiley and Sons36
Like other nanoparticle candidates, mesoporous silica based nanoparticle (MSNPs) for siHER2 delivery need to be evaluated extensively for acute and chronic toxicity prior to clinical applications. In mice, the adverse reaction above maximum tolerated dose (MTD) of IV administered MSNPs is typically linked to mechanical obstruction of MSNPs in the vasculature that leads to congestion in multiple vital organs and subsequent organ failure.96 This could be attributed to very high local concentration during bolus injection into small veins of mice, which should be largely mitigated with infusion in humans. In general, the toxicity (in vitro and in vivo) of MSNPs depends largely on porosity, size, shape, and surface characteristic, as extensively reviewed elsewhere.97,98 For example, aminated-MSNPs96 or PEGylated-MSNPs99 were found to be safer than non-modified MSNPs. While some generalization regarding toxicity may be made, the toxicity of specific material must be evaluated with the final constructs as we have done with our material. Cargos like siRNAs can neutralize cationic charged particles, making them safer. Owing to small and uniform particle size of MSNP, steric effect of PEG, and charge neutralization with siRNA, we have reported outstanding blood compatibility, low immune response,36 and low cytotoxicity of our material.36, 91 Nevertheless, extensive efficacy and toxicity profile of this drug candidate in mice and primates needs to be established prior to clinical evaluation.
Conclusion and Future directions
For nanoparticle-based siRNA therapeutics to be successful, aside from overcoming the general bottleneck associated with effective systemic bioavailability and intracellular delivery as outlined in this Review, their ability to avoid adverse toxicity associated with immune response, organ accumulation, their manufacturability (scale-up with high reproducibility) and their storability are just as vital. However, limited data have been reported on these subjects. Co-delivery of multiple siRNAs or siRNAs and chemotherapies may be needed to address cancer heterogeneity or modified over time with different disease stages. Lastly, to help realize the goals of personalized medicine, adaptable nanoconstructs that can facilitate this concept are highly beneficial (e.g., siRNAs is loaded last, by simple mixing in saline,36 upon identification of target genes).
Search strategy and selection criteria
References for this Review were identified through searches of PubMed with the search terms “siRNA in vivo”, “systemic/i.v./tail vein”, “nanoparticles”, and “cancer” from 2000 until May, 2015. Articles were also identified through searches of the authors’ own files. Only papers published in English were reviewed. The final reference list was generated on the basis of originality and relevance to the broad scope of this Review.
Highlights.
Oligonucleotides as alternative candidates for targeted therapies.
siRNAs open doors to hitting ‘undruggable’ targets.
Translation of siRNAs to clinics is dependent on delivery platform.
Nanoparticles have been widely studied for siRNA delivery.
siRNAs have great potential in drug-resistant HER2-positive breast cancer.
Acknowledgments
We are grateful to Dr. Oleh Taratula of Oregon State University for independently reviewing this manuscript. This work was supported by National Cancer Institute of NIH under a contract HHSN261201300078C, the Prospect Creek Foundation, and OHSU’s Office of the Vice President for Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interests
OHSU, W.N., D.J.C., J.M., J.W.G., and W.Y. have a significant financial interest in PDX Pharmaceuticals, LLC. This potential personal and institutional conflict of interest has been reviewed and managed by OHSU.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 4.Burnett JC, Rossi JJ. RNA-based Therapeutics-Current Progress and Future Prospects. Chemistry & biology. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes-da-Silva LC, Simoes S, Moreira JN. Challenging the future of siRNA therapeutics against cancer: the crucial role of nanotechnology. Cellular and molecular life sciences : CMLS. 2014;71:1417–38. doi: 10.1007/s00018-013-1502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bramsen JB, Laursen MB, Nielsen AF, Hansen TB, Bus C, Langkjaer N, et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009;37:2867–81. doi: 10.1093/nar/gkp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X. MicroRNA: Function, Detection, and Bioanalysis. Chemical Reviews. 2013;113:6207–33. doi: 10.1021/cr300362f. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Developmental Biology. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 9.Daige CL, Wiggins JF, Priddy L, Nelligan-Davis T, Zhao J, Brown D. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol Cancer Ther. 2014;13:2352–60. doi: 10.1158/1535-7163.MCT-14-0209. [DOI] [PubMed] [Google Scholar]
- 10.Eckstein F. Phosphorothioate oligodeoxynucleotides: what is their origin and what is unique about them? Antisense & nucleic acid drug development. 2000;10:117–21. doi: 10.1089/oli.1.2000.10.117. [DOI] [PubMed] [Google Scholar]
- 11.Bijsterbosch MK, Manoharan M, Rump ET, De Vrueh RL, van Veghel R, Tivel KL, et al. In vivo fate of phosphorothioate antisense oligodeoxynucleotides: predominant uptake by scavenger receptors on endothelial liver cells. Nucleic Acids Res. 1997;25:3290–6. doi: 10.1093/nar/25.16.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett CF, Swayze EE. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annual Review of Pharmacology and Toxicology. 2010;50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 13.Herbst RS, Frankel SR. Oblimersen Sodium (Genasense bcl-2 Antisense Oligonucleotide): A Rational Therapeutic to Enhance Apoptosis in Therapy of Lung Cancer. Clinical Cancer Research. 2004;10:4245s–8s. doi: 10.1158/1078-0432.CCR-040018. [DOI] [PubMed] [Google Scholar]
- 14.Frantz S. Lessons learnt from Genasense’s failure. Nat Rev Drug Discov. 2004;3:542. doi: 10.1038/nrd1464. [DOI] [PubMed] [Google Scholar]
- 15.Inman S. Custirsen Combination Misses Goal in Phase III mCRPC Trial. OncLive; Plainsboro, NJ: 2014. [Google Scholar]
- 16.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nature medicine. 2005;11:623–9. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto Y, Loriot Y, Beraldi E, Zhang F, Wyatt AW, AL NAKOUZI N, et al. Generation 2.5 Antisense Oligonucleotides Targeting the Androgen Receptor and its Splice Variants Suppress Enzalutamide Resistant Prostate Cancer Cell Growth. Clinical Cancer Research. 2015 doi: 10.1158/1078-0432.CCR-14-1108. [DOI] [PubMed] [Google Scholar]
- 18.Hong D, Kim Y, Younes A, Nemunaitis J, Fowler N, Hsu J, et al. Abstract LB-227: Preclinical pharmacology and clinical efficacy of AZD9150 (ISIS-STAT3Rx), a potent next-generation antisense oligonucleotide inhibitor of STAT3. Cancer Research. 2014;74:LB–227. [Google Scholar]
- 19.Weng DE, Masci PA, Radka SF, Jackson TE, Weiss PA, Ganapathi R, et al. A phase I clinical trial of a ribozyme-based angiogenesis inhibitor targeting vascular endothelial growth factor receptor-1 for patients with refractory solid tumors. Molecular Cancer Therapeutics. 2005;4:948–55. doi: 10.1158/1535-7163.MCT-04-0210. [DOI] [PubMed] [Google Scholar]
- 20.Morrow PK, Murthy RK, Ensor JD, Gordon GS, Margolin KA, Elias AD, et al. An open-label, phase 2 trial of RPI.4610 (Angiozyme) in the treatment of metastatic breast cancer. Cancer. 2012;118:4098–104. doi: 10.1002/cncr.26730. [DOI] [PubMed] [Google Scholar]
- 21.Rayburn ER, Zhang R. Antisense, RNAi, and gene silencing strategies for therapy: mission possible or impossible? Drug discovery today. 2008;13:513–21. doi: 10.1016/j.drudis.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg JE, Bambury RM, Van Allen EM, Drabkin HA, Lara PN, Jr, Harzstark AL, et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investigational new drugs. 2014;32:178–87. doi: 10.1007/s10637-013-0045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hickerson RP, Vlassov AV, Wang Q, Leake D, Ilves H, Gonzalez-Gonzalez E, et al. Stability Study of Unmodified siRNA and Relevance to Clinical Use. Oligonucleotides. 2008;18:345–54. doi: 10.1089/oli.2008.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bramsen JB, Laursen MB, Nielsen AF, Hansen TB, Bus C, Langkjær N, et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Research. 2009 doi: 10.1093/nar/gkp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pascut D, Bedogni G, Tiribelli C. Silencing efficacy prediction: a retrospective study on target mRNA features. Bioscience Reports. 2015;35:e00185. doi: 10.1042/BSR20140147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinn PL, Sauter SL, McCray PB., Jr Gene therapy progress and prospects: development of improved lentiviral and retroviral vectors–design, biosafety, and production. Gene therapy. 2005;12:1089–98. doi: 10.1038/sj.gt.3302570. [DOI] [PubMed] [Google Scholar]
- 27.Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-Layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-Negative Breast Cancer Treatment. ACS Nano. 2013;7:9571–84. doi: 10.1021/nn4047925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song E, Zhu P, Lee S-K, Chowdhury D, Kussman S, Dykxhoorn DM, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotech. 2005;23:709–17. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 29.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–57. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 30.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical Studies To Understand Nanoparticle Interaction with the Immune System and Its Potential Effects on Nanoparticle Biodistribution. Molecular pharmaceutics. 2008;5:487–95. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Advanced drug delivery reviews. 2003;55:403–19. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 32.Salmaso S, Caliceti P. Stealth Properties to Improve Therapeutic Efficacy of Drug Nanocarriers. Journal of Drug Delivery. 2013;2013:19. doi: 10.1155/2013/374252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen KB, Northeved H, Kumar Ek P, Permin A, Gjetting T, Andresen TL, et al. In vivo toxicity of cationic micelles and liposomes. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11:467–77. doi: 10.1016/j.nano.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24:1121–31. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 35.O’Mahony AM, Ogier J, Darcy R, Cryan JF, O’Driscoll CM. Cationic and PEGylated Amphiphilic Cyclodextrins: Co-Formulation Opportunities for Neuronal Sirna Delivery. PloS one. 2013;8:e66413. doi: 10.1371/journal.pone.0066413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ngamcherdtrakul W, Morry J, Gu S, Castro DJ, Goodyear SM, Sangvanich T, et al. Cationic Polymer Modified Mesoporous Silica Nanoparticles for Targeted siRNA Delivery to HER2+ Breast Cancer. Advanced Functional Materials. 2015;25:2646–59. doi: 10.1002/adfm.201404629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabrielson NP, Lu H, Yin L, Kim KH, Cheng J. A Cell-penetrating Helical Polymer For siRNA Delivery to Mammalian Cells. Mol Ther. 2012;20:1599–609. doi: 10.1038/mt.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. Journal of controlled release : official journal of the Controlled Release Society. 2000;65:271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 39.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Advanced drug delivery reviews. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG, et al. Effective Targeting of Solid Tumors in Patients With Locally Advanced Cancers by Radiolabeled Pegylated Liposomes. Clinical Cancer Research. 2001;7:243–54. [PubMed] [Google Scholar]
- 41.Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer discovery. 2013;3:406–17. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- 43.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, et al. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Research. 2013;73:2412–7. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toporkiewicz M, Meissner J, Matusewicz L, Czogalla A, Sikorski AF. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: principles, hopes, and challenges. International journal of nanomedicine. 2015;10:1399–414. doi: 10.2147/IJN.S74514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li S-D, Chen Y-C, Hackett MJ, Huang L. Tumor-targeted Delivery of siRNA by Self-assembled Nanoparticles. Mol Ther. 2007;16:163–9. doi: 10.1038/sj.mt.6300323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seib FP, Jones AT, Duncan R. Comparison of the endocytic properties of linear and branched PEIs, and cationic PAMAM dendrimers in B16f10 melanoma cells. Journal of controlled release : official journal of the Controlled Release Society. 2007;117:291–300. doi: 10.1016/j.jconrel.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 47.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. The Journal of Cell Biology. 2010;188:759–68. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer cell. 2009;16:510–20. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. Journal of Cell Science. 2010;123:1183–9. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 50.Xu Y, Szoka FC. Mechanism of DNA Release from Cationic Liposome/DNA Complexes Used in Cell Transfection. Biochemistry. 1996;35:5616–23. doi: 10.1021/bi9602019. [DOI] [PubMed] [Google Scholar]
- 51.Tam YYC, Chen S, Cullis PR. Advances in Lipid Nanoparticles for siRNA Delivery. Pharmaceutics. 2013;5:498–507. doi: 10.3390/pharmaceutics5030498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. Journal of controlled release : official journal of the Controlled Release Society. 2015;203:1–15. doi: 10.1016/j.jconrel.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 53.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Molecular pharmaceutics. 2009;6:659–68. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 54.Vader P, van der Aa LJ, Storm G, Schiffelers RM, Engbersen JF. Polymeric carrier systems for siRNA delivery. Current topics in medicinal chemistry. 2012;12:108–19. doi: 10.2174/156802612798919123. [DOI] [PubMed] [Google Scholar]
- 55.Hayashi K, Nakamura M, Sakamoto W, Yogo T, Miki H, Ozaki S, et al. Superparamagnetic nanoparticle clusters for cancer theranostics combining magnetic resonance imaging and hyperthermia treatment. Theranostics. 2013;3:366–76. doi: 10.7150/thno.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Quantum dots versus organic dyes as fluorescent labels. Nat Meth. 2008;5:763–75. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 57.Austin LA, Mackey MA, Dreaden EC, El-Sayed MA. The optical, photothermal, and facile surface chemical properties of gold and silver nanoparticles in biodiagnostics, therapy, and drug delivery. Archives of toxicology. 2014;88:1391–417. doi: 10.1007/s00204-014-1245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morry J, Ngamcherdtrakul W, Gu S, Goodyear SM, Castro DJ, Reda MM, et al. Dermal delivery of HSP47 siRNA with NOX4-modulating mesoporous silica-based nanoparticles for treating fibrosis. Biomaterials. 2015;66:41–52. doi: 10.1016/j.biomaterials.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X. Gold Nanoparticles: Recent Advances in the Biomedical Applications. Cell biochemistry and biophysics. 2015 doi: 10.1007/s12013-015-0529-4. [DOI] [PubMed] [Google Scholar]
- 60.Ehlerding EB, Chen F, Cai W. Biodegradable and Renal Clearable Inorganic Nanoparticles. Advanced Science. 2016;3:n/a–n/a. doi: 10.1002/advs.201500223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Chen Y-C, Tseng Y-C, Huang L. Biodegradable Calcium Phosphate Nanoparticle with Lipid Coating for Systemic siRNA Delivery. Journal of controlled release : official journal of the Controlled Release Society. 2010;142:416–21. doi: 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ. Mesoporous silica nanoparticle nanocarriers: biofunctionality and biocompatibility. Accounts of chemical research. 2013;46:792–801. doi: 10.1021/ar3000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung TH, Wu SH, Yao M, Lu CW, Lin YS, Hung Y, et al. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3T3-L1 cells and human mesenchymal stem cells. Biomaterials. 2007;28:2959–66. doi: 10.1016/j.biomaterials.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 64.Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Science translational medicine. 2014;6:260ra149. doi: 10.1126/scitranslmed.3009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen J, Kim HC, Su H, Wang F, Wolfram J, Kirui D, et al. Cyclodextrin and Polyethylenimine Functionalized Mesoporous Silica Nanoparticles for Delivery of siRNA Cancer Therapeutics. Theranostics. 2014;4:487–97. doi: 10.7150/thno.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin D, Cheng Q, Jiang Q, Huang Y, Yang Z, Han S, et al. Intracellular cleavable poly(2-dimethylaminoethyl methacrylate) functionalized mesoporous silica nanoparticles for efficient siRNA delivery in vitro and in vivo. Nanoscale. 2013;5:4291–301. doi: 10.1039/c3nr00294b. [DOI] [PubMed] [Google Scholar]
- 67.Meng H, Mai WX, Zhang H, Xue M, Xia T, Lin S, et al. Codelivery of an Optimal Drug/siRNA Combination Using Mesoporous Silica Nanoparticles To Overcome Drug Resistance in Breast Cancer in Vitro and in Vivo. ACS Nano. 2013;7:994–1005. doi: 10.1021/nn3044066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chertok B, Moffat BA, David AE, Yu F, Bergemann C, Ross BD, et al. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials. 2008;29:487–96. doi: 10.1016/j.biomaterials.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu C, Gong F, Pang P, Shen M, Zhu K, Cheng D, et al. An RGD-modified MRI-visible polymeric vector for targeted siRNA delivery to hepatocellular carcinoma in nude mice. PloS one. 2013;8:e66416. doi: 10.1371/journal.pone.0066416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang Y, Hu Y, Wang Y, Li J, Liu F, Huang L. Nanoparticle Delivery of Pooled siRNA for Effective Treatment of Non-Small Cell Lung Caner. Molecular pharmaceutics. 2012;9:2280–9. doi: 10.1021/mp300152v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, et al. Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Science translational medicine. 2013:5. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Y, Zhao Y, Sun B, Chen C. Understanding the toxicity of carbon nanotubes. Accounts of chemical research. 2013;46:702–13. doi: 10.1021/ar300028m. [DOI] [PubMed] [Google Scholar]
- 73.Podesta JE, Al-Jamal KT, Herrero MA, Tian B, Ali-Boucetta H, Hegde V, et al. Antitumor Activity and Prolonged Survival by Carbon-Nanotube-Mediated Therapeutic siRNA Silencing in a Human Lung Xenograft Model. Small (Weinheim an der Bergstrasse, Germany) 2009;5:1176–85. doi: 10.1002/smll.200801572. [DOI] [PubMed] [Google Scholar]
- 74.Nguyen KC, Rippstein P, Tayabali AF, Willmore WG. Mitochondrial Toxicity of Cadmium Telluride Quantum Dot Nanoparticles in Mammalian Hepatocytes. Toxicological sciences : an official journal of the Society of Toxicology. 2015;146:31–42. doi: 10.1093/toxsci/kfv068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin G, Ouyang Q, Hu R, Ding Z, Tian J, Yin F, et al. In vivo toxicity assessment of non-cadmium quantum dots in BALB/c mice. Nanomedicine: Nanotechnology, Biology and Medicine. 11:341–50. doi: 10.1016/j.nano.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 76.Molitoris BA, Dagher PC, Sandoval RM, Campos SB, Ashush H, Fridman E, et al. siRNA Targeted to p53 Attenuates Ischemic and Cisplatin-Induced Acute Kidney Injury. Journal of the American Society of Nephrology : JASN. 2009;20:1754–64. doi: 10.1681/ASN.2008111204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuckerman JE, Gritli I, Tolcher A, Heidel JD, Lim D, Morgan R, et al. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proceedings of the National Academy of Sciences. 2014;111:11449–54. doi: 10.1073/pnas.1411393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuckerman JE, Choi CHJ, Han H, Davis ME. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proceedings of the National Academy of Sciences. 2012;109:3137–42. doi: 10.1073/pnas.1200718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aleku M, Schulz P, Keil O, Santel A, Schaeper U, Dieckhoff B, et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008;68:9788–98. doi: 10.1158/0008-5472.CAN-08-2428. [DOI] [PubMed] [Google Scholar]
- 80.Schultheis B, Strumberg D, Santel A, Vank C, Gebhardt F, Keil O, et al. First-in-Human Phase I Study of the Liposomal RNA Interference Therapeutic Atu027 in Patients With Advanced Solid Tumors. Journal of Clinical Oncology. 2014;32:4141–8. doi: 10.1200/JCO.2013.55.0376. [DOI] [PubMed] [Google Scholar]
- 81.Ramanathan RK, Hamburg SI, Borad MJ, Seetharam M, Kundranda MN, Lee P, et al. A phase I dose escalation study of TKM-080301, a RNAi therapeutic directed against PLK1, in patients with advanced solid tumors. AACR 104th Annual Meeting 2013; Washington DC. 2013. [Google Scholar]
- 82.Dudek H, Wong DH, Arvan R, Shah A, Wortham K, Ying B, et al. Knockdown of [beta]-catenin with Dicer-Substrate siRNAs Reduces Liver Tumor Burden In vivo. Mol Ther. 2014;22:92–101. doi: 10.1038/mt.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tolcher AW, Papadopoulos KP, Patnaik A, Rasco DW, Martinez D, Wood DL, et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. 2015 ASCO Annual Meeting; 2015. [Google Scholar]
- 84.Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–8. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 85.Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14:843–56. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 86.ACS, Cancer Facts & Figures 2015. Atlanta: American Cancer Society; 2015. [Google Scholar]
- 87.Nassar A, Khoor A, Radhakrishnan R, Radhakrishnan A, Cohen C. Correlation of HER2 overexpression with gene amplification and its relation to chromosome 17 aneuploidy: a 5-year experience with invasive ductal and lobular carcinomas. International Journal of Clinical and Experimental Pathology. 2014;7:6254–61. [PMC free article] [PubMed] [Google Scholar]
- 88.Prat A, Carey LA, Adamo B, Vidal M, Tabernero J, Cortes J, et al. Molecular features and survival outcomes of the intrinsic subtypes within HER2-positive breast cancer. Journal of the National Cancer Institute. 2014;106 doi: 10.1093/jnci/dju152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alvarez RH, Cortés J, Mattos-Arruda LD, Falzon M, Fasolo A, Gandy M, et al. Handbook of HER2-Targeted Agents in Breast Cancer. London, UK: Springer Healthcare Communications; 2013. [Google Scholar]
- 90.Spector NL, Blackwell KL. Understanding the Mechanisms Behind Trastuzumab Therapy for Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. Journal of Clinical Oncology. 2009;27:5838–47. doi: 10.1200/JCO.2009.22.1507. [DOI] [PubMed] [Google Scholar]
- 91.Gu S, Hu Z, Ngamcherdtrakul W, Castro DJ, Morry J, Reda MM, et al. Therapeutic siRNA for Drug-resistant HER2-Positive Breast Cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.7409. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Inoue S, Ding H, Portilla-Arias J, Hu J, Konda B, Fujita M, et al. Polymalic Acid– Based Nanobiopolymer Provides Efficient Systemic Breast Cancer Treatment by Inhibiting both HER2/neu Receptor Synthesis and Activity. Cancer research. 2011;71:1454–64. doi: 10.1158/0008-5472.CAN-10-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Degenhardt Y, Lampkin T. Targeting Polo-like Kinase in Cancer Therapy. Clinical Cancer Research. 2010;16:384–9. doi: 10.1158/1078-0432.CCR-09-1380. [DOI] [PubMed] [Google Scholar]
- 94.Yao YD, Sun TM, Huang SY, Dou S, Lin L, Chen JN, et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Science translational medicine. 2012;4:130ra48. doi: 10.1126/scitranslmed.3003601. [DOI] [PubMed] [Google Scholar]
- 95.Dou S, Yang XZ, Xiong MH, Sun CY, Yao YD, Zhu YH, et al. ScFv-decorated PEG-PLA-based nanoparticles for enhanced siRNA delivery to Her2(+) breast cancer. Adv Healthc Mater. 2014;3:1792–803. doi: 10.1002/adhm.201400037. [DOI] [PubMed] [Google Scholar]
- 96.Yu T, Greish K, McGill LD, Ray A, Ghandehari H. Influence of geometry, porosity, and surface characteristics of silica nanoparticles on acute toxicity: their vasculature effect and tolerance threshold. ACS Nano. 2012;6:2289–301. doi: 10.1021/nn2043803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen Y, Chen H, Shi J. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv Mater. 2013;25:3144–76. doi: 10.1002/adma.201205292. [DOI] [PubMed] [Google Scholar]
- 98.Shi Y, Miller ML, Di Pasqua AJ. Biocompatibility of Mesoporous Silica Nanoparticles? Comments on Inorganic Chemistry. 2016;36:61–80. [Google Scholar]
- 99.He Q, Zhang J, Shi J, Zhu Z, Zhang L, Bu W, et al. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials. 2010;31:1085–92. doi: 10.1016/j.biomaterials.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 100.Isaac O-J, Olivia T, Julia L, Victor FP. Engineered nonviral nanocarriers for intracellular gene delivery applications. Biomedical Materials. 2012;7:054106. doi: 10.1088/1748-6041/7/5/054106. [DOI] [PubMed] [Google Scholar]