

Abstract
Polycomb group proteins represent a global silencing system involved in development regulation. In specific, they regulate the transition from proliferation to differentiation, contributing to stem-cell maintenance and inhibiting an inappropriate activation of differentiation programs. Enhancer of Zeste Homolog 2 (EZH2) is the catalytic subunit of Polycomb repressive complex 2, which induces transcriptional inhibition through the tri-methylation of histone H3, an epigenetic change associated with gene silencing. EZH2 expression is high in precursor cells while its level decreases in differentiated cells. EZH2 is upregulated in various cancers with high levels associated with metastatic cancer and poor prognosis. Indeed, aberrant expression of EZH2 causes the inhibition of several tumor suppressors and differentiation genes, resulting in an uncontrolled proliferation and tumor formation. This editorial explores the role of Polycomb repressive complex 2 in cancer, focusing in particular on EZH2. The canonical function of EZH2 in gene silencing, the non-canonical activities as the methylation of other proteins and the role in gene transcriptional activation, were summarized. Moreover, mutations of EZH2, responsible for an increased methyltransferase activity in cancer, were recapitulated. Finally, various drugs able to inhibit EZH2 with different mechanism were described, specifically underscoring the effects in several cancers, in order to clarify the role of EZH2 and understand if EZH2 blockade could be a new strategy for developing specific therapies or a way to increase sensitivity of cancer cells to standard therapies.
Keywords: Enhancer of Zeste Homolog 2, Polycomb group proteins, Histone methyltransferase, Enhancer of Zeste Homolog 2 inhibitors, Anticancer drugs, Cancer therapy, Epigenetics
Core tip: Epigenetics modifications are key players in differentiation programs and are frequently altered in cancer. Since chromatin changes can be reversed with specific drugs, in the last years several studies explored the possibility to target epigenetics alteration as a new strategy for cancer treatment. This editorial focuses on Enhancer of Zeste Homolog 2 (EZH2), the catalytic subunit of Polycomb repressive complex 2 in cancer, analyzing different roles of this protein in various cancers. Several different classes of EZH2 inhibitors are also highlighted, giving distinct thoughtfulness to small molecules that are now under consideration as potential candidates for cancer treatment alone or in combination with other drugs.
INTRODUCTION
The chromatin structure in eukaryotes depends on covalent modifications that distinguish transcriptionally active and silent regions of the genome and affects the genetic material functionality. Histone post-translational modifications play a key role in the regulation of chromatin structure and gene expression. For instance, acetylation of histone H3 tail, on several lysines, is associated with gene expression, whereas the trimethylation of histone H3 on Lys27 (H3K27me3) or trimethylation of histone H3 on Lys9 is a mark of silent chromatin[1,2].
Several enzymes dynamically deposit or remove specific marks on the chromatin regulating gene expression and drive numerous biological processes. For instance, the differentiation of distinctive cell types in an organism is rigorously related to the establishment, while the maintenance of the correct epigenetic status and alteration of chromatin structure is frequently associated with different disease disorders, including cancer[1-3].
Numerous cancer types are associated with specific patterns of histones H3 and H4 modification and several epigenetic patterns enable to distinguish disease subtypes[4,5].
Epigenetic changes are reversible and specific drugs are capable to recover the correct chromatin status of a normal cell and, in turn, promote differentiation, cellular senescence or apoptosis. Therefore, the inhibition of epigenetic enzyme could be a good strategy for cancer treatment[6].
This editorial focuses on the role of the Enhancer of Zeste Homolog 2 (EZH2), the catalytic subunit of Polycomb repressive complex 2, which catalyzes the addition of methyl groups to lysine 27 of the N-tail of histone H3. This enzyme is responsible for the silencing of various genes involved in several processes as cell cycle progression, apoptosis and differentiation and it is frequently deregulated in cancer[7].
POLYCOMB GROUP PROTEINS
Polycomb group proteins (PcG) are a family of proteins highly conserved among eukaryotes, involved in development, stem cell biology and cancer[8-13]. They are regulators of the epigenetic gene silencing required in many processes, like mammalian X-chromosome inactivation and imprinting[14,15]. Moreover, the PcG-dependent epigenetic silencing controls the timely expression of genes involved in stem cell fate and lineage commitment[9-11,16], ensuring the establishment and maintenance of the correct transcriptome during development[17].
In mammals, PcG proteins form two main complexes: Polycomb-repressive complex 1 (PRC1) and 2 (PRC2)[18-22]. PRC1 is formed by BMI1, RING1A/B, CBX, and PHC subunits[23]. As of now, the mechanism of PRC1 dependent gene silencing is not completely clear. RING1A/B is an ubiquitin E3 ligases which catalyze the monoubiquitylation of histone H2A at lysine 119 (H2AK119ub1), an histone post translational modification associated with gene silencing[13,24]. Nevertheless, transcriptional silencing has been detected also in absence of ubiquitylation[18]. Other studies in vitro demonstrated that PRC1 prevents the SWI/SNF-dependent chromatin remodeling, competing for the binding with target nucleosomes. Indeed PRC1 complex is able to bind three nucleosomes, resulting in the chromatin compaction[25]. PRC1 affects also the transcription while inhibiting a correct assembly of RNA Polymerase II preinitiation complex[26].
PRC2 is composed by EZH2 or EZH1, EED, SUZ12 and RbAp46. EZH2 is the most studied catalytic subunit of PRC2 and contains the SET domain responsible for the histone methyltransferase activity on lysine-27 of histone H3[19-22]. SUZ12 and EED stimulates H3K27 histone methyltransferas increasing more than 1000 fold the catalytic activity of EZH2 alone whereas RbAp46 is responsible for the histone binding[13]. EZH1 is a homologue of EZH2, which originates an alternative PRC2 complex, however, data about this protein are sometimes contrasting[27]. It has been demonstrated that EZH1, in embryonic stem (ES) cells, is able to tri-methylates H3K27, contributing to the silencing of a subset of developmental genes. Its activity partially complements EZH2 role in the maintenance of the ES cells pluripotency[28]. On the other hand, other findings showed that EZH1 and EZH2 are recruited at the same set of target genes but EZH1 is ubiquitously expressed, whereas EZH2 expression is associated with proliferating cells[29]. There is also evidence that EZH1, compared to EZH2, exhibits a weaker histone H3 methyltransferase activity, while its depletion does not affect the global H3K27me2/3 levels; despite this, it is able to efficiently compact the chromatin through a mechanism independent of the presence of the methyltransferase cofactor S-Adenosyl methionine (SAM)[29].
Remarkably, during muscle differentiation several evidences showed a role of EZH1 in transcriptional activation. Indeed, EZH1 occupies transcriptionally active genes marked with H3K4me3 and interacts with RNA Pol II, promoting transcriptional elongation[30]. It has also been shown that the interchange between PRC2-EZH2 and PRC2-EZH1 complexes controls the correct timing of transcriptional activation of muscle specific genes such as myogenin[31]. A similar mechanism has been discovered at the promoter of PSD-95 gene during the development of hippocampal neurons[32].
The most common PcG-dependent gene silencing mechanism is the cooperation between PRC1 and PRC2; in fact, the establishment of H3K27me3 by PRC2 complex induces the recruitment of PRC1 by binding the chromodomain of the PHC subunits[21,33]. Once recruited, PRC1 brings transcriptional repression of target genes through the mechanisms described above.
PRC2 is also able to cooperate with other epigenetic silencing enzymes, for instance, it acts upstream of DNA methyltransferases (DNMTs) in order to induce a more stable transcriptional silencing characterized by the methylation of di-nucleotides CG. Although the mechanism is not completely clarified, essential proofs supported that DNMTs recruitment depends on the presence of the active form of EZH2, suggesting a context-dependent crosstalk between EZH2 and DNMTs. However, much remains unknown about this interaction, particularly, it is not clear if DNMTs bind directly EZH2 or H3K27me3 or if other factors are involved[34-37].
PRC2 is also able to associate with histone deacetylases, reinforcing transcriptional repression of target genes[6,19-22,38-40].
Other than transcriptional repression, EZH2 has a role in the promotion of gene activation[41-43], this mechanism has been discovered in breast and prostate cancer and it is described below.
EZH2 AND CANCER
Epigenetic modifications have a key role in the normal mammalian development and are required in all somatic cells. In ES cells and in precursor, PRC2 contributes to silence the principal genes involved in the differentiation promotion, preventing the premature activation of the differentiation processes and maintaining their pluripotency[9-11]. In addition, the main targets of EZH2 are genes involved in cell cycle regulation, as for instance Ink4b/Arf/Ink4alocus; its inhibition impedes cell cycle arrest and contributes to preserve the proliferative potential[9,44-50].
Because of its importance in various aspects of cellular development and tissue differentiation, EZH2 expression is strictly regulated. For instance high levels are detected in stem cells and undifferentiated cell progenitors, while its expression decreases during the differentiation process[6,7,51]. EZH2 activity could be regulated, other than transcriptionally, also by different mechanisms as several miRNA and post translational modifications (reviewed in[7]). Furthermore, the recruitment of PRC2 complex at target promoters covers a very important role: PRC2 binds DNA with low affinity and recruiting factors are supposed to be necessary to drive the complex to target genes[52]. This hypothesis could also explain why EZH2 is recruited, in different tissue, at different set of genes.
Epigenetic abnormalities result in an inappropriate gene expression that drives to an altered cellular physiology in several diseases. The first evidences of the involvement of EZH2 in cancer were found in breast and prostate[39,53] but a number of human tumors are nowadays associated with EZH2 alteration[7]. Frequently, EZH2 expression is correlated with metastatic cancer cells and poor prognosis[6,7,51].
The role of EZH2 in cancer could be linked to its activity in self-renewal promotion and in the maintenance of undifferentiated state of cells.
EZH2 target genes are generally crucial regulators of the balance between cellular differentiation and cell cycle progression, and their deregulation is able to promote cancer progression[6]. For instance, EZH2-dependent silencing of Ink4b/Arf/Ink4alocus leads to the downregulation of p16, p15 and p14, resulting in uncontrolled proliferation and inhibition of apoptosis[54,55]. Furthermore, EZH2 inhibits other tumor suppressor genes such as p21, PTEN, DAB2IP, and Bim[56-60].
PRC2 complex inhibits also several miRNA involved in cell cycle regulation, for instance mir-31 in melanoma[61], miR-139-5p, miR-125b, miR-101, let-7c, and miR-200b in metastatic liver cancers, promoting cell motility and metastasis[62].
The other class of EZH2 target genes is composed by differentiation-related factors. Genome wide assays showed that factors as Gata, Sox, Fox, Pou, Pax, components of Wnt, TGF-β, Notch, FGF and retinoic acid pathways are silenced by EZH2. The activity of EZH2 inhibits differentiation and promotes carcinogenesis[8-12]. In embryonal rhabdomyosarcoma, for example, high levels of EZH2 inhibit the activation of muscle specific genes and its depletion promotes muscle specific genes transcription and a partial recovery of the muscle differentiation program[63].
Activity of EZH2 independent of H3K27me3
EZH2 activity is not restricted to H3K27 trimethylation, in fact several studies reported that it is also able to methylate other proteins[64-68].
EZH2 and other PRC2 subunits have been found in the cytoplasm, where they control actin polymerization and cell proliferation of T-lymphocytes and fibroblasts[64]. Aberrant EZH2 overexpression has been detected in both nuclei and cytoplasm of human prostate cancer cells. The cytoplasmic fraction, responsible for the reduction of the pool of insoluble F-actin, influences cell adhesion and migration, therefore contributes to invasiveness and metastatic ability of tumor cells[65].
Previous studies showed that EZH2 is able also to methylate other histones as the histone H1 at lysine 26 when associated with a different isoform of EED[66]. Recently it has been discovered that EZH2 is also able to methylate GATA4, inhibiting its transcriptional activity in heart[67]. This is the first evidence that PRC2 influences the function of transcription factors involved in the developmental processes not only modulating their expression levels but also regulating their post-translational modifications.
This evidence is also supported by another study showing that in breast cancer, EZH2, in association with other PRC2 components, plays an essential role in the regulation of p38 pathway. p38 mitogen-activated protein kinase signaling pathway is involved in the promotion of epithelial-to-mesenchymal transition, cell invasion and motility. EZH2 is able to bind the phosphorylated and activated p38 counterpart, increasing its downstream signaling. This study highlighted a novel fundamental role of EZH2 in breast cancer. EZH2 overexpression enhances the levels of phospho-p38 while EZH2 knockdown induces a mesenchymal-to-epithelial transition and decreases cell motility. Clinical breast cancer specimens reveal that EZH2 is overexpressed, and co-expressed with phospho-p38 in about two-third of cases, while EZH2 inhibition results in a reduction of spontaneous breast cancer metastasis in vivo[68].
Finally, EZH2 is also able to promote transcriptional activation interacting with different transcription factors[41,42,69]. In breast cancer, for instance, EZH2 interacts with ERα, Wnt signaling components TCF, and β-catenin at the promoter of target genes, enhancing transcriptional activation of c-Myc and cyclin D1 genes; this mechanism is independent of the methyltransferase activity[41]. Still in breast cancer, EZH2, independently from other PRC2 subunits, is also able to activate NF-κB signaling, interacting with its components Rel A and Rel B, and inducing the activation of genes implicated in oncogenesis such as IL6 and TNF[42]. Similarly, in castration-resistant prostate cancer the oncogenic functions of EZH2 are not dependent on its transcriptional silencing activity but on the transcriptional activation of a subset of genes. EZH2 does not bind these genes by recruiting other PRC2 components, but rather through the association with the androgen receptor (AR), which in turn, after EZH2 dependent methylation, leads to increase the transcriptional activation of these genes. It has been proposed that the methylation of AR is dependent on AKT that phosphorylates EZH2 at serine 21, promoting the binding with AR[43,70]. Interestingly, it has been shown that AKT-dependent phosphorylation decreases the affinity of EZH2 with histone H3, resulting in a reduction of the H3K27 methylation[71]; this event can promote the binding of EZH2 with AR and the role of the methyltrasferase as transcriptional activator.
Finally, EZH2 is also able to promote cyclin A transcription[72]. Cyclin A gene transcription is inhibited by pRb2/p130, a member of Rb family with an onco-suppressor role[73]. pRb2/p130 is able to recruits HDAC1 at cyclin A gene inducing gene silencing and G1 arrest[74]. EZH2 competes with HDAC1 for its binding with pRb2/p130, disrupting the occupancy of both proteins on cyclin A promoter and inducing gene activation and cell cycle progression[72,75].
Mutations
The activity of EZH2 in cancer is also influenced by mutations. In diffuse large B-cell lymphoma, an heterozygous mutation of EZH2 at Tyrosine 641, (Y641), which affects its catalytic domain, was initially associated with a loss of functions, but other studies showed that this mutation results in a limited capacity to carry out H3K27 monomethylation but augmented ability for di- and tri-methylation. In these tumors, wild type Tyrosine can be substituted with different amino acids (Phenylalanine Y641F, Histidine Y641H, Asparagine Y641N and Serine Y641S) and mutants cooperate with wild type protein to increase EZH2 activity[76-78]. Another mutation, called A677G, has been discovered in lymphoma cell lines and primary tumors. This mutation, that replaces Alanine with Glycine, as the mutation in Y641, increases the trymethylation of H3K27 but, on the contrary, displays similar affinity for all three substrates: Unmethylated, mono-edy-methylated H3K27[79]. A687V is another gain-of-function (GOF) mutation discovered in lymphoma, it substitutes Alanine 687 with Valine, and it is similar to other mutations since enhances EZH2 ability to perform dimethylations, whereas the ability of catalyzing trimethylations remains the same[80]. Parallel mutations have been discovered also in melanoma, where they contribute to the promotion of tumor growth[81,82].
EZH2 INHIBITORS
The peculiar role of PRC2 in the promotion of tumor growth, deregulation of apoptosis, and alteration of proper proliferation and differentiation programs, suggests that EZH2 can be a good target for therapy in cancer. Several inhibitors of EZH2 have been designed and they can be classified based on the different mechanism of inhibition (Figure 1 and Table 1).
Figure 1.
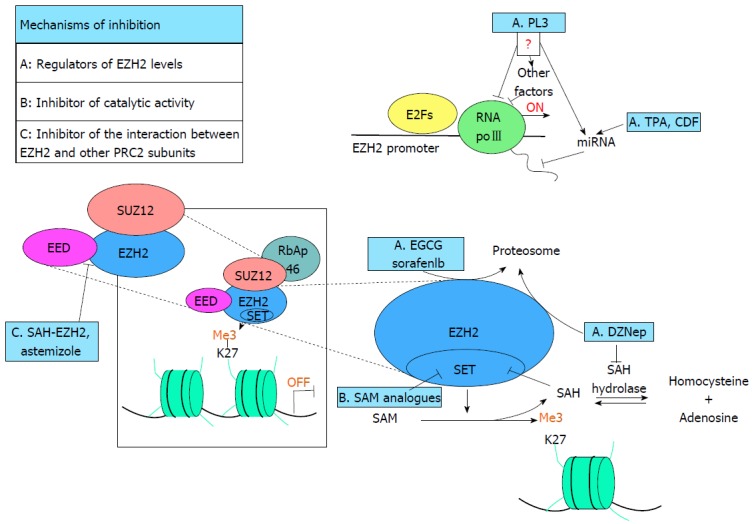
Schematic representation of polycomb-repressive complex 2 inhibition mechanisms. The image enclosed in the square represent the PRC2 active complex, able to methylate the lysine 27 of the histone H3 (H3K27me3), suppressing the transcription of target genes. Dashed lines pointed a part of the complex or the methylation pathway. The main PRC2 inhibitors (blue square) are included in the figure and labeled with the letters A, B or C depending on the mechanism of action (drugs marked with A are responsible for EZH2 depletion, with B are inhibitors of the catalytic activity of EZH2 and with C are able to destroy PRC2 complex. In group A PL3 is able to repress EZH2 expression but is not clear if the mechanism is direct or involves other factors or miRNAs. TPA and CDF reduce levels of EZH2 inducing the expression of some miRNA. EGCG, Sorafenib and DZNep promote proteasome-dependent EZH2 degradation. On the other hand DZNep, inhibiting SAH-Hydrolase, leads to an accumulation of SAH that, in turn, hampers try-methylation of H3K27me3 inhibiting EZH2 SET domain. Group B, SAM analogues compete with SAM for the binding with the substrate pocket blocking SET domain activity. Finally in the group C both the peptide SAH-EZH2 and Astemizole destroy the binding between EZH2 and EED event required for the activity of PRC2 complex. SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine; PRC2: Polycomb-repressive complex 2; EZH2: Enhancer of Zeste Homolog 2; EGCG: (-)-epigallocatechin-3-gallate; DZNep: 3-Deazaneplanocin A; SAH-EZH2: Stabilized alpha-helix of EZH2.
Table 1.
Enhancer of Zeste Homolog 2 inhibitors classified for Enhancer of Zeste Homolog 2 inhibition mechanism
| Mechanism of inhibition | Drug | Level of development | Ref. |
| Regulators of EZH2 levels | DZNep | [83-85,87] | |
| EGCG | [86,87] | ||
| PL3 | [93] | ||
| CDF | [94] | ||
| TPA | [98] | ||
| Sorafenib | [99] | ||
| SAM analogues | EPZ005687 | [100] | |
| EI1 | [101] | ||
| GSK126 | [102,109] | ||
| GSK343 | [103] | ||
| GSK926 | [103] | ||
| EPZ-6438 | Phase 2 (ClinicalTrials.gov Identifier: NCT01897571) | [104,110] | |
| UNC199 | [105,109] | ||
| Tanshindiols | [106] | ||
| 5-Methoxyquinoline Derivatives | [107] | ||
| Tetramethylpiperidinyl Benzamides | [108] | ||
| Inhibition of the interaction between EZH2 with other PRC2 subunits | SAH-EZH2 Astemizole | [111] [112] | |
| Unknown | CPI-1205 | Phase1 (ClinicalTrials.gov Identifier: NCT02395601) | |
| GSK2816126 | Phase 1 (ClinicalTrials.gov Identifier: NCT02082977 |
PRC2: Polycomb-repressive complex 2; EZH2: Enhancer of Zeste Homolog 2; PL3: 16-Hydroxycleroda-3,13-dien-15,16-olide; CDF: Diflourinated-curcumin; SAM: S-Adenosyl methionine; DZNep: 3-Deazaneplanocin A; EGCG: (-)-epigallocatechin-3-gallate; TPA: 12-O-tetradecanoylphorbol-13-acetate; SAH-EZH2: Stabillized alpha-helix of EZH2.
Regulators of EZH2 levels
3-Deazaneplanocin A (DZNep) is a S-adenosyl-l-homocysteine (AdoHcy) hydrolase inhibitor able to deplete EZH2 and to reduce H3K27me3 at PRC2 target genes. The mechanism is not completely understood but it seems that an absent or reduced activity of AdoHcy hydrolase and the successive AdoHcy accumulation causes an inhibition of SAM-dependent methyltransferases. DZNep treatment reduces levels of EZH2 and other PRC2 components thought a proteosome-mediated degradation, while RNA transcription does not change[83]. DZNep treatment induces apoptosis in breast, colorectal, prostate cancer and hepatocellular carcinoma whereas apparently, it is able to discriminate between cancerous and non-cancerous cell lines; consequently, it does not induce cell death in breast and lung epithelial cells, primary lung fibroblast, and human skin fibroblast cells[83]. Other studies, focusing on several non-small cell lung cancer (NSCLC) cell lines, showed that DZNep induces p27 accumulation, cell cycle arrest and apoptosis, while immortalized bronchial epithelial and fibroblast cell lines are less sensitive to apoptosis[84]. Interestingly, studies performed in several gastric cancer cell lines and in primary human gastric cancer cells showed that the effects of DZNep are related to p53 status, and cells with p53 wild type are more sensible to the treatment. p53 genomic status could be a potential predictive marker of DZNep response in this specific cell type[85].
Effects on the EZH2 protein levels have been detected also during the treatment with (-)-epigallocatechin-3-gallate (EGCG), a green tea-derived bioactive polyphenol. In skin cancer cells, treatment with EGCG induces a global reduction of H3K27me3 and reduces the levels of two PcG proteins: Bmi-1 and EZH2. Reduced levels of these PcG proteins are associated with decreased expression of several cyclin dependent kinases (CDKs) and cyclins (CDK1, CDK2, CDK4, cyclin D1, cyclin E, cyclin A and cyclin B1) and increased levels of p21 and p27 that, in turn, induce cell cycle arrest. Apoptosis is also stimulated by the treatment, indeed levels of caspase 3, 8 and 9 and PARP cleaved and Bax are higher compared with not treated cells whereas level of Bcl-xL expression decreased. Bmi-1 over expression reverses these EGCG-dependent changes[86] indicating that effects are dependent on PcG proteins. Similarly to DZNep, EGCG reduces the levels of EZH2 and Bmi-1 through a mechanism proteasome-dependent but, in skin cancer cells, the combination of EGCG with DZNep is more effective than each single agent[87]; indicating that the two molecules can also cause responses in different pathways.
The functions of PRC2 are tissue-specific and its expression is strictly regulated during normal development. The main regulators of PRC2 expression are proteins of pRb/E2F pathway; it has been demonstrated that E2F factors are required for the expression of EZH2 and EED in mouse embryonic fibroblasts and ectopic expression of pRb and p16, that are involved in E2F target gene repression, induces transcriptional repression of PRC2 subunits; silencing of pRb, on the contrary, increases their transcription[88-90]. Also c-Myc binds EZH2 promoter and induces transcription through the acetylation of histones H3 and H4[91]. Furthermore, several miRNA are also involved in EZH2 regulation[92].
It has been discovered that a natural compound isolated from the bark of Polyalthia longifolia, 16-Hydroxycleroda-3,13-dien-15,16-olide (PL3), induces apoptosis in leukemia cells[93]. The effects of this compound, already known for its anti-inflammatory activity and for its potential cytotoxicity in breast cancer cells and hepatocellular carcinoma cells, seem dependent on the repression of EZH2 and Suz12 expression. PL3 treatment reactivates the tumor suppressor targeted by PRC2 while inducing apoptosis[93]. The concentration required for pro-apoptotic effects in cancer cells is high, therefore additional studies focused in designing novel PL3-derived compound are required to improve the pharmacological properties and decrease the side effects of the molecules. Alternatively, the compound can be used in combination with other drugs to increase the effects at lower concentration[93].
Another natural compound called diflourinated-curcumin (CDF), a synthetic derivative of curcumin characterized by natural antitumor activity, is able to decrease EZH2 expression in pancreatic cancer cells. This molecule exhibits a particular mechanism of action, which involves the upregulation of a set of microRNAs (miRNA) such as let-7a,b,c,d, miR-26a, miR-101, miR-146a, and miR-200b,c, typically downregulated in pancreatic cancer[94]. Some of these miRNA are, at the same time, target and regulator of EZH2[7]. Although it has been confirmed that the re-expression of miR-101 is able to significantly decrease the expression of EZH2[95,96], however the peculiar ability of CDF to inhibit pancreatic tumor growth remains controversial[94]. Numerous evidences underscored that miR-101 plays a key role in the regulation of EZH2 in a wide panel of cancer and that miR-101 is linked to EZH2 through a negative mutual feedback loop. Indeed re-expression of miR-101 downregulates EZH2 expression, while the inactivation of EZH2 leads to the upregulation of miR-101[94]. In hepatocellular carcinoma, EZH2 is a direct target of miR-101, the expression level of this latter is negatively correlated with the protein level of EZH2; in fact, miR-101 is frequently underexpressed. MiR-101 overexpression downregulates EZH2, repressing proliferation, invasion, colony formation and cell cycle progression in vitro, while suppresses tumorigenicity in vivo[97]. miR-101 is also able to increase sensitivity to doxorubicin or fluorouracil, improving the activity of chemotherapeutic drugs in liver cancer cells[97]. Remarkably, in hepatocellular carcinoma, treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA) promotes miR-101 expression reducing levels of EZH2, EED and H3K27me3. In liver cancer cells, TPA induces G0/G1 cell cycle arrest with a mechanism mediated by PKCα and ERK pathways[98].
Finally, Sorafenib, a multikinase inhibitor used for the treatment of advanced-stage hepatocellular carcinoma, reduces level of EZH2, accelerating the proteasome-mediated EZH2 degradation. Sorafenib induces cell growth arrest and apoptosis and its effects can be reversed by the overexpression of EZH2. The combination of Sorafenib with DZNep has synergic effects in cell growth arrest and apoptosis and these properties could be evaluated in the future as a new combination therapy for the treatment of advanced hepatocellular carcinoma[99].
SAM analogues
The development of indirect methods inhibiting EZH2 protein levels could be recognized as a promising strategy for therapy, although it is crucial to consider the potential involvement of other pathways that may lead to a decrease of the specificity and an increase of the side effects.
The specific targeting of EZH2 catalytic domain could prove to be a powerful tool in gene based therapy, which could solve the problems related to the indirect mechanisms. Specific molecules, competitors of S-adenosyl-methionine (SAM), have been developed and are able to inhibit methyltransferase activity of EZH2 competing for the binding with the active site of EZH2, without affecting its expression levels.
One of these compounds called EPZ005687 has been designed in 2012 and it specifically inhibits H3K27me3 in lymphoma cells. Specificity for EZH2 is greater than 500-fold compared to a panel of 15 other methyltransferases analyzed, and 50-fold compared to EZH1. Particularly, this inhibitor is able to induce specifically G1 arrest and apoptosis in lymphoma cell lines, carrying point mutations of Y641 and A677 residues within the catalytic domain of EZH2, but it establishes minimal effects on the cell lines containing wild-type EZH2[100].
In 2012, Novartis developed an EZH2 inhibitor, EI1, which showed a strong selectivity for EZH2 (more than 10000-fold compared to other methyltransferases and about 90-fold compared to EZH1). This inhibitor decreases the global level of H3K27me3 in both EZH2 wild type and mutant lymphoma cell lines; however, it triggers cell cycle arrest and apoptosis specifically in mutant cells[101].
GSK126, another EZH2 inhibitor discovered in 2012, is more than 1000-fold selective for EZH2 compared to 20 other methyltransferases and more than 150-fold compared to EZH1. This inhibitor has a strong effect in EZH2 mutated diffuse large B-cell lymphoma cell lines and also inhibits the growth of EZH2 mutant large B-cell lymphoma in xenografts models[102].
Other SAM-competitive inhibitors have been proposed in the last years and their activity has been tested in several tumors, for instance GSK343 and GSK926[103], EPZ-6438[104], UNC199[105], Tanshindiols[106], 5-Methoxyquinoline Derivatives[107], Tetramethylpiperidinyl Benzamides[108]. UNC199 is the first orally bio-available inhibitor characterized by having a strong activity in vitro. Although it is an analogue of GSK126, it is less selective for EZH1[105,109]. Remarkably, EPZ-6438 is recently going through clinical testing for Non-Hodgkin Lymphomas patients[110].
Inhibition of the interaction between EZH2 with other PRC2 subunits
Interactions with other components as EED or Suz12 are necessary for the canonical activity of EZH2[6]. Another strategy for EZH2 inhibition is to block the interactions between the methyltransferase and other PRC2 subunits.
A peptide called SAH-EZH2 has been designed starting from the alpha-helical domain of EZH2 (aa 40-68), a 27-mer-peptide domain responsible for the binding between EZH2 and EED. This peptide, short enough to cross the cellular membranes, is able to disrupt the EED/EZH2 complex and to inhibit H3K27me3 in a dose-dependent manner; moreover, it decreases levels of EZH2 possibly impairing its protein stability. MLL-AF9 leukemia cells treated with SAH-EZH2 underwent cell cycle arrest and monocyte/macrophage differentiation, while were not driven to apoptosis[111].
Peptides or peptidomimetic inhibitors are generally considered metabolically unstable and have poor bioavailability to be suitable as therapeutic drugs. Recently, it has been identified that Astemizole, a drug previously used in the treatment of seasonal allergic rhinitis, is an inhibitor of the EED/EZH2 protein-protein interaction. Astemizole competes with EZH2 for the binding with EED, destabilizing PRC2 complex and inducing cell cycle arrest in leukemia cells[112]. Interestingly, the combination treatment of SAH-EZH2 or astemizole and various SAM analogues produced a significant synergistic effect on lymphoma cells[111,112], indicating that the suppression of catalytic activity and the disruption of PRC2 complex could influence different pathways and a combinatorial drug therapy can be a more effective therapeutic strategy.
Combination of EZH2 inhibitor with other drugs
Chromatin alterations are considered excellent candidates to explain how different factors may increase the risk of cancer, for that reason they represent an important aspect of tumor biology and may constitute good targets for future epigenetic-based therapies. For instance, several histone deacetylase and DNA methyltransferase inhibitors are already under evaluation as potential anticancer drugs, and several clinical trials are underway[113,114]. Various studies are exploring the possibility to combine the effects of EZH2 inhibitors with other epigenetic drugs in order to develop new strategies for cancer therapy. The combination of 5-AZA-2’-deoxycytidine (5-AZA-CdR), a DNA methyltranserase inhibitor with DZNep to treat human and murine leukemia cells, revealed similar synergic effects accompanied by a meaningful reduction in clonogenicity. Additionally, microarray analysis showed that the combination therapy increases the expression of more than 150 genes, including CDKN1A and FBXO32[115]. Histone deacetylases are also frequently altered in cancer and can contribute to tumor progression. A synergic interaction was observed with a three-drug combination of trichostatin-A (TSA: A histone deacetylase inhibitor) plus DZNep and 5-AZA-CdR in acute myeloid leukemia, showing a significant activation of several tumor suppressor genes and inhibition of cell growth and cell survival[116].
EZH2 inhibitors can be also used in combination with classical chemotherapeutic drugs to solve problems related to side effects (combined treatment consent to reduce concentration of drugs) or resistance of cancer cells often associated to the treatments with the only chemotherapeutic agents. The discovery of heterozygous EZH2 GOF mutants, identified in non-Hodgkin Lymphomas, which proved to be more sensitive to EZH2 inhibitors, leaded to the hypothesis that combination of classical treatment with PRC2 inhibitors could improve the efficacy of the cancer therapy. Standard Non-Hodgkin Lymphoma therapy is a combination of several drugs, called CHOP (Cyclophosphamide, Hydroxyldaunorubicin, Oncovin, and Prednisone). Studies in vitro and in vivo demonstrated that the combination of EPZ-6438 with CHOP increases anti-proliferative benefits compared to treatment with the EZH2 inhibitor alone, and this effect is thought to be mediated by glucocorticoid receptor agonists such as Prednisolone, the active metabolite of Prednisone[110].
Moreover, other studies investigated the inhibition of EZH2 in combination with conventional therapies in prostate cancer. Etoposide, an inhibitor of topoisomerase IIα (Top2a), is used in combination with other drugs in the standard therapy for castrate resistant prostate cancer. Levels of Top2a are higher in aggressive cancer and in these patients EZH2 expression correlates positively with Top2a expression. It has been demonstrated that the combined inhibition of EZH2 and Top2a, increases anti-tumor response both in vitro and in vivo models of prostate cancer, indicating that combination therapy can be a new strategy for aggressive prostate cancer[117].
Finally, various studies explored the interplay between Myc and EZH2, both involved in cancer. In Myc-driven prostate cancer, EZH2 directly suppresses interferon-γ receptor 1 (IFNGR1) in a Myc-dependent manner. EZH2 depletion restores the expression of IFNGR1, increasing the sensitivity of these cells to interferon-γ with consequent activation of IFN-JAK-STAT1 tumor-suppressor signaling that leads to apoptosis. Activity of EZH2 in Myc-driven tumors is strictly correlated to Myc amplification. Myc knockdown reduces levels of EZH2 and H3K27me3 at IFNGR1 promoter only in Myc-amplified prostate cell lines; while in Myc-independent tumor growth, IFNGR1 inhibition seems to be related on DNA hypermethylation[118]. This evidence suggested that combination of EZH2 inhibition with interferon-γ could be a specific strategy for Myc-driven prostate tumors and can help to improve the outcome of patients.
Numerous evidences confirm that EZH2 activity is strongly correlated with Myc. Definitely, Myc induce the expression of EZH2, downregulating miR-26a/b[119]; inhibits AKT-dependent phosphorylation of EZH2 at Serine 21 increasing its H3K27me3 activity[120]. Remarkably, Myc recruits EZH2 to miR-26a promoter and cooperatively suppresses miR-26a expression with consequent EZH2 upregulation. EZH2 in turn, inhibits miR-494, a repressor of Myc. Consequently, Myc and EZH2 generate a positive feedback loop to assure persistent high protein levels of both proteins[121]. In several hematological malignancies, Myc expression is inhibited by a BET bromodomain inhibitor, JQ1[122,123]. In B-cell Lymphoma cells, the pharmacological inhibition of EZH2 with DZNep in combination with JQ1 has a synergic effect in the suppression of cell growth and clonogenicity with a mechanism mediated by miR26a re-expression. The combined inhibition of Myc and EZH2 expression levels could result in an effective therapeutic strategy to successively suppress tumor growth in aggressive B-cell Lymphoma[124].
Synthetic lethality
Synthetic lethality occurs when two mutations result in cell death when acting in combination, but when acting separately they do not have any effect on cell viability[125]. Based on the distinctive genomic features of each tumor, current cancer research focus on finding targets that are able to kill exclusively cancer cells. Synthetic lethality screenings such as Synthetic genetic array, synthetic lethality by microarray, and genetic interaction mapping, are high-throughput methods to identify tumor mutations, or altered pathways that can lead to the synthetic lethality. Consequently, the presence of one of these mutations in cancer cells, but not in normal tissues, can make them ready to be selectively killed by mimicking the effect of the second alteration with targeted therapy[126]. Inhibition of EZH2 is under evaluation as a strategy to induce synthetic lethality. Recent findings have underlined that inactivating mutations in the gene encoding the AT-rich interacting domain containing protein 1A (ARID1A), a SWI/SNF complex subunit, are frequently detected in a large selection of cancers[127,128]. The first evidence, capable of proving a synthetic lethality between EZH2 inhibition and ARID1A mutations, has been reported in ARID1A-deficient ovarian clear cell carcinomas (OCCCs), an aggressive human cancer that commonly develops resistance to treatments[129,130]. ARID1A mutated OCCCs treated with EZH2 inhibitor GSK126 exhibited significantly cell growth arrest and apoptosis, however ARID1A wild type cells are not sensitive to the treatment, even if the reduction of H3K27me3 is comparable[131]. The same data set demonstrated that gene PIK3IP1, an inhibitor of PI3K–AKT signaling, is a direct target of both ARID1A and EZH2. These results suggested a specific implication of the PI3K/AKT signaling in ARID1A-mutated cells. ARID1A-deficient tumors appeared to be addicted to EZH2 activity, and the pharmacological inhibition of EZH2 promoted the upregulation of PIK3IP1 and contributed to the synthetic lethality through the inhibition of the PI3K–AKT pathway. Remarkably, the EZH2 inhibitor GSK126 induced regression of ARID1A-mutated ovarian tumors also in vivo[131]. Similarly, in rhabdoid tumors pharmacological inhibition of EZH2 induced apoptosis in SMARD1 (another subunits of SWI/SNF) mutated cells[104].
CONCLUSION
In the last years many researchers focused their studies on the design of new EZH2 inhibitors and in trying to clarify if EZH2 modulation can be a specific therapeutic strategy for cancer treatment, alone or in combination with other standard drugs or other epigenetics inhibitors. Three EZH2 inhibitors are currently under observation in clinical trials: CPI-1205 in B-Cell Lymphomas (ClinicalTrials.gov Identifier: NCT02395601) E7438 (previously called EPZ-6438) in Advanced Solid Tumors or B Cell Lymphomas (ClinicalTrials.gov Identifier: NCT01897571) and GSK2816126 in Relapsed/Refractory Diffuse Large B Cell and Transformed Follicular Lymphomas (ClinicalTrials.gov Identifier: NCT02082977). These studies will help to clarify if EZH2 inhibition is a good strategy for cancer treatment. Indeed the importance of polycomb protein during development and differentiation processes can lead to side effect. Data obtained in vitro in breast cancer showed that EZH2 specifically induces cell death in cancer cells but not in normal cells[83]. Moreover, in vivo studies showed that, treatments with some EZH2 inhibitors alone or in combination with other chemotherapeutic agents are well tolerated by mice resulting in minimal toxicity or modest weight loss of 10%[105,117,118].
Nevertheless, the studies described above demonstrated that EZH2 activity can behave differently in distinguishing neoplasms, and a singular inhibitor can be effective in certain kind of cancer but not in others. For instance, it is well known that Top2a inhibitor Etoposide is actually effective in a minority of patients with non-small-cell lung cancer. Recent findings validated that in this particular type of cancer, EZH2 inhibition drives differential effects in response to the Top2a inhibitors in vitro and in vivo. BRG1 (LOF) and EGFR (GOF) mutant cancer cells in response to EZH2 inhibition increased Top2a inhibitor sensitivity, thus shrinking and fragmenting into apoptotic bodies. On the contrary, EGFR and BRG1 wild-type NSCLC cells responded to EZH2 inhibition with upregulation of BRG1 and ultimately become more resistant to the Top2a inhibitor[132]. Furthermore, in rhabdomyosarcomas, treatment with EZH2 inhibitors has different effects in embryonic or alveolar subtypes where induces respectively differentiation or apoptosis[133,134].
Another interesting example concerning the selective inhibition of EZH2 was displayed in prostate cancer. It has been shown that in Myc-driven prostate cancer, EZH2 exhibited a significantly Myc-dependent downregulation of IFNGR1. The pharmacological depletion of EZH2 by the inhibitor DZNep restored the expression of IFNGR1 and, when treated in combination of interferon-γ, as discussed previously, provided a remarkable synergic antitumor effect with interferon-γ. Conversely, EZH2 catalytic inhibitors, although they efficiently reduced H3K27me3, failed to mimic EZH2 depletion. Therefore, patients with advanced prostate cancer driven by Myc could have benefits from a therapeutic depletion of EZH2, indicating that the ability of EZH2 to increase the sensitivity of cancer cells to interferon-γ is independent by its catalytic activity[118].
An important point for consideration, in order to understand if a drug is suitable for therapy, concerns taking into account its penetration into the organs. A panel of five EZH2 inhibitors was recently tested to evaluate the affinity with P-glycoprotein (P-gp/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) two members of ABC transporter superfamily, both present in blood-brain barrier. All of them were able to bind the transporters, resulting in a limited brain penetration[135].
Another key point to be contemplated concerns the real possibility that cancer cells could develop new mutations, making themselves resistant to EZH2 inhibitors as it was highlighted recently for SAM analogues[136].
Moreover, the role of EZH1 in tumorigenesis is not completely understood and because functions change in different tissues, it will be necessary to evaluate, depending form the tumor, inhibitors that target both EZH1 and EZH2, or drugs that specifically target EZH2 maintaining EZH1 activity.
For all these reasons, further studies are needed to better clarify the different roles of EZH2 in cancer, in addition noteworthy efforts are required in developing new generation of EZH2 inhibitors in order to design specific therapeutic strategies that alone or in combination with classical therapy could provide novel potential clinical approaches aimed at eradicating a wide variety of human cancers.
Footnotes
Conflict-of-interest statement: Authors have not conflict of interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 30, 2015
First decision: September 17, 2015
Article in press: February 16, 2016
P- Reviewer: Pajares MA, Vlachostergios P, Wiemer E S- Editor: Kong JX L- Editor: A E- Editor: Li D
References
- 1.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Fiorentino FP, Marchesi I, Giordano A. On the role of retinoblastoma family proteins in the establishment and maintenance of the epigenetic landscape. J Cell Physiol. 2013;228:276–284. doi: 10.1002/jcp.24141. [DOI] [PubMed] [Google Scholar]
- 4.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 5.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 6.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Marchesi I, Bagella L. Role of Enhancer of Zeste Homolog 2 Polycomb Protein and Its Significance in Tumor Progression and Cell Differentiation. In: Radzioch D. Chromatin Remodeling. InTech, 2013: 119-152 [Google Scholar]
- 8.Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, Farnham PJ. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–1605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 11.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Squazzo SL, O’Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, Margueron R, Reinberg D, Green R, Farnham PJ. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006;16:890–900. doi: 10.1101/gr.5306606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 14.Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 15.Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet. 2004;36:1296–1300. doi: 10.1038/ng1467. [DOI] [PubMed] [Google Scholar]
- 16.Pietersen AM, van Lohuizen M. Stem cell regulation by polycomb repressors: postponing commitment. Curr Opin Cell Biol. 2008;20:201–207. doi: 10.1016/j.ceb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 18.Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, Kingston RE. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98:37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 19.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 20.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 21.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 23.Surface LE, Thornton SR, Boyer LA. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell. 2010;7:288–298. doi: 10.1016/j.stem.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 26.Lehmann L, Ferrari R, Vashisht AA, Wohlschlegel JA, Kurdistani SK, Carey M. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J Biol Chem. 2012;287:35784–35794. doi: 10.1074/jbc.M112.397430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchesi I, Giordano A, Bagella L. Roles of enhancer of zeste homolog 2: from skeletal muscle differentiation to rhabdomyosarcoma carcinogenesis. Cell Cycle. 2014;13:516–527. doi: 10.4161/cc.27921. [DOI] [PubMed] [Google Scholar]
- 28.Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32:503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mousavi K, Zare H, Wang AH, Sartorelli V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol Cell. 2012;45:255–262. doi: 10.1016/j.molcel.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 31.Stojic L, Jasencakova Z, Prezioso C, Stützer A, Bodega B, Pasini D, Klingberg R, Mozzetta C, Margueron R, Puri PL, et al. Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin. 2011;4:16. doi: 10.1186/1756-8935-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henriquez B, Bustos FJ, Aguilar R, Becerra A, Simon F, Montecino M, van Zundert B. Ezh1 and Ezh2 differentially regulate PSD-95 gene transcription in developing hippocampal neurons. Mol Cell Neurosci. 2013;57:130–143. doi: 10.1016/j.mcn.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 33.Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, van Lohuizen M. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat Genet. 2006;38:694–699. doi: 10.1038/ng1792. [DOI] [PubMed] [Google Scholar]
- 34.Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 35.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 36.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 37.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet. 1999;23:474–478. doi: 10.1038/70602. [DOI] [PubMed] [Google Scholar]
- 39.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 40.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 41.Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27:5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798–810. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gil J, Bernard D, Martínez D, Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol. 2004;6:67–72. doi: 10.1038/ncb1077. [DOI] [PubMed] [Google Scholar]
- 45.Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, Van Lohuizen M, Marino S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 46.Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E, Hulsman D, Leung C, Arsenijevic Y, Marino S, et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005;19:1438–1443. doi: 10.1101/gad.1299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 48.Dietrich N, Bracken AP, Trinh E, Schjerling CK, Koseki H, Rappsilber J, Helin K, Hansen KH. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 2007;26:1637–1648. doi: 10.1038/sj.emboj.7601632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23:975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23:906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knudsen ES, Dervishaj O, Kleer CG, Pajak T, Schwartz GF, Witkiewicz AK. EZH2 and ALDH1 expression in ductal carcinoma in situ: complex association with recurrence and progression to invasive breast cancer. Cell Cycle. 2013;12:2042–2050. doi: 10.4161/cc.25065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, Voigt P, Martin SR, Taylor WR, De Marco V, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richly H, Aloia L, Di Croce L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2011;2:e204. doi: 10.1038/cddis.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aldiri I, Vetter ML. PRC2 during vertebrate organogenesis: a complex in transition. Dev Biol. 2012;367:91–99. doi: 10.1016/j.ydbio.2012.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280:22437–22444. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- 57.Wu ZL, Zheng SS, Li ZM, Qiao YY, Aau MY, Yu Q. Polycomb protein EZH2 regulates E2F1-dependent apoptosis through epigenetically modulating Bim expression. Cell Death Differ. 2010;17:801–810. doi: 10.1038/cdd.2009.162. [DOI] [PubMed] [Google Scholar]
- 58.Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, Hornyak TJ. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res. 2011;9:418–429. doi: 10.1158/1541-7786.MCR-10-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen J, Li J, Han Q, Sun Z, Wang J, Wang S, Zhao RC. Enhancer of zeste homolog 2 is overexpressed and contributes to epigenetic inactivation of p21 and phosphatase and tensin homolog in B-cell acute lymphoblastic leukemia. Exp Biol Med (Maywood) 2012;237:1110–1116. doi: 10.1258/ebm.2012.012075. [DOI] [PubMed] [Google Scholar]
- 60.Smits M, van Rijn S, Hulleman E, Biesmans D, van Vuurden DG, Kool M, Haberler C, Aronica E, Vandertop WP, Noske DP, et al. EZH2-regulated DAB2IP is a medulloblastoma tumor suppressor and a positive marker for survival. Clin Cancer Res. 2012;18:4048–4058. doi: 10.1158/1078-0432.CCR-12-0399. [DOI] [PubMed] [Google Scholar]
- 61.Asangani IA, Harms PW, Dodson L, Pandhi M, Kunju LP, Maher CA, Fullen DR, Johnson TM, Giordano TJ, Palanisamy N, et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget. 2012;3:1011–1025. doi: 10.18632/oncotarget.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Au SL, Wong CC, Lee JM, Fan DN, Tsang FH, Ng IO, Wong CM. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology. 2012;56:622–631. doi: 10.1002/hep.25679. [DOI] [PubMed] [Google Scholar]
- 63.Marchesi I, Fiorentino FP, Rizzolio F, Giordano A, Bagella L. The ablation of EZH2 uncovers its crucial role in rhabdomyosarcoma formation. Cell Cycle. 2012;11:3828–3836. doi: 10.4161/cc.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, Viale A, Reinberg D, Wülfing C, Tarakhovsky A. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell. 2005;121:425–436. doi: 10.1016/j.cell.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 65.Bryant RJ, Winder SJ, Cross SS, Hamdy FC, Cunliffe VT. The Polycomb Group protein EZH2 regulates actin polymerization in human prostate cancer cells. Prostate. 2008;68:255–263. doi: 10.1002/pros.20705. [DOI] [PubMed] [Google Scholar]
- 66.Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14:183–193. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 67.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, Pu WT. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moore HM, Gonzalez ME, Toy KA, Cimino-Mathews A, Argani P, Kleer CG. EZH2 inhibition decreases p38 signaling and suppresses breast cancer motility and metastasis. Breast Cancer Res Treat. 2013;138:741–752. doi: 10.1007/s10549-013-2498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu C, Bian C, Yang W, Galka M, Ouyang H, Chen C, Qiu W, Liu H, Jones AE, MacKenzie F, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci USA. 2010;107:19266–19271. doi: 10.1073/pnas.1008937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cavalli G. Molecular biology. EZH2 goes solo. Science. 2012;338:1430–1431. doi: 10.1126/science.1232332. [DOI] [PubMed] [Google Scholar]
- 71.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 72.Tonini T, Bagella L, D’Andrilli G, Claudio PP, Giordano A. Ezh2 reduces the ability of HDAC1-dependent pRb2/p130 transcriptional repression of cyclin A. Oncogene. 2004;23:4930–4937. doi: 10.1038/sj.onc.1207608. [DOI] [PubMed] [Google Scholar]
- 73.Giordano A, Rossi A, Romano G, Bagella L. Tumor suppressor pRb2/p130 gene and its derived product Spa310 spacer domain as perspective candidates for cancer therapy. J Cell Physiol. 2007;213:403–406. doi: 10.1002/jcp.21225. [DOI] [PubMed] [Google Scholar]
- 74.Stiegler P, De Luca A, Bagella L, Giordano A. The COOH-terminal region of pRb2/p130 binds to histone deacetylase 1 (HDAC1), enhancing transcriptional repression of the E2F-dependent cyclin A promoter. Cancer Res. 1998;58:5049–5052. [PubMed] [Google Scholar]
- 75.Tonini T, D’Andrilli G, Fucito A, Gaspa L, Bagella L. Importance of Ezh2 polycomb protein in tumorigenesis process interfering with the pathway of growth suppressive key elements. J Cell Physiol. 2008;214:295–300. doi: 10.1002/jcp.21241. [DOI] [PubMed] [Google Scholar]
- 76.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA. 2010;107:20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proc Natl Acad Sci USA. 2012;109:2989–2994. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Majer CR, Jin L, Scott MP, Knutson SK, Kuntz KW, Keilhack H, Smith JJ, Moyer MP, Richon VM, Copeland RA, et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012;586:3448–3451. doi: 10.1016/j.febslet.2012.07.066. [DOI] [PubMed] [Google Scholar]
- 81.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barsotti AM, Ryskin M, Zhong W, Zhang WG, Giannakou A, Loreth C, Diesl V, Follettie M, Golas J, Lee M, et al. Epigenetic reprogramming by tumor-derived EZH2 gain-of-function mutations promotes aggressive 3D cell morphologies and enhances melanoma tumor growth. Oncotarget. 2015;6:2928–2938. doi: 10.18632/oncotarget.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kikuchi J, Takashina T, Kinoshita I, Kikuchi E, Shimizu Y, Sakakibara-Konishi J, Oizumi S, Marquez VE, Nishimura M, Dosaka-Akita H. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer. 2012;78:138–143. doi: 10.1016/j.lungcan.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng LL, Itahana Y, Lei ZD, Chia NY, Wu Y, Yu Y, Zhang SL, Thike AA, Pandey A, Rozen S, et al. TP53 genomic status regulates sensitivity of gastric cancer cells to the histone methylation inhibitor 3-deazaneplanocin A (DZNep) Clin Cancer Res. 2012;18:4201–4212. doi: 10.1158/1078-0432.CCR-12-0036. [DOI] [PubMed] [Google Scholar]
- 86.Balasubramanian S, Adhikary G, Eckert RL. The Bmi-1 polycomb protein antagonizes the (-)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis. 2010;31:496–503. doi: 10.1093/carcin/bgp314. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 87.Choudhury SR, Balasubramanian S, Chew YC, Han B, Marquez VE, Eckert RL. (-)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis. 2011;32:1525–1532. doi: 10.1093/carcin/bgr171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol. 2001;21:6820–6832. doi: 10.1128/MCB.21.20.6820-6832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Müller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15:267–285. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neri F, Zippo A, Krepelova A, Cherubini A, Rocchigiani M, Oliviero S. Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol Cell Biol. 2012;32:840–851. doi: 10.1128/MCB.06148-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benetatos L, Voulgaris E, Vartholomatos G, Hatzimichael E. Non-coding RNAs and EZH2 interactions in cancer: long and short tales from the transcriptome. Int J Cancer. 2013;133:267–274. doi: 10.1002/ijc.27859. [DOI] [PubMed] [Google Scholar]
- 93.Lin YH, Lee CC, Chang FR, Chang WH, Wu YC, Chang JG. 16-hydroxycleroda-3,13-dien-15,16-olide regulates the expression of histone-modifying enzymes PRC2 complex and induces apoptosis in CML K562 cells. Life Sci. 2011;89:886–895. doi: 10.1016/j.lfs.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 94.Bao B, Ali S, Banerjee S, Wang Z, Logna F, Azmi AS, Kong D, Ahmad A, Li Y, Padhye S, et al. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res. 2012;72:335–345. doi: 10.1158/0008-5472.CAN-11-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Banerjee R, Mani RS, Russo N, Scanlon CS, Tsodikov A, Jing X, Cao Q, Palanisamy N, Metwally T, Inglehart RC, et al. The tumor suppressor gene rap1GAP is silenced by miR-101-mediated EZH2 overexpression in invasive squamous cell carcinoma. Oncogene. 2011;30:4339–4349. doi: 10.1038/onc.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Friedman JM, Liang G, Liu CC, Wolff EM, Tsai YC, Ye W, Zhou X, Jones PA. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009;69:2623–2629. doi: 10.1158/0008-5472.CAN-08-3114. [DOI] [PubMed] [Google Scholar]
- 97.Xu L, Beckebaum S, Iacob S, Wu G, Kaiser GM, Radtke A, Liu C, Kabar I, Schmidt HH, Zhang X, et al. MicroRNA-101 inhibits human hepatocellular carcinoma progression through EZH2 downregulation and increased cytostatic drug sensitivity. J Hepatol. 2014;60:590–598. doi: 10.1016/j.jhep.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 98.Chiang CW, Huang Y, Leong KW, Chen LC, Chen HC, Chen SJ, Chou CK. PKCalpha mediated induction of miR-101 in human hepatoma HepG2 cells. J Biomed Sci. 2010;17:35. doi: 10.1186/1423-0127-17-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang S, Zhu Y, He H, Liu J, Xu L, Zhang H, Liu H, Liu W, Liu Y, Pan D, et al. Sorafenib suppresses growth and survival of hepatoma cells by accelerating degradation of enhancer of zeste homolog 2. Cancer Sci. 2013;104:750–759. doi: 10.1111/cas.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 101.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 103.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woo J, Kim HY, Byun BJ, Chae CH, Lee JY, Ryu SY, Park WK, Cho H, Choi G. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg Med Chem Lett. 2014;24:2486–2492. doi: 10.1016/j.bmcl.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 107.Xiang P, Jie H, Zhou Y, Yang B, Wang HJ, Hu J, Hu J, Yang SY, Zhao YL. 5-Methoxyquinoline Derivatives as a New Class of EZH2 Inhibitors. Molecules. 2015;20:7620–7636. doi: 10.3390/molecules20057620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nasveschuk CG, Gagnon A, Garapaty-Rao S, Balasubramanian S, Campbell R, Lee C, Zhao F, Bergeron L, Cummings R, Trojer P, et al. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med Chem Lett. 2014;5:378–383. doi: 10.1021/ml400494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, Liu Y, Li F, Vedadi M, Frye SV, Garcia BA, Zheng D, Jin J, Wang GG. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–357. doi: 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter Scott M, Smith JJ, Moyer MP, et al. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014;9:e111840. doi: 10.1371/journal.pone.0111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–650. doi: 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kong X, Chen L, Jiao L, Jiang X, Lian F, Lu J, Zhu K, Du D, Liu J, Ding H, et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J Med Chem. 2014;57:9512–9521. doi: 10.1021/jm501230c. [DOI] [PubMed] [Google Scholar]
- 113.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97:1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 114.Federico M, Bagella L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J Biomed Biotechnol. 2011;2011:475641. doi: 10.1155/2011/475641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Momparler RL, Idaghdour Y, Marquez VE, Momparler LF. Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk Res. 2012;36:1049–1054. doi: 10.1016/j.leukres.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 116.Momparler RL, Côté S, Momparler LF, Idaghdour Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2’-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenetics. 2014;6:19. doi: 10.1186/1868-7083-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kirk JS, Schaarschuch K, Dalimov Z, Lasorsa E, Ku S, Ramakrishnan S, Hu Q, Azabdaftari G, Wang J, Pili R, et al. Top2a identifies and provides epigenetic rationale for novel combination therapeutic strategies for aggressive prostate cancer. Oncotarget. 2015;6:3136–3146. doi: 10.18632/oncotarget.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014;8:204–216. doi: 10.1016/j.celrep.2014.05.045. [DOI] [PubMed] [Google Scholar]
- 119.Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Möller P, Stilgenbauer S, Pollack JR, Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 120.Kaur M, Cole MD. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res. 2013;73:695–705. doi: 10.1158/0008-5472.CAN-12-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22:506–523. doi: 10.1016/j.ccr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhao JC, Yu J, Runkle C, Wu L, Hu M, Wu D, Liu JS, Wang Q, Qin ZS, Yu J. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22:322–331. doi: 10.1101/gr.131508.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tucker CL, Fields S. Lethal combinations. Nat Genet. 2003;35:204–205. doi: 10.1038/ng1103-204. [DOI] [PubMed] [Google Scholar]
- 126.Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10:351–364. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schmoldt A, Benthe HF, Haberland G. Digitoxin metabolism by rat liver microsomes. Biochem Pharmacol. 1975;24:1639–1641. [PubMed] [Google Scholar]
- 129.Jones S, Wang TL, Shih IeM, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IeM, Conejo-Garcia JR, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520:239–242. doi: 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ciarapica R, De Salvo M, Carcarino E, Bracaglia G, Adesso L, Leoncini PP, Dall’Agnese A, Walters ZS, Verginelli F, De Sio L, et al. The Polycomb group (PcG) protein EZH2 supports the survival of PAX3-FOXO1 alveolar rhabdomyosarcoma by repressing FBXO32 (Atrogin1/MAFbx) Oncogene. 2014;33:4173–4184. doi: 10.1038/onc.2013.471. [DOI] [PubMed] [Google Scholar]
- 134.Ciarapica R, Carcarino E, Adesso L, De Salvo M, Bracaglia G, Leoncini PP, Dall’agnese A, Verginelli F, Milano GM, Boldrini R, et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer. 2014;14:139. doi: 10.1186/1471-2407-14-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang P, de Gooijer MC, Buil LC, Beijnen JH, Li G, van Tellingen O. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-Inhibitors. Int J Cancer. 2015;137:2007–2018. doi: 10.1002/ijc.29566. [DOI] [PubMed] [Google Scholar]
- 136.Gibaja V, Shen F, Harari J, Korn J, Ruddy D. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558–566. doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]