

Two familial gastrointestinal neoplasia syndromes are linked with increased incidence of brain tumors. Hereditary nonpolyposis colorectal carcinoma (HNPCC), or Lynch syndrome, is an autosomal dominant disorder caused by inactivating germline mutations in mismatch repair genes MLH1, MSH2, MSH6, or PMS2 (OMIM 609310, 120435, 614350, and 614337). Patients with Lynch syndrome have markedly increased incidence of carcinoma of the colon, stomach, endometrium, and biliary system. A subset of Lynch syndrome patients also develop malignant gliomas and are said to have Turcot syndrome type 1. Familial adenomatous polyposis type 1 (FAP1) is an autosomal dominant syndrome caused by inactivating germline mutation in the APC gene (OMIM 175100). Patients with FAP1 invariably develop numerous gastrointestinal adenomas and carcinoma. A subset of patients with FAP1 also develop medulloblastomas and are said to have Turcot syndrome type 2.1
Here we report two pediatric patients with high-grade midline gliomas found to have inactivating germline mutations in the MUTYH gene on chromosome 1p34, which encodes a DNA repair enzyme involved in base excision repair.2 Biallelic inactivating MUTYH germline mutations, either homozygous or compound heterozygous, cause markedly increased incidence of gastrointestinal adenomas and carcinomas termed familial adenomatous polyposis type 2 (FAP2, OMIM 608456), but this syndrome has not been previously linked with brain tumors.3–6 Our two patients, however, strongly suggest that monoallelic germline MUTYH mutations may increase risk of malignant brain tumors.
Patient #1 was a 6-year old boy who presented with lower extremity weakness. MR imaging revealed a heterogeneously enhancing intramedullary mass in the thoracic spinal cord (Fig. 1). Pathology from a subtotal resection demonstrated glioblastoma, WHO grade IV, with histone H3-K27M mutant protein expression. He received adjuvant radiation with concurrent temozolomide. However, 8 months after diagnosis he was found to have disseminated recurrence within the posterior fossa. Repeat irradiation was administered, followed with trials of temozolomide, lomustine, erlotinib, bevacizumab, and a PARP inhibitor. Unfortunately, he had progressive disease and died 18 months after diagnosis. Targeted next-generation sequencing of 510 cancer-associated genes was performed that identified a germline nonsense mutation in the MUTYH gene at codon 466, causing early truncation of the encoded protein. The tumor demonstrated retention of the remaining wild-type allele, but the possibility of epigenetic inactivation could not be excluded. Additionally, the tumor harbored somatic missense mutations in H3F3A, ACVR1, and PIK3CA, all known to be recurrently mutated in pediatric high-grade gliomas.7 Interestingly, activating ACVR1 mutations have been recurrently found in diffuse intrinsic pontine gliomas with histone H3 p.K27M mutations,7,8 but this is the first report of ACVR1 mutation in a pediatric glioma outside of the pons.
Fig. 1.
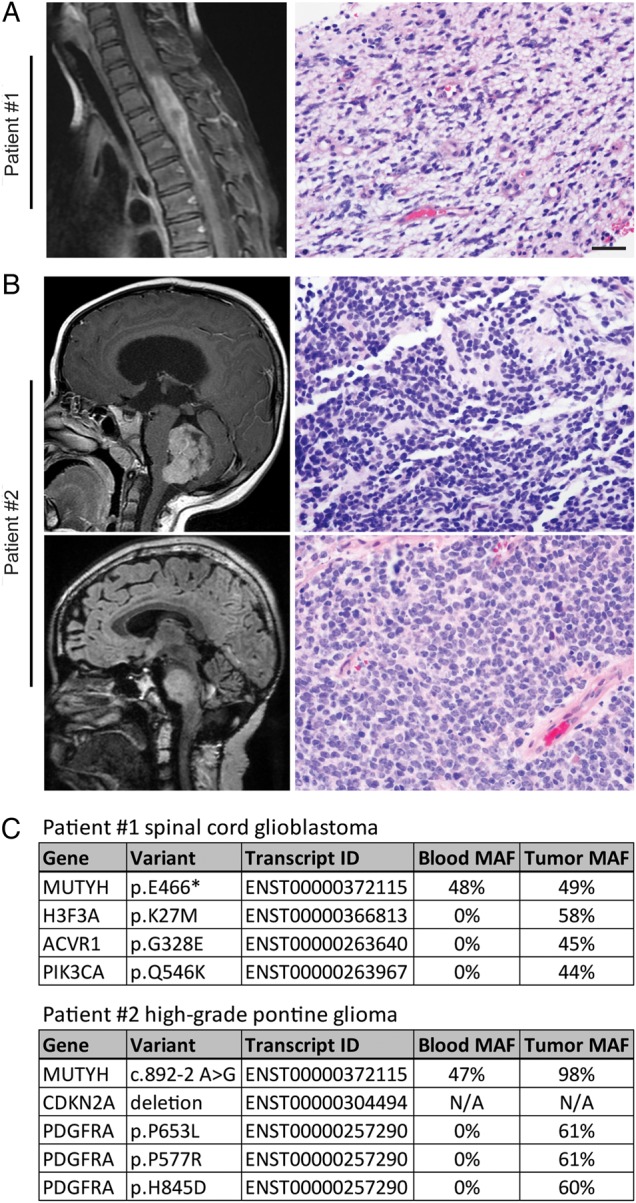
(A) T1 postcontrast MR imaging and hematoxylin and eosin (H&E) stained section from the resection of a glioblastoma in the thoracic spinal cord from patient #1 at 6 years of age. (B) T1 postcontrast MR imaging and H&E stained section from the resection of a medulloblastoma from patient #2 at 5 years of age (top). Fluid-attenuated inversion recovery MR imaging and H&E stained section from the biopsy of a high-grade pontine glioma from patient #2 at 15 years of age (bottom). (C) List of pathogenic germline and somatic mutations that were identified upon targeted next-generation sequencing of 510 cancer-associated genes on genomic DNA isolated from peripheral blood and tumor tissue. MAF, mutant allele frequency. Scale bar, 20 µm.
Patient #2 initially presented at 5 years of age with emesis, headaches, and seizure. He was found to have an enhancing mass in the inferior vermis of the cerebellum. Pathology from gross total resection demonstrated medulloblastoma, WHO grade IV. He underwent irradiation followed by chemotherapy with vincristine, cisplatin, and lomustine. The patient was stable for 10 years until he presented with worsening ataxia, dysarthria, and diplopia. He was found to have an expansile nonenhancing lesion in the pons. Biopsy demonstrated high-grade infiltrative astrocytoma, at least WHO grade III. Targeted next-generation sequencing was performed that identified a germline mutation in MUTYH at the canonical splice acceptor of exon 11, predicted to disrupt appropriate splicing of the encoded mRNA. The tumor demonstrated loss of heterozygosity due to deletion of chromosome 1p eliminating the remaining wild-type MUTYH allele. The tumor also contained homozygous deletion of CDKN2A and 3 missense mutations in PDGFRA likely to be activating. Sanger sequencing over the MUTYH mutation in the patient's prior medulloblastoma also revealed loss of heterozygosity in this tumor. The patient was treated with focal irradiation and nivolumab. Follow-up imaging 3 months later revealed improvement of the brainstem lesion but new onset of metastatic disease. Targeted therapy with dasatinib was attempted based on the genomic profiling but was discontinued due to anaphylactic shock. He is currently receiving lomustine and bevacizumab.
These patients both exhibited inactivating MUTYH germline mutations known to cause a gastrointestinal polyposis syndrome (FAP2) that has not been previously reported in brain tumor patients. Neither patient had family history of gastrointestinal neoplasia, as expected given the lack of biallelic mutation. Inactivating germline mutations in 2 unrelated patients with high-grade gliomas and one with prior medulloblastoma strongly suggest that germline MUTYH mutations increase risk of malignant brain tumors. Of note, a recent study identified germline MUTYH variants in 9 pediatric cancer patients with tumors including Ewing sarcoma, B-lineage acute lymphoblastic leukemia, medulloblastoma, and osteosarcoma, suggesting that MUTYH may function as a broad spectrum tumor suppressor.9
Funding
C.N.K. is supported by an NIH T32 training grant (CA128583). S.M. is supported by the NIH National Center for Advancing Translational Sciences through UCSF-CTSI (KL2TR000143). D.A.S. is supported by a Career Development Award from the UCSF Brain Tumor SPORE and NIH Director's Early Independence Award (DP5 OD021403).
References
- 1.Hamilton SR, Liu B, Parsons RE et al. . The molecular basis of Turcot's syndrome. N Engl J Med. 1995;332(13):839–847. [DOI] [PubMed] [Google Scholar]
- 2.McGoldrick JP, Yeh YC, Solomon M, Essigmann JM, Lu AL. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol Cell Biol. 1995;15(2):989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Tassan N, Chmiel NH, Maynard FJ et al. . Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nature Genet. 2002;30(2):227–232. [DOI] [PubMed] [Google Scholar]
- 4.Jones S, Emmerson P, Maynard J et al. . Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Hum Mol Genet. 2002;11(23):2961–2967. [DOI] [PubMed] [Google Scholar]
- 5.Sieber OM, Lipton L, Crabtree M et al. . Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348(9):791–799. [DOI] [PubMed] [Google Scholar]
- 6.Sampson JR, Dolwani S, Jones S et al. . Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362(9377):39–41. [DOI] [PubMed] [Google Scholar]
- 7.Wu G, Diaz AK, Paugh BS et al. . The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nature Genet. 2014;46(5):444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor KR, Mackay A, Truffaux N et al. . Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nature Genet. 2014;46(5):457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Walsh MF, Wu G et al. . Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]