

Abstract
Objective
Lesch–Nyhan disease (LND) is caused by congenital deficiency of the purine recycling enzyme, hypoxanthine-guanine phosphoribosyltransferase (HGprt). Affected patients have a peculiar neurobehavioral syndrome linked with reductions of dopamine in the basal ganglia. The purpose of the current studies was to determine the anatomical basis for the reduced dopamine in human brain specimens collected at autopsy.
Methods
Histopathological studies were conducted using autopsy tissue from 5 LND cases and 6 controls. Specific findings were replicated in brain tissue from an HGprt-deficient knockout mouse using immunoblots, and in a cell model of HGprt deficiency by flow-activated cell sorting (FACS).
Results
Extensive histological studies of the LND brains revealed no signs suggestive of a degenerative process or other consistent abnormalities in any brain region. However, neurons of the substantia nigra from the LND cases showed reduced melanization and reduced immunoreactivity for tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine synthesis. In the HGprt-deficient mouse model, immunohistochemical stains for TH revealed no obvious loss of midbrain dopamine neurons, but quantitative immunoblots revealed reduced TH expression in the striatum. Finally, 10 independent HGprt-deficient mouse MN9D neuroblastoma lines showed no signs of impaired viability, but FACS revealed significantly reduced TH immunoreactivity compared to the control parent line.
Interpretation
These results reveal an unusual phenomenon in which the neurochemical phenotype of dopaminergic neurons is not linked with a degenerative process. They suggest an important relationship between purine recycling pathways and the neurochemical integrity of the dopaminergic phenotype.
Lesch–Nyhan disease (LND) is an inherited disorder with a characteristic neurobehavioral phenotype that includes a movement disorder dominated by generalized dystonia, intellectual disability, and recurrent self-injurious behavior.1–4 The disorder is caused by mutations in the HPRT1 gene, leading to deficiency of the purine recycling enzyme, hypoxanthine-guanine phosphoribosyltransferase (HGprt).5,6
The mechanisms by which HGprt deficiency leads to the neurological and behavioral problems are not well understood. However, there is strong evidence that they arise from dysfunction of basal ganglia circuits, and particularly dopaminergic pathways.7,8 Neurochemical studies of LND brains collected at autopsy have revealed 60 to 80% loss of dopamine throughout the basal ganglia.9–11 Positron emission tomography studies have demonstrated similar reductions of dopamine transporters and dopamine uptake.12,13 These studies have led to suggestions that dopamine neurons or their axonal projections are damaged.9,13 However, several histopathological studies of autopsied brains have not revealed any consistent loss of neurons in the substantia nigra.1,11,14 The reason for profound loss of dopamine-related measures with apparently preserved nigral dopamine neurons has never been established.
Dysfunction of dopaminergic pathways also is observed in animal and cell models of HGprt deficiency.15 The HGprt knockout (HGprt−) mouse model has a 30 to 60% loss of striatal dopamine and associated biochemical markers such as homovanillic acid, dihydroxyphenylacetic acid, tyrosine hydroxylase (TH), aromatic amino acid decarboxylase, and dopamine transporters.16–18
However, quantitative stereological studies of these mutant mice have revealed no loss of midbrain dopamine neurons or their axonal projections.19 Several HGprt-deficient cell models also have shown loss of dopaminergic markers with no apparent loss of viability.20–25 In these cell models, mRNA expression profiling has revealed broad disruption of the neurotransmitter phenotype. These findings from cell and animal models have led to suggestions that HGprt deficiency disrupts early developmental programs that lead to the expression of the dopaminergic neurochemical phenotype. This hypothesis was explored in the current studies by examining the integrity of midbrain dopamine neurons in the brains of 5 LND brains collected at autopsy. Key findings were confirmed in the HGprt− mouse model19 and the MN9D cell model21 of HGprt deficiency.
Materials and Methods
Human Brain Tissue
Formalin-fixed brains were collected at autopsy from 5 males with LND and 6 male controls spanning the same age range (Table 1). The diagnosis was confirmed in each LND case by the occurrence of the classical clinical phenotype together with either biochemical evidence of reduced HGprt enzyme activity or molecular evidence for a pathological mutation in the HPRT1 gene. Tissue blocks were collected from multiple regions of the cerebral cortex, hippocampus, amygdala, entorhinal cortex, basal ganglia, hypothalamus, and thalamus including subthalamic nucleus, midbrain, brainstem, and cerebellum. Tissue was embedded in paraffin and cut via microtome at 8μm. A complete neuropathological survey was conducted with hematoxylin/eosin stains to assess tissue quality and identify any overt defects. Immunostains for TH and ubiquitin were performed on sections from the basal ganglia (focusing on the putamen), midbrain (focusing on the substantia nigra), and brainstem (focusing on the locus coeruleus).
Table 1. LND Cases and Controls Used in the Autopsy Study.
| Case | Age at Death, yr | PMI, h | Additional Diagnoses | Cause of Death | Findings |
|---|---|---|---|---|---|
| LND1 | 5 | 8 | Renal insufficiency | Respiratory arrest | Mild gliosis and pigmentary incontinence in SN |
| LND2 | 11 | 50 | Pneumonia, nephrolithiasis | Cardiorespiratory arrest | Ubiquitin + cytoplasmic staining and inclusions in SN |
| LND3 | 21 | <2 | None | Accidental asphyxiation | Mild gliosis and pigmentary incontinence in SN |
| LND4 | 31 | 24 | Candida sepsis, asthma | Respiratory failure | Candida infection; subacute contusion in cortex; diffuse superficial spongiosis |
| LND5 | 34 | <4 | Chronic respiratory distress | Seizure with aspiration | Vascular proliferation |
| CON1 | 5 | 17 | None | Cardiac arrest | No significant pathology |
| CON2 | 12 | 18 | Clotting disorder | Stroke | No significant pathology |
| CON3 | 40 | 31 | Pancreatitis | Peritonitis and sepsis | No significant pathology |
| CON4 | 45 | 28 | Metastatic colon cancer | Metastatic colon cancer | No significant pathology |
| CON5 | 70 | 24 | Cardiovascular disease | Myocardial infarction | No significant pathology |
| CON6 | 92 | <12 | Parkinson disease | N/A | No significant pathology |
Data are given for 5 Lesch–Nyhan disease (LND) cases (LND1–LND5) and for 6 controls (CON1–CON6).
N/A = not available; PMI = postmortem interval; SN = substantia nigra.
Immunohistochemistry was performed following deparaffinization and rehydration of the sections. The sections were first preheated at 60°C for 5 to 30 minutes, followed by rinses in Histo-Clear (National Diagnostics, Atlanta, GA) or xylenes 3× for 5 to 10 minutes each. They then were immersed in acetone for 20 to 30 seconds followed by 1 wash in 100% ethanol and 2 washes in 95% ethanol for 20 to 30 seconds each. The slides were microwaved twice for 2.5 minutes in citrate buffer (pH 6.0) and allowed to cool to room temperature for 30 minutes. The sections were next pretreated in 3% H2O2 in methanol for 5 minutes at 40°C followed by 3 rinses in 0.075% Brij 35 solution (Sigma-Aldrich, St Louis, MO) and equilibrated for 10 minutes in TS Brij buffer consisting of 100mM Tris-Cl (pH 7.5), 100mM NaCl, 5mM MgCl2, and Brij 35. The sections then were incubated for 15 minutes at 40°C in 2% normal serum in TS Brij buffer (pH 7.5).
Immunostaining was performed using the T1299 mouse monoclonal anti-TH primary antibody (Sigma, St Louis, MO) at a dilution of 1:100 with the Vectastain alkaline phosphatase ABC kit (Vector Laboratories, Burlingame, CA). Negative controls, consisting of sections incubated without primary or secondary antibodies, were included in each experiment. The primary anti-TH antibody was diluted in 15mM NaCl, 5mM MgCl2, and 100mM Tris-HCl at pH 7.5, applied at 4°C overnight. The biotinylated secondary antibody was diluted 1:200 in blocking solution. The tissues were rinsed 3× in TS Brij buffer, incubated with the secondary antibody for 30 minutes at 37°C, and then rinsed again 3× with TS Brij buffer. The sections were next incubated in the Vector avidin-biotin complex mix for 1 hour at 37°C, and the stain was developed by incubating for 5 minutes at room temperature with the Vector Red alkaline phosphatase substrate (Vector Laboratories). Sections were rinsed and counterstained with aqueous hematoxylin (Genetex, Irvine, CA).
Sections were viewed using an Olympus (Tokyo, Japan) BX51 microscope. Photomicrographs were taken using Olympus CellSens v1.5 in TIFF format. The images were imported into Illustrator v15.0 (Adobe Systems, San Jose, CA), cropped to focus on the elements of interest, and annotated with text or arrows. No adjustments were made in the TIFF images regarding brightness, contrast, or color balance.
Knockout Mouse Model
The animals used in these studies were HGprt-competent (HGprt+) and congenic HGprt− mutant mice bred on a C57BL/6J background for >20 generations (C57BL/6JHPRTBM3).19 Normal males were mated with heterozygous females to generate HGprt− males and HGprt+ littermate controls. All animals were housed on a 14:10-hour light:dark cycle with food and water ad libitum. Mutants were distinguished from normal mice by molecular confirmation of the mutation by polymerase chain reaction applied to a DNA sample isolated from a tail clip. All procedures conformed to guidelines recommended by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Emory University Animal Care and Use Committee.
For TH immunostains, animals were deeply anesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde in 0.1M phosphate buffer and stored in fixative overnight. Tissue then was placed in 30% sucrose in 0.1M phosphate buffer for at least 1 week. Brains were sectioned serially on a microtome into 6 series in the coronal plane at a thickness of 30μm and stored at −20°C in a cryoprotectant solution consisting of 30% glycerol, 30% ethylene glycol, and 0.1M phosphate buffer. One series of brain sections of each animal was immunostained for TH. Free-floating sections were washed in 50mM Tris with 150mM NaCl at pH 7.4, then treated in 0.4% Triton X-100 (TX-100) in the same buffer for 30 minutes at room temperature. Sections were then rinsed with the buffer, and blocked in 5% normal horse serum and 0.1% TX-100 in buffer for 1 hour. Next, sections were incubated in mouse monoclonal antibody to TH (1:100, T1299, Sigma-Aldrich) containing 1% normal horse serum, 0.1% TX-100, and 0.01% sodium azide for 3 days at 4°C. After rinsing 3× in 0.1% TX-100, sections were incubated in biotinylated horse anti-mouse secondary antibody (1:500, BA-2000, Vector Laboratories) with 1% normal horse serum, 0.1% TX-100, and 0.01% sodium azide overnight at 4°C. Sections were rinsed in buffer once again, and the stain was developed using the Vectastain Alkaline Phosphatase ABC kit and the Vector Red alkaline phosphatase substrate kit at pH 7.9. Stained sections were mounted onto glass Superfrost slides, dehydrated, and coverslipped.
For quantitative immunoblots, striatum and midbrain tissues were dissected from 8 HGprt− mice and 8 age- and sex-matched littermate controls. The experiment was repeated independently with an additional 4 mutants and 4 controls, for a total of 12 HGprt− mutants and 12 controls. Samples were sonified (Sonifier 450; Branson, Danbury, CT; duty cycle = 30%; output control level = 3) in 20mM Tris-HCl (pH 7.4) containing protease inhibitor (Complete Mini, EDTA-free; Roche, Mannheim, Germany) and centrifuged at 12,000rpm for 20 minutes at 4°C. Protein concentrations were determined in the supernatants according to the BCA method (Thermo Fisher Scientific, Logan, UT). Equivalent amounts of total protein (5μg) were loaded and separated on 12% mini-protean TGX precast gels (Bio-Rad, Hercules, CA) and electroblotted to nitrocellulose membranes (GE Healthcare Biosciences, Pittsburgh, PA). Membranes were blocked at room temperature in 5% nonfat dry milk in phosphate-buffered saline (PBS) containing 0.05% Tween-20 (PBST). Membranes were then incubated overnight at 4°C with 1:1,000 of rabbit anti-TH polyclonal antibody (Pel-Freez, Rogers, AR), 1:500 of rabbit anti-actin polyclonal antibody (Abcam, Cambridge, MA), and 1:1,000 of rabbit anti-HPRT polyclonal antibody (Aviva Systems Biology, San Diego, CA) in PBST containing 5% dry milk. After washing 3× in PBST, the membranes were incubated with 1:1,000 of anti-rabbit immunoglobulin G horseradish peroxidase–linked secondary antibody (Cell Signaling, Danvers, MA) in PBST containing 5% dry milk for 1 hour at room temperature, followed by 3 additional washing steps in PBST. Signals were visualized using the Immun-Star chemiluminescent kit (Bio-Rad) and scanned with an Image Reader LAS-3000 (Fujifilm, Valhalla, NY). Quantification of signal intensities was performed with Multi Gauge v3.1 software (Fujifilm). Statistical analysis was performed using Sigmaplot (Systat Software, San Jose, CA).
MN9D Cell Culture Model
Immunostains for TH were applied to 10 previously developed mouse MN9D neuroblastoma cell lines with complete HGprt deficiency.21 The original MN9D line was developed through somatic fusion of primary midbrain dopaminergic neurons from an embryonic day 14 mouse with the mouse neuroblastoma line N18TG2.26 The parent MN9D line and all 10 HGprt− mutant lines were immunostained simultaneously for TH, and fluorescence intensities were quantified by flow-activated cell sorting (FACS). A sample of ∼106 cells was washed 3× with cold PBS at pH 7.4 and fixed with 1% paraformaldehyde for 20 minutes at room temperature. Cells were washed once with PBS and incubated with 0.5% saponin in PBS for 10 minutes. Cells were incubated with P4010 anti-TH primary antibody (Pel-Freez) diluted at 1:100 for 30 minutes at 4°C. Cells then were washed 3× with PBS and next incubated with goat antirabbit Alexa Fluor 488 secondary antibody (Invitrogen, Carlsbad, CA) at a dilution of 1:100 for 30 minutes at 4°C. Finally, the cells were washed and examined by FACS in a FACSort analyzer (Becton Dickinson, Franklin Lakes, NJ) with CellQuest software (BD Biosciences, Franklin Lakes, NJ).
Results from FACS-based measurements of fluorescence intensities of TH immunostains were not normally distributed, so pairwise comparisons of the HGprt+ parent cell line with each of the HGprt− mutant cell lines were performed via the Wilcoxon 2-sample test and SAS 9.3 software (SAS Institute, Cary, NC).
Results
Hematoxylin and Eosin Surveys of Autopsy Material
LND brains collected from patients spanning the ages from 5 to 34 years and controls in the range from 5 to 92 years old were investigated (see Table 1). In keeping with prior reports,1,14 there were no obvious abnormalities in the cerebral cortex, hippocampus, amygdala, entorhinal cortex, basal ganglia, hypothalamus, thalamus, subthalamic nucleus, or brainstem. None of the LND cases showed evidence for a neurodegenerative process, and no Lewy bodies were found in any brain region. The patterned loss of cerebellar neurons described in a prior report14 was not apparent in our cases.
The most consistent abnormality in the LND brains was a reduction of pigmentation in the midbrain, evident at both the gross and microscopic levels (Fig 1). Four of 5 LND brains had clearly reduced melanin, whereas 1 was judged to have normal levels (LND3). The reduced pigmentation in the LND brains appeared to reflect a combination of processes. First, nigral pigmentation due to melanin deposition is an age-dependent process.27–30 The first neuromelanin granules typically appear around the third year of life, and they increase with age. The lower pigmentation thus could reflect the relatively young ages of some of the LND cases. Despite the age-dependence of melanin deposition, most LND cases had less pigmentation than age-matched controls. Another process that contributed to reduced pigmentation was that 3 LND cases were judged to have mild to moderate reduction of nigral neurons (LND1, LND2, and LND4). This reduction was not age-dependent, as it was observed in the youngest case (LND1, age 5 years) yet not the oldest case (LND5, age 34 years). Three LND cases also appeared to have relatively small nigral neurons (LND1, LND4, and LND5). Nigral neurons were normal in number and size for LND3. These results suggest that reduced melanization might reflect a combination of processes including young age, a reduction in nigral neurons, and/or a reduction in nigral neuron sizes. However, subjective impressions of mild neuronal loss or changes in neuronal size could not be confirmed quantitatively because of the small numbers of brains available.
Figure 1.
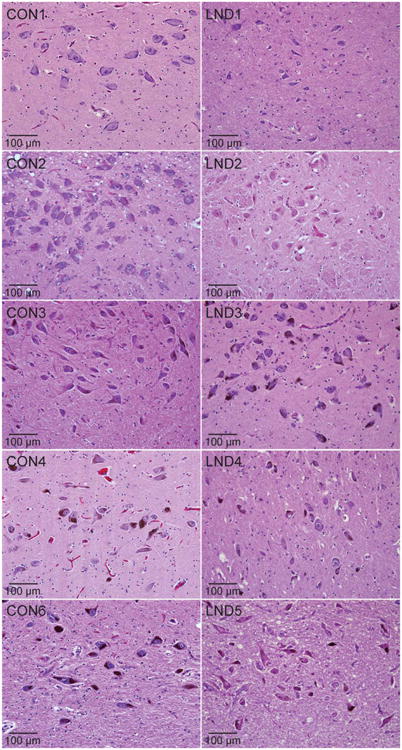
Histopathology in the substantia nigra. Representative hematoxylin and eosin stains of midbrain sections from control (CON1–CON4, CON6) and LND (LND1–LND5) brains are shown. In at least 3 independently stained sections at different levels of the midbrain, there was an impression of reduced neuromelanin pigmentation in 4 cases (LND1, LND2, LND4, LND5), mild or moderate cell loss in 3 cases (LND1, LND4, LND5), and somewhat small soma sizes in 4 cases (LND1, LND2, LND4, LND5).
In addition to the relatively consistent reductions in midbrain melanization, individual cases showed a few incidental abnormalities (see Table 1). In LND1 and LND3, mild gliosis and pigmentary incontinence were noted in the substantia nigra. These changes were not seen in the other cases. In LND2, ubiquitin immunohistochemistry revealed a faint cytoplasmic label with round or linear inclusions in several neurons in the substantia nigra. These changes were not identified in other regions of LND2 or in any of the other LND cases. In LND4, there was moderate spongiform change and scattered microglial nodules in many regions, with evidence for Candida invasion, consistent with the history of Candida sepsis and respiratory failure. LND4 also showed an area of subacute or old contusion with hemorrhage in the inferior frontal cortex. LND5 showed extensive vascular proliferation throughout the brain, in agreement with the history of chronic respiratory distress before death. In summary, the most consistent abnormality was a reduction in melanization, although some cases showed additional incidental findings.
TH Immunostains of Autopsy Material
Because melanin deposition is a byproduct of dopamine metabolism, the lower levels of melanin might also reflect chronically low levels of dopamine synthesis, consistent with prior biochemical studies showing reduced dopamine and related metabolites in the LND brain.9–11 To address this possibility, midbrain sections were immunostained for TH, the rate-limiting enzyme in the synthesis of dopamine. Controls consistently showed robust immunoreactivity for TH in melanized nigral neurons, including the youngest case (5 years old) and a 92-year-old with Parkinson disease (Fig 2). However, there was a consistent reduction in staining intensity in all LND cases. Because each LND case was stained simultaneously with at least 1 age-matched control, it is unlikely that these differences in staining intensity reflected day-to-day variation in staining efficiency. Careful inspection of the LND cases always revealed a few midbrain neurons that stained normally for TH, interspersed with a larger population of neurons that stained poorly. These few well-stained cells imply that relatively normal TH staining is achievable in the LND tissue and not related to poor tissue quality from inadequate fixation, overfixation, or poor storage conditions. TH immunostaining of fibers in the putamen also was weaker in the LND cases compared to controls (not shown), consistent with findings in the substantia nigra. However, it was not possible to determine whether this loss of staining was due to loss of staining intensity in individual axons, or actual loss of axons.
Figure 2.
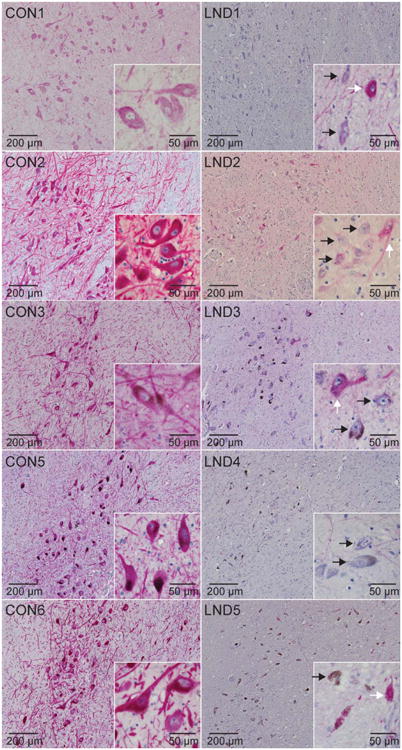
Tyrosine hydroxylase (TH) immunostains of the substantia nigra. Representative TH immunostains of midbrain sections from control (CON1–CON3, CON5, CON6) and LND (LND1–LND5) brains are shown. In at least 3 independently stained sections at different levels of the midbrain, there was an impression that all LND cases showed markedly reduced stain intensity in most pigmented neurons, mixed with a few scattered neurons that stained normally. Melanized neurons with low TH stain intensity (black arrows) can be seen adjacent to neurons with normal staining in the LND cases (white arrows).
The possibility of reduced TH immunoreactivity due to issues related to tissue quality was further considered by examining brainstem sections containing locus coeruleus neurons from each case (Fig 3). Hematoxylin and eosin immunostains revealed normal numbers of locus coeruleus neurons in all LND cases. All LND cases also had normal staining intensity for TH in the locus coeruleus, although the youngest case had reduced melanin pigmentation (LND1). These results demonstrate regional selectivity of TH deficits, and they argue against the concept that low TH immunoreactivity in the midbrain was due to poor tissue quality.
Figure 3.
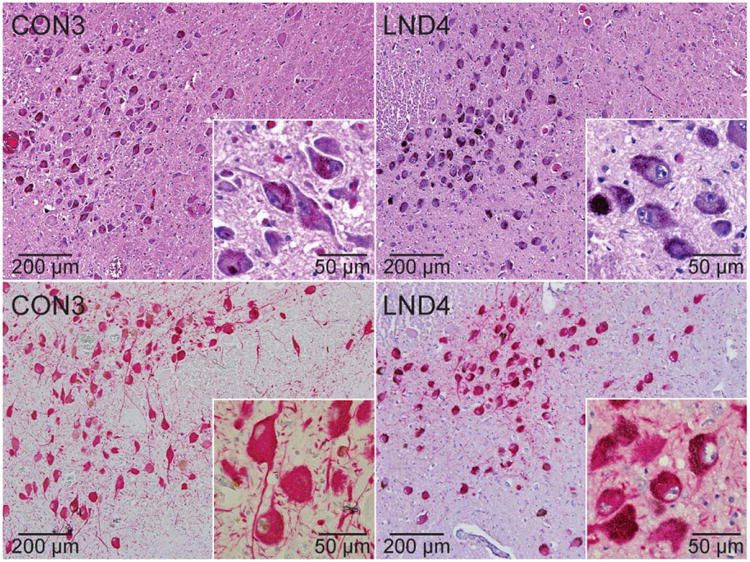
Brainstem locus coeruleus. Representative stains of brainstem locus coeruleus sections from a control (CON3) and LND patient (LND4) are shown. In at least 3 independently stained sections, the LND brains showed normal numbers and sizes of neurons in hematoxylin and eosin (upper panels) or tyrosine hydroxylase (TH)-immunostained sections (lower panels), normal melanin pigmentation, and normal TH immunostain intensity.
TH Immunoreactivity in HGprt− Knockout Mice
Histopathological studies of human autopsy material are valuable for revealing abnormalities in the human brain, but rigorous quantitative studies of formalin-fixed human brains are rarely feasible. To determine whether the reductions of TH might be replicated in a more rigorously controlled experimental model, we turned to the HGprt− knockout mouse model of LND. Prior biochemical studies of these mice have revealed 30 to 60% loss of dopamine and its metabolites.16–18,31 Conversely, quantitative stereological studies of the same mouse revealed no structural abnormalities of TH-immunopositive cells or axonal projections in the basal ganglia, even at the electron microscopic level.19 Because enzyme-amplified immunohistochemical methods are not ideally suited for detecting quantitatively small reductions in the target antigen, an explanation for this discrepancy is that intact dopaminergic neurons have lower levels of TH, analogous to findings in the LND brain described above. To address this possibility, we quantified TH expression.
In keeping with prior studies,19 there were no obvious reductions in the numbers of TH-immunopositive midbrain neurons in the HGprt− mice (Fig 4). Also consistent with prior studies, these neurons had a normal size and morphology. Immunoblots of tissue from the midbrain and striatum (Fig 5) revealed bands with the expected molecular weights for TH (60kDa) and actin (42kDa). A band for HGprt at 25kDa was seen only for the normal control mice, as expected. Results for TH/actin ratios were examined via 2-way analysis of variance with HGprt status and brain region as explanatory variables. This analysis revealed significant differences between the normal and HGprt− mice for the striatum (F = 27.6, p < 0.001) but not the midbrain (F = 2.4, p > 0.10). These results show reduced TH expression in the HGprt− mouse model, although the reductions are not as robust as those observed in the human LND brain and can be measued only in the striatum.
Figure 4.
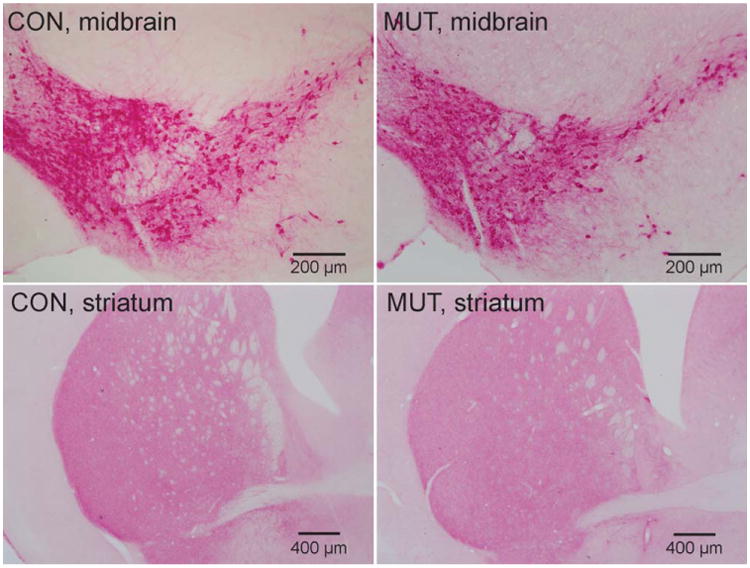
Tyrosine hydroxylase immunohistochemical stains of the midbrain and striatum in control mice (CON; left column) and hypoxanthine-guanine phosphoribosyltransferase–negative (HGprt−) mice (MUT; right column). Representative sections from the midbrain are shown in the top panels, whereas sections from the striatum are shown in the bottom panels. Consistent results were obtained on tissue from 4 control animals and 4 HGprt− animals.
Figure 5.
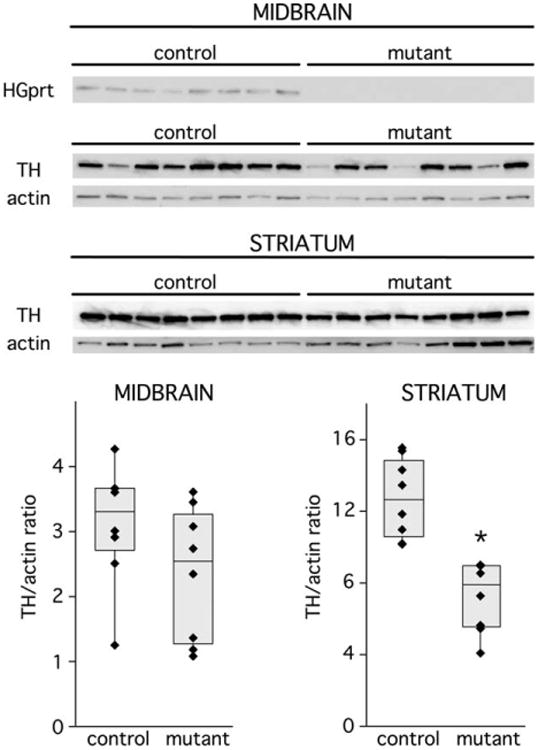
Quantitative immunoblots for tyrosine hydroxylase (TH) in midbrain and striatum. The relative abundance of TH and actin as a loading standard are shown for immunoblots of midbrain and striatum. The left 8 lanes show results for 8 independent hypoxanthine-guanine phosphoribosyltransferase–positive (HGprt+) control mice, and the remaining 8 lanes show results for independent HGprt− mice. TH band intensities were normalized to actin in midbrain (bottom left) and striatum (bottom right); each value for each animal is presented as a separate symbol, with superimposed box-whisker plots to show median (center line), confidence intervals (top and bottom bar), and entire data range (whiskers). Results were examined by 2-way analysis of variance with brain region and HGprt status as explanatory variables. This analysis revealed significant differences between the normal and HGprt− mice for the striatum (*p < 0.001) but not the midbrain (p > 0.10). This experiment was repeated with 4 additional mutants and controls on an independent occasion for a total of 12 different control and HGprt− mice, with results similar to those shown here.
TH Immunoreactivity of MN9D Culture Model
We next turned to a previously developed MN9D tissue culture model of HGprt deficiency, where prior studies again have shown significant loss of dopamine without signs of impaired viability.21 Here we immunostained the cells for TH and conducted FACS analyses to obtain rigorously quantifiable results at the single-cell level. Quantitative comparisons of fluorescence intensities calculated from >20,000 gated cells per line revealed significant variation between and within each cell line. However, each of the HGprt− MN9D lines had lower median TH intensity values than the parent HGprt+ line (Table 2). Pairwise statistical comparisons with the Wilcoxon 2-sample test confirmed each of the 10 HGprt− lines to be significantly lower than the parent line (p < 0.001).
Table 2. TH Stain Intensity for Parent and 10 HGprt− MN9D Sublines.
| Cell Line | HGprt Status | Cells Counted | Median Fluorescence |
|---|---|---|---|
| MN9D parent | Normal | 24,700 | 36.5 |
| MN9DA2 | Deficient | 26,900 | 6.9a |
| MN9DA5 | Deficient | 26,800 | 7.8a |
| MN9DA6 | Deficient | 24,200 | 23.1a |
| MN9DA8 | Deficient | 23,800 | 16.8a |
| MN9DA10 | Deficient | 24,400 | 8.1a |
| MN9DT1 | Deficient | 26,500 | 8.6a |
| MN9DT2 | Deficient | 26,300 | 35.5a |
| MN9DT3 | Deficient | 23,900 | 9.8a |
| MN9DT4 | Deficient | 27,200 | 7.4a |
| MN9DT5 | Deficient | 28,300 | 11.0a |
Median tyrosine hydroxylase (TH) fluorescence intensity values were calculated using FlowJo software (Tree Star, Ashland, OR). The parent and each mutant (A2, A5, A6, A8, A10, T1, T2, T3, T4, and T5) were compared using the Wilcoxon 2-sample test. p < 0.001. HGprt = hypoxanthine-guanine phosphoribosyltransferase.
All 10 HGprt− MN9D lines had higher TH immunoreactivity than negative controls without primary antibody, indicating that TH was low but not absent (Fig 6). These results confirm significant reductions of TH expression in the cell model. They imply that the effect of HGprt is a cell-intrinsic phenomenon that does not depend on interactions among other cell populations in an intact brain.
Figure 6.
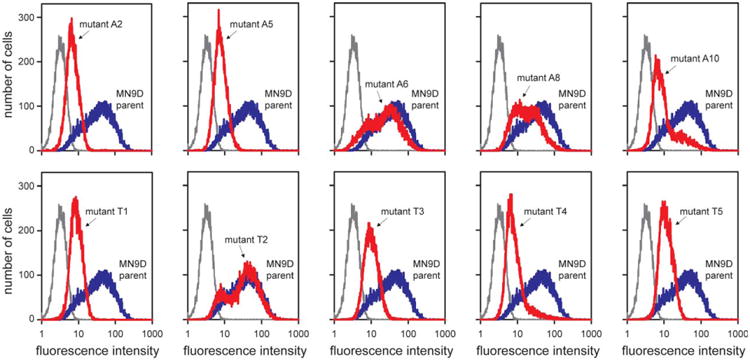
Tyrosine hydroxylase immunostains of the MN9D cell model of hypoxanthine-guanine phosphoribosyltransferase (HGprt) deficiency. Fluorescence intensity histograms from flow-activated cell sorting analyses are shown as separate panels for each of the HGprt− MN9D subclones (red traces) along with the HGprt+ MN9D parent cell line as control (blue traces) and a negative control without primary antibody (gray traces). Summary statistics for each mutant line (A2, A5, A6, A8, A10, T1, T2, T3, T4, and T5) are provided in Table 2. The results show representative traces, with the entire experiment being repeated at least 3 independent times.
To determine whether the reduction of TH expression might be reversible by restoration of HGprt activity in the HGprt− MN9D lines, each of the 10 mutants was transfected with a cDNA encoding HGprt and revertants isolated by growing in hypoxanthine-aminopterinthymidine tissue culture medium as previously described.32 The dopamine phenotype was not restored to levels observed in the parental control MN9D line. These results imply one of two possibilities. The first is that loss of HGprt activity results in a permanent reduction in TH and dopamine expression in the MN9D cell model due to irreversible developmental processes. The second is that isolation of HGprt− MN9D subclones selectively favors survival of those with low TH expression.
Discussion
The present study is the largest autopsy evaluation of LND to date. In keeping with prior case reports and smaller series, no obvious abnormalities were identified in most brain regions. However, midbrain substantia nigra neurons showed reduced pigmentation and significantly reduced staining for TH, the rate-limiting enzyme for dopamine synthesis. The reduced staining did not appear to reflect poor tissue preservation in the LND brains, because sporadic substantia nigra neurons and most locus coeruleus neurons stained normally for TH. The reduction of TH in these neurons is consistent with the reduction of pigmentation, because neuromelanin is a byproduct of dopamine metabolism. The reduction of TH observed in the human brains is consistent with results from the HGprt− knockout mouse model, where there are no reductions in the numbers of THimmunopositive midbrain neurons or striatal fibers, yet there are reductions in TH staining on quantitative immunoblots. The reductions of TH in the human autopsy material also are consistent with a similar phenomenon in the HGprt− MN9D cell model, where there is significantly reduced TH, without reduced cell viability. Finally, the reductions in TH are consistent with reductions in dopamine levels that have been documented for the human LND brain,9–11 the HGprt− knockout mouse,16–18 and the MN9D cell model.21
Several prior studies have suggested that abnormal function of dopamine neurons plays an important role in the clinical phenotype of LND by affecting specific sub-circuits of the basal ganglia.7,33 For example, the major motor problem is dystonia, a movement disorder that has been linked with early developmental dysfunction of dopamine pathways and the motor circuit of the basal ganglia involving the putamen and motor cortex.34,35 The behavioral phenotype, in particular self-injurious behavior, has been linked with early brain dopamine loss in the limbic circuit involving the ventral striatum and orbitofrontal cortex.36–38 The pattern of cognitive deficits shows prominent problems with attention and executive functions, defects that have been proposed to result from dysfunction of pathways involving the caudate and dorsolateral prefrontal cortex.3,39 The clinical problems are not likely to be related to a simple deficiency of dopamine in these pathways, because administration of the dopamine precursor L-dopa or direct-acting dopamine agonists is not therapeutic.40 One potential explanation is that dopamine neurons fail to mature properly, a possibility that is consistent with several gene expression studies of HGprt− cell models showing disruption of molecular pathways for dopamine neuron development.20,25,41–43 If this is the case, the loss of dopamine or TH reflect an aspect of a broader developmental defect in these neurons, and restoration of dopamine alone may be insufficient.
Although most attention has focused on basal ganglia dopamine neurons in LND, recent studies have shown that downstream targets of these neurons in the basal ganglia and associated cortical regions also are affected.39 Because dopamine itself plays a significant role in neuronal development, independent from its role as a neurotransmitter,44 abnormalities in the basal ganglia and cortex may be secondary to dopamine loss. However, it is also possible that the basal ganglia and cortex are affected directly by the loss of HGprt. Although we did not detect any obvious neuropathological abnormalities in these regions of our LND brains, subtle changes in neuronal numbers or morphologies in these or other regions could have been overlooked.
Limitations of the Current Studies
Although studies of autopsy material are important for revealing abnormalities occurring directly in the human brain, they also have certain limitations. The first limitation involves the relatively small number of cases studied, which is related to the rarity of LND. In this regard, it is important to emphasize that our study is the largest ever reported. Altogether, there are only 25 prior neuropathological studies including a total of 33 brains of LND reported in the literature, with no consistent abnormalities.1,14 Signs of a degenerative process are notably absent from these reports. Our cases did not show the multifocal atrophy of the cerebellar granular layer described in a prior study.14 Only 1 prior study reported a detailed examination of midbrain dopamine neurons for 2 LND cases,11 and the small number of cases and variability in staining made it difficult to identify any consistent defects. Despite the small number of cases, our findings were robust and relatively consistent across our cases.
The second limitation is that tissue quality in autopsy studies is difficult to control because of variable antemortem and postmortem conditions. A lengthy or critical illness can affect tissue quality, and lengthy postmortem intervals or prolonged fixation can affect staining intensity. However, the tissue evaluated for the current studies was of suitable quality for identifying most common types of pathology. The findings in the LND brain are not likely to be related to poor tissue quality or poor staining for several reasons. First, melanization is a relatively stable phenomenon that is not sensitive to tissue fixation and is relatively insensitive to postmortem interval. Second, reduced TH staining in the midbrain was not likely to reflect poor tissue preservation or overfixation, because TH staining in the locus coeruleus of the same brains was normal. Finally, the key finding of reduced TH could be replicated in the HGprt− knockout mouse model and a tissue culture model, where technical issues related to tissue quality can be excluded.
More specifically, we confirmed key findings from the human LND brains using experimental models where experimental conditions could be precisely controlled and rigorous quantitative methods could be applied. Studies of the HGprt− knockout mouse model of LND, with both TH immunostains and quantitative immunoblots, imply a reduction in TH expression, although the differences are not as great as those observed in the human LND brain or the cellular models. The results provide an explanation of prior conflicting studies of the mouse model, which has significant loss of striatal dopamine, with no obvious reduction in the numbers of midbrain dopamine neurons.17,19 Studies of the MN9D model of HGprt deficiency also confirmed the human autopsy finding of significantly reduced TH immunoreactivity.
Of course, surrogate animal or tissue culture models of disease have their own limitations. The most important limitation involves uncertainty regarding whether results from the mouse brain or rodent cell lines examined in vitro can be extrapolated to human brain. Another limitation is the lack of melanin deposition in the mouse brain and cell models, making it impossible to verify the finding of reduced pigmentation in the human LND brain. Finally, we did not evaluate our animal or cell models for subtle signs of neuronal injury that might disclose poor neuronal health without overt cell death. Despite some differences and limitations of the MN9D and knockout mouse models, they provide converging evidence from different experimental approaches that the loss of HGprt has a significant influence on expression of TH in substantia nigra neurons, with no obvious degenerative process.
Cell Death versus Loss of Neurotransmitter Phenotype
The relative preservation of substantia nigra neurons with apparent loss of neurochemical phenotype observed in our LND cases is unusual, but it is not unprecedented. A prior study described a similar phenomenon involving melanin-positive but TH-negative substantia nigra neurons in Parkinson disease and related Parkinsonian syndromes.45 Such neurons were found with a frequency of 17% in Parkinson diseases and an even higher frequency of 64% in progressive supranuclear palsy. The authors attributed this phenomenon to an agonal state, in which unhealthy and dying nigral neurons were unable to maintain expression of TH. This explanation does not seem applicable to LND, where there is no evidence for a neurodegenerative process. An alternative explanation is that the mechanisms controlling the expression of the neurochemical phenotype of a dopamine neuron are separable from those controlling viability. Another study of Parkinson disease showed that a substantial proportion of melanin-positive substantia nigra neurons did not stain normally for Nurr1, a transcription factor known to control the neurochemical phenotype of dopamine neurons.46 Some lines of Nurr1-deficient mice also show preservation of substantia nigra neurons that have lost most of the usual biochemical markers of the dopamine neuron phenotype.47,48 These results from Parkinson disease and related models are consistent with the concept that molecular controls of the neurochemical phenotype may be separable from cell death. That is, a dopamine neuron may not die, but may lose its ability to communicate with its usual neurotransmitter. Either mechanism would result in failure of dopaminergic signaling.
Reductions of TH and nigral pigmentation also have been reported for dopa-responsive dystonia, which is most often due to defects in the gene encoding guanosine triphosphate cyclohydrolase.49–51 This enzyme is involved in the synthesis of the biopterin cofactor that is essential for the function of TH during dopamine production. In this disorder, the loss of TH may reflect coregulation of biopterin and TH, or a reduced half-life of TH due to lack of substrate stabilization.52 These mechanisms cannot explain the reductions of TH in LND, where there is no evidence for loss of biopterin.31
Our results, combined with similar observations from other disorders, imply that otherwise viable dopaminergic neurons may lose their neurochemical functionality. Understanding the mechanisms responsible for dissociating the neurotransmitter phenotype from cell viability has important implications for therapy, because restoring the neurochemical phenotype may be easier than replenishing lost neurons or preventing neuronal death. The relationship between loss of HGprt-mediated purine recycling and loss of dopamine or TH suggests that these mechanisms involve purine pathways.
Acknowledgments
This work was supported by the Lesch–Nyhan Syndrome Children's Research Foundation (HAJ); grants from the NIH National Institute of Neurological Disorders and Stroke (NINDS; R01 NS40470; HAJ), the NIH National Institute of Child Health and Human Development (NICHD; R01 HD053312; HAJ), and the Office of Rare Diseases Research at the National Center for Advancing Translational Studies of the NIH (U54 NS065701; HAJ); the Emory Alzheimer's Disease Research Center (P50 AG025688; JDG, MG); an Emory University NINDS Neuroscience Core Facilities grant (P30 NS055077); and the Netherlands Organization for Scientific Research (NWO/ZonMw, VENI 916.12.167; JEV).
Tissue from controls was obtained from the Johns Hopkins Brain Resource Center, the Emory Alzheimer's Disease Research Center, the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland (contract HHSN275200900011C, #N01-HD-9–0011), and Children's Healthcare of Atlanta Pediatric Hospital.
We thank R. Vigorito and R. Zielke for their expert assistance in obtaining specimens from the NICHD Brain and Tissue Bank for Developmental Disorders.
Footnotes
Potential Conflicts of Interest: H.A.J.: travel expenses, Dystonia Medical Research Foundation, Bachmann-Strauss Dystonia and Parkinson's Foundation, American Academy of Neurology, Movement Disorders Society; consultancy, Psyadon Pharmaceuticals; grants/grants pending, NIH, Lesch–Nyhan Syndrome Children's Research Foundation; speaking fees, Georgia Neurological Society.
References
- 1.Jinnah HA, Visser JE, Harris JC, et al. Delineation of the motor disorder of Lesch-Nyhan disease. Brain. 2006;129:1201–1217. doi: 10.1093/brain/awl056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schretlen DS, Ward J, Meyer SM, et al. Behavioral aspects of Lesch-Nyhan disease and its variants. Dev Med Child Neurol. 2005;47:673–677. doi: 10.1017/S0012162205001374. [DOI] [PubMed] [Google Scholar]
- 3.Schretlen DS, Harris JC, Park KS, et al. Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc. 2001;7:805–812. doi: 10.1017/s135561770177703x. [DOI] [PubMed] [Google Scholar]
- 4.Jinnah HA, Friedmann T. Lesch-Nyhan disease and its variants. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th. New York, NY: McGraw-Hill; 2001. pp. 2537–2570. [Google Scholar]
- 5.Fu R, Ceballos-Picot I, Torres RJ, et al. Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain. 2014;137(pt 5):1282–1303. doi: 10.1093/brain/awt202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jinnah HA, DeGregorio L, Harris JC, et al. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res. 2000;463:309–326. doi: 10.1016/s1383-5742(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 7.Visser JE, Baer PR, Jinnah HA. Lesch-Nyhan syndrome and the basal ganglia. Brain Res Rev. 2000;32:449–475. doi: 10.1016/s0165-0173(99)00094-6. [DOI] [PubMed] [Google Scholar]
- 8.Baumeister AA, Frye GD. The biochemical basis of the behavioral disorder in the Lesch-Nyhan syndrome. Neurosci Biobehav Rev. 1985;9:169–178. doi: 10.1016/0149-7634(85)90043-0. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd KG, Hornykiewicz O, Davidson L, et al. Biochemical evidence of dysfunction of brain neurotransmitters in the Lesch-Nyhan syndrome. N Engl J Med. 1981;305:1106–1111. doi: 10.1056/NEJM198111053051902. [DOI] [PubMed] [Google Scholar]
- 10.Saito Y, Takashima S. Neurotransmitter changes in the pathophysiology of Lesch-Nyhan syndrome. Brain Dev. 2000;22(suppl 1):S122–S131. doi: 10.1016/s0387-7604(00)00143-1. [DOI] [PubMed] [Google Scholar]
- 11.Saito Y, Ito M, Hanaoka S, et al. Dopamine receptor upregulation in Lesch-Nyhan syndrome: a postmortem study. Neuropediatrics. 1999;30:66–71. doi: 10.1055/s-2007-973462. [DOI] [PubMed] [Google Scholar]
- 12.Wong DF, Harris JC, Naidu S, et al. Dopamine transporters are markedly reduced in Lesch-Nyhan disease in vivo. Proc Natl Acad Sci U S A. 1996;93:5539–5543. doi: 10.1073/pnas.93.11.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst M, Zametkin AJ, Matochik JA, et al. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N Engl J Med. 1996;334:1568–1572. doi: 10.1056/NEJM199606133342403. [DOI] [PubMed] [Google Scholar]
- 14.Del Bigio MR, Halliday WC. Multifocal atrophy of cerebellar internal granular neurons in Lesch-Nyhan disease: case reports and review. J Neuropathol Exp Neurol. 2007;66:346–353. doi: 10.1097/nen.0b013e3180515319. [DOI] [PubMed] [Google Scholar]
- 15.Jinnah HA. Lesch-Nyhan disease: from mechanism to model and back again. Dis Model Mech. 2009;2:116–121. doi: 10.1242/dmm.002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jinnah HA, Langlais PJ, Friedmann T. Functional analysis of brain dopamine systems in a genetic mouse model of Lesch-Nyhan syndrome. J Pharmacol Exp Ther. 1992;263:596–607. [PubMed] [Google Scholar]
- 17.Jinnah HA, Wojcik BE, Hunt MA, et al. Dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. J Neurosci. 1994;14:1164–1175. doi: 10.1523/JNEUROSCI.14-03-01164.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jinnah HA, Jones MD, Wojcik BE, et al. Influence of age and strain on striatal dopamine loss in a genetic mouse model of Lesch-Nyhan disease. J Neurochem. 1999;72:225–229. doi: 10.1046/j.1471-4159.1999.0720225.x. [DOI] [PubMed] [Google Scholar]
- 19.Egami K, Yitta S, Kasim S, et al. Basal ganglia dopamine loss due to defect in purine recycling. Neurobiol Dis. 2007;26:396–407. doi: 10.1016/j.nbd.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang TH, Park Y, Bader JS, Friedmann T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS One. 2013;8:e74967. doi: 10.1371/journal.pone.0074967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewers JC, Ceballos-Picot I, Shirley TL, et al. Consequences of impaired purine recycling in dopaminergic neurons. Neuroscience. 2008;152:761–772. doi: 10.1016/j.neuroscience.2007.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh J, Zheng S, Howard BD. Impaired differentiation of HPRT-deficient dopaminergic neurons: a possible mechanism underlying neuronal dysfunction in Lesch-Nyhan syndrome. J Neurosci Res. 1998;53:78–85. doi: 10.1002/(SICI)1097-4547(19980701)53:1<78::AID-JNR8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 23.Bitler CM, Howard BD. Dopamine metabolism in hypoxanthineguanine phosphoribosyltransferase-deficient variants of PC12 cells. J Neurochem. 1986;47:107–112. doi: 10.1111/j.1471-4159.1986.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 24.Goettle M, Burhenne H, Sutcliffe D, Jinnah HA. Purine metabolism during neuronal differentiation: the relevance of purine synthesis and recycling. J Neurochem. 2013;127:805–818. doi: 10.1111/jnc.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ceballos-Picot I, Mockel L, Potier MC, et al. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum Mol Genet. 2009;18:2317–2327. doi: 10.1093/hmg/ddp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi HK, Won LA, Kontur PJ, et al. Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 1991;552:67–76. doi: 10.1016/0006-8993(91)90661-e. [DOI] [PubMed] [Google Scholar]
- 27.Bazelon M, Fenichel GM, Randall J. Studies on neuromelanin. I. A melanin system in the human adult brainstem. Neurology. 1967;17:512–519. doi: 10.1212/wnl.17.5.512. [DOI] [PubMed] [Google Scholar]
- 28.Fenichel GM, Bazelon M. Studies on neuromelanin. II. Melanin in the brainstems of infants and children. Neurology. 1968;18:817–820. doi: 10.1212/wnl.18.8.817. [DOI] [PubMed] [Google Scholar]
- 29.Zecca L, Fariello R, Riederer P, et al. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson's disease. FEBS Lett. 2002;510:216–220. doi: 10.1016/s0014-5793(01)03269-0. [DOI] [PubMed] [Google Scholar]
- 30.Fedorow H, Halliday GM, Rickert CH, et al. Evidence for specific phases in the development of human neuromelanin. Neurobiol Aging. 2006;27:506–512. doi: 10.1016/j.neurobiolaging.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Hyland K, Kasim S, Egami K, et al. Tetrahydrobiopterin and brain dopamine loss in a genetic mouse model of Lesch-Nyhan disease. J Inherit Metab Dis. 2004;27:165–178. doi: 10.1023/B:BOLI.0000028728.93113.4d. [DOI] [PubMed] [Google Scholar]
- 32.Fujimoto WY, Subak-Sharpe JH, Seegmiller JE. Hypoxanthine-gua-nine phosphoribosyltransferase deficiency: chemical agents selective for mutant or normal cultured fibroblasts in mixed and heterozygote cultures. Proc Natl Acad Sci U S A. 1971;68:1516–1519. doi: 10.1073/pnas.68.7.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nyhan WL. Dopamine function in Lesch-Nyhan disease. Environ Health Perspect. 2000;108:409–411. doi: 10.1289/ehp.00108s3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson DA, Sejnowski TJ, Poizner H. Convergent evidence for abnormal striatal synaptic plasticity in dystonia. Neurobiol Dis. 2010;37:558–573. doi: 10.1016/j.nbd.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perlmutter JS, Mink JW. Dysfunction of dopaminergic pathways in dystonia. Adv Neurol. 2004;94:163–170. [PubMed] [Google Scholar]
- 36.Moy SS, Criswell HE, Breese GR. Differential effects of bilateral dopamine depletion in neonatal and adult rats. Neurosci Biobehav Rev. 1997;21:425–435. doi: 10.1016/s0149-7634(96)00040-1. [DOI] [PubMed] [Google Scholar]
- 37.Breese GR, Criswell HE, Duncan GE, Mueller RA. Dopamine deficiency in self-injurious behavior. Psychopharmacol Bull. 1989;25:353–357. [PubMed] [Google Scholar]
- 38.Taira T, Kobayashi T, Hori T. Disappearance of self-mutilating behavior in a patient with Lesch-Nyhan syndrome after bilateral chronic stimulation of the globus pallidus interna. J Neurosurg. 2003;98:414–416. doi: 10.3171/jns.2003.98.2.0414. [DOI] [PubMed] [Google Scholar]
- 39.Schretlen DJ, Varvaris M, Ho TE, et al. Cross-sectional analyses of regional brain volume abnormalities in Lesch-Nyhan disease and its variants. Lancet Neurol. 2013;12:1151–1158. doi: 10.1016/s1474-4422(13)70238-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visser JE, Schretlen DJ, Bloem BR, Jinnah HA. Levodopa is not a useful treatment for Lesch-Nyhan disease. Mov Disord. 2011;26:746–749. doi: 10.1002/mds.23478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang TH, Guibinga GH, Jinnah HA, Friedmann T. HPRT deficiency coordinately dysregulates canonical Wnt and presenilin-1 signaling: a neuro-developmental regulatory role for a housekeeping gene? PLoS One. 2011;6:e16572. doi: 10.1371/journal.pone.0016572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guibinga GH, Hrustanovic G, Bouic K, et al. MicroRNA-mediated dysregulation of neural developmental genes in HPRT deficiency: clues for Lesch-Nyhan disease? Hum Mol Genet. 2011;21:609–622. doi: 10.1093/hmg/ddr495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guibinga GH, Hsu S, Friedmann T. Deficiency of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) dysregulates neurogenesis. Mol Ther. 2010;18:54–62. doi: 10.1038/mt.2009.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Money KM, Stanwood GD. Developmental origins of brain disorders: roles for dopamine. Front Cell Neurosci. 2013;7:260. doi: 10.3389/fncel.2013.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirsch E, Graybiel AM, Agid Y. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 46.Alavian KN, Scholz C, Simon HH. Transcriptional regulation of mesencephalic dopaminergic neurons: the full circle of life and death. Mov Disord. 2008;23:319–328. doi: 10.1002/mds.21640. [DOI] [PubMed] [Google Scholar]
- 47.Jiang C, Wan X, He Y, et al. Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp Neurol. 2005;191:154–162. doi: 10.1016/j.expneurol.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 48.Witta J, Baffi J, Palkovits M, et al. Nigrostriatal innervation is preserved in Nurr1-null mice, although dopaminergic neuron precursors are arrested from terminal differentiation. Mol Brain Res. 2000;84:67–78. doi: 10.1016/s0169-328x(00)00211-4. [DOI] [PubMed] [Google Scholar]
- 49.Furukawa Y, Nygaard TG, Gutlich M, et al. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology. 1999;53:1032–1041. doi: 10.1212/wnl.53.5.1032. [DOI] [PubMed] [Google Scholar]
- 50.Furukawa Y, Kapatos G, Haycock JW, et al. Brain biopterin and tyrosine hydroxylase in asymptomatic dopa-responsive dystonia. Ann Neurol. 2002;51:637–641. doi: 10.1002/ana.10175. [DOI] [PubMed] [Google Scholar]
- 51.Grotzsch H, Pizzolato GP, Ghika J, et al. Neuropathology of a case of dopa-responsive dystonia associated with a new genetic locus, DYT14. Neurology. 2002;58:1839–1842. doi: 10.1212/wnl.58.12.1839. [DOI] [PubMed] [Google Scholar]
- 52.Hyland K, Surtees RAH, Rodeck C, Clayton PT. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology. 1992;42:1980–1988. doi: 10.1212/wnl.42.10.1980. [DOI] [PubMed] [Google Scholar]