

Abstract
Advanced glycation end-products (AGEs) are a diverse group of molecules produced by the non-enzymatic addition of glucose to proteins, lipids, and nucleic acids. AGE levels have been associated with hyperglycemia and diabetic complications, especially in animal models, but less clearly in human studies. We measured total serum AGEs using an enzyme linked immunosorbant assay (ELISA) in 506 subjects from 246 families in the Diabetes Heart Study (DHS)/DHS MIND Study (n= 399 type 2 diabetes(T2D)-affected). Single nucleotide polymorphisms (SNPs) in several candidate genes, including known AGE receptors, were tested for their influence on circulating AGE levels. The genetic analysis was expanded to include an exploratory genome-wide association study (GWAS) and exome chip analysis of AGEs (≈440,000 SNPs). AGEs were found to be highly heritable (h2 = 0.628, p=8.96×10-10). While no SNPs from candidate genes were significantly associated after Bonferroni correction, rs1035798 in the gene AGER was the most significantly associated (p=0.007). Additionally, rs7198427, in MT1A, showed a nominally significant p-value (p=0.0099). No SNPs from the GWAS or exome studies were identified after correction for multiple comparisons; however, rs17054480 in the PALLD2 gene on chromosome 4 showed the strongest association (p=7.77×10−7). Five SNPs at two loci (ISCA2/NPC2 and FBXO33) had p-values of less than 2.0×10−5 and three additional SNPs (rs716326 in MACROD2, and rs6795197 and rs6765857 in ZBTB38) showed a nominal association with p-values of less than 1.0×10−5. These findings provide a foundation for further investigation into the genetic component of circulating AGEs.
Keywords: Genome-Wide Association Study, type 2 diabetes, AGE receptor
Introduction
Advanced glycation end products (AGEs) are a diverse, heterogenous class of molecules, formed through the non-enzymatic glycation and oxidation of proteins, lipids, and nucleic acids. AGEs have been implicated in a wide range of diseases and phenotypes and are a major pathway of interest in complications of type 2 diabetes (T2D) [1]. AGE formation is accelerated in T2D- Affected individuals due to increased concentration of circulating glucose [2]. Previous investigations in diabetic cohorts have associated AGEs with cardiovascular outcomes [3], diabetic nephropathy [4], retinopathy [5], and neuropathy [6]. In addition, AGEs have been shown to be associated with a number of clinical measures and behaviors including estimated glomerular filtration rate (eGFR) [8], soluble vascular cell adhesion molecule 1 [van Eupen, et al. 2013], body mass index (BMI), and smoking [7-9].
While there are numerous association studies investigating the relationship between AGE levels and diabetic complications, there are relatively few prior studies investigating the genetics influencing AGE levels. AGEs were previously reported to be heritable [10] and, in addition, single nucleotide polymorphisms (SNPs) in AGER (RAGE; Receptor for Advanced Glycation End Products) and MT1A (Metallothionein-1A) were observed to be associated with AGE levels [11-13]. However, there are additional AGE receptors that represent potential candidates for genetic analyses, including AGE-R1 (Oligosaccharyl Transferase-48), AGE-R2 (PRKCSH protein kinase C substrate 80K-H), AGE-R3/ Galectin-3, and SR-A (macrophage scavenger receptors type I and type II). We have evaluated heritability of AGEs and then conducted genetic association analyses with AGE levels in the Diabetes Heart Study (DHS)/DHS Mind Study cohort, an ongoing genetic and epidemiological analysis of families enriched for type 2 diabetes with extensive genetic data [14, 15].
Materials and Methods
Study Population
The DHS is a family-based study examining risk for macrovascular and other complications in T2D. Briefly, the DHS includes siblings concordant for T2D but without advanced renal insufficiency. When possible, unaffected siblings were also recruited. T2D was clinically defined as diabetes developing after the age of 35 years and initially treated with oral agents and/or diet and exercise, in the absence of historical evidence of ketoacidosis. Diagnoses were confirmed by measurement of fasting blood glucose and glycosylated hemoglobin (HbA1c). Extensive measurements of cardiovascular disease (CVD) risk factors were obtained during baseline exams, which occurred from 1998 to 2006. Ascertainment and recruitment have been previously described in detail [14, 15].
The DHS MIND is an ancillary study to the DHS that included a cognitive testing component and brain magnetic resonance imaging (MRI). The purpose was to investigate the relationships between cognitive function, brain imaging, and vascular disease in T2D. Participants from the original DHS investigation were re-examined on average 6.7 ± 1.6 years after their initial visit. Participant examinations were conducted in the General Clinical Research Center of the Wake Forest Baptist Medical Center. Study protocols were approved by the Institutional Review Board at Wake Forest School of Medicine and all study procedures were carried out in accordance with the Declaration of Helsinki. All participants provided written informed consent before participation.
Advanced Glycation End Products
Total serum AGEs were measured using a competitive enzyme linked immunosorbant assay (ELISA) (Lifeome Biolabs; Oceanside, CA). The ELISA was run according to the manufacturer's recommendations using stored serum. The ELISA uses a monoclonal antibody that is specific to AGEs. This ELISA has a minimum detectable dose of AGEs less than 35.2 ng/ml. Intra-assay and inter-assay precision was 2.0% and 22.5%, respectively.
Exome Chip
Additional SNPs, predominately low-frequency and rare coding SNPs, were also captured by the Illumina HumanExome BeadChips v.1.0 (Illumina Inc., San Diego, CA) for which genotype data was available in the DHS [16]. For DHS exome chip data, genotype calling was completed using Genome Studio Software v1.9.4 (Illumina). Samples failing to meet a minimum acceptable call rate of 98% (n=3) were excluded from further analyses. An additional 58 samples were included as blind duplicates within the genotyping set to serve as QC samples; the concordance rate for blind duplicates was 99.9 ±0.0001% (mean± standard deviation (SD)). Exclusion criteria for SNP performance included call rate < 95% (n=972), SNPs with 5 or fewer observances (n=204,273) and Hardy-Weinberg Equilibrium (HWE) p-value <1×10−6 (n=26); 41,961 SNPs were retained for analysis. Additional QC of exome chip data set was completed to exclude samples with poor quality genotype calls, gender errors, or unclear/unexpected sibling relationships.
Genome-Wide Association Study
Genomic DNA was purified from whole-blood samples obtained from subjects using the PUREGENE DNA isolation kit (Gentra Systems., Minneapolis, MN). DNA was quantitated using standardized fluorometric readings on a Hoefer DyNA Quant 200 fluorometer (Hoefer Pharmacia Biotech, Inc., San Francisco, CA).
A genome-wide association study (GWAS) was completed using the Affymetrix Genome-wide Human SNP Array 5.0 (Affymetrix, CA, USA) as reported [17]. Genotype calling was completed using the BRLLM-P algorithm in Genotyping Console v4.0 (Affymetrix). Samples failing to meet an intensity quality control (QC) threshold and those failing to meet a minimum acceptable call rate of 95% were excluded from further analyses (n=7). An additional 39 samples were included as blind duplicates within the genotyping set to serve as QC samples; the concordance rate for these blind duplicates was 99.0 ±0.72% (mean± SD). Exploratory analyses of genotype data were performed using PLINK v. 1.07 (http://pngu.mgh.harvard.edu/purcell/plink/) and samples with poor quality genotype calls, gender errors, or unclear/unexpected sibling relationships were excluded from further analysis. Exclusion criteria for SNP performance included call rate < 95% (n=11,085), HWE p-value < 1×10−6 (n=332), and minor allele frequency (MAF) < 0.01 (n=57,382); 371,951 SNPs were retained for analysis.
Heritability Analysis
Heritability estimates for AGEs were assessed in 506 related individuals from 245 families that had AGE measures. The AGE measurements were natural log transformed to approximate the normality assumptions of the analysis. To determine the contribution of genetic factors to AGE levels, the data in family members were analyzed using Sequential Oligogenic Linkage Analysis Routines (SOLAR) v6.3.4 (Texas Biomedical Research Institute, San Antonio, TX, USA). SOLAR performs a variance components analysis of family data where the total phenotypic variation is partitioned into genetic and nongenetic sources of variation. This approach has been used previously in the DHS [18, 19]. To minimize the bias associated with shared environmental factors, the estimates of heritability (h2) were based on all available family data and were controlled for covariates related to AGEs including age, gender, BMI, eGFR, T2D affected status, and smoking status (current or former). Five models were developed that incorporated an increasing number of covariates to determine the extent that genetic factors contribute to variation in AGE levels independent of other confounding variables. The first model was unadjusted. The second model was adjusted for age and gender. The third model was adjusted for age, gender, and BMI. The fourth was adjusted for age, gender, BMI, and eGFR. The fifth model was the fully adjusted model and included age, gender, BMI, eGFR, T2D-affected status, and a history of smoking. The significance of the heritability estimates was obtained by likelihood ratio tests.
Genetic Association Analysis
Genetic association was run using SOLAR v6.3.4 (Texas Biomedical Research Institute, San Antonio, TX) which uses variance component methods to account for family structure. AGE measures were natural log transformed prior to analysis to approximate conditional normality. Covariates used in the analysis were age, gender, BMI, T2D affected status, eGFR, and smoking history. All analyses were run using the additive model of inheritance. The Bonferroni corrected p - value for significance for the 121 candidate gene SNPs was set at 0.00041 (α=0.05/121). Similarly, the p - value for significance for the GWAS and exome chip associations was set at 1.21×10−7 after correcting for multiple comparisons based upon the number of SNPs (413,912) investigated.
Results
Demographics
Demographic characteristics of the 506 individuals included in this study are shown in Table 1. Briefly, the average age was 67 years old and just over half of the participants were female (55.3%). The individuals were on average overweight or obese with an average BMI of 31.5kg/m2. Approximately 79% of the individuals were affected by T2D with average diabetes duration of 16.6 years. The DHS cohort is broadly representative of T2D-affected patients in the general population: older, relatively obese, and with significant risk factors and history of CVD and kidney function decline.
Table 1.
Demographics characteristics of the Diabetes Heart Study participants
| All | T2D | Non-T2D | |
|---|---|---|---|
| N | 506 | 399 | 107 |
| Age (years) ± SD | 67.7±9.0 | 68.0±8.7 | 66.5±9.8 |
| Female (%) | 280 (55.3) | 210 (52.6) | 70 (65.4) |
| Body Mass Index (m2/kg) ± SD | 31.5±6.6 | 32.3±6.6 | 28.5±5.6 |
| T2D Affected (%) | 399 (78.9) | 399 (100) | 0 (0.0) |
| T2D Duration (years) ± SD | 16.6±6.6 | 16.6±6.6 | N/A |
| Coronary Artery Calcification ± SD | 1101±2140 | 1245±2215 | 561±1737 |
| Carotid Artery Calcification± SD | 226±597 | 252±626 | 131±464 |
| Abdominal Aortic Calcification ± SD | 7922±11912 | 8648±12139 | 5175±10636 |
| Carotid Intima-Media Thickness ± SD | 0.67±0.13 | 0.67±0.13 | 0.63±0.10 |
| Prior History of CVD (%) | 164 (32.4) | 149 (37.3) | 15 (14.0) |
| Cholesterol ± SD | 180.1±47.9 | 180.1±47.9 | 180.1±47.9 |
| High Density Lipoprotein ± SD | 41.5±12.2 | 41.5±12.2 | 41.5±12.2 |
| Low Density Lipoprotein ± SD | 99.5±34.7 | 99.5±34.7 | 99.5±34.7 |
| Triglycerides ± SD | 198.8±142.2 | 198.8±142.2 | 198.8±142.2 |
| Pulse Pressure (mm Hg)± SD | 61.1±16.1 | 62.7±16.1 | 55.1±14.7 |
| Hypertension (%) | 440 (87.0) | 363 (91.0) | 77 (72.0) |
| Estimated Glomerular Filtration Rate ± SD | 69.2±19.6 | 69.5±20.5 | 68.3±15.8 |
| Albumin/Creatine Ratio ± SD | 77.4±465.7 | 94.2±522.7 | 14.2±22.7 |
| Fasting Glucose ± SD | 133.8±49.5 | 143.6±51.3 | 97.2±10.3 |
| HbA1c ± SD | 7.11±1.34 | 7.45±1.30 | 5.85±0.31 |
| Insulin Use (%) | 148 (29.2) | 147 (36.8) | 1 (0.09) |
| Diabetes Medication Use (%) | 327 (64.6) | 323 (81.0) | 4 (3.7) |
| Statin Use (%) | 235 (46.4) | 200 (50.1) | 35 (32.7) |
| Lipid Medication Use (%) | 240 (47.4) | 204 (51.1) | 36 (33.6) |
| Blood Pressure Medication Use (%) | 363 (71.7) | 307 (76.9) | 56 (52.3) |
| History of Smoking (%) | 276 (54.5) | 220 (55.1) | 56 (52.3) |
| Advanced Glycation End Products ± SD | 40.7±20.2 | 41.1±21.3 | 49.2±15.3 |
SD: Standard Deviation, T2D: Type 2 Diabetes
Heritability
A heritability analysis was performed on AGEs (Table 2). This analysis was run using a variety of covariates. First, with no covariates included, AGEs were not found to be heritable (h2 =0.045 p=0.291). However, as applicable covariates were added, the heritability estimate became increasingly more significant. When age and sex were included in the analysis as covariates the h2 increased to 0.084 (p=0.167); addition of BMI increased the heritability estimate further to 0.114 (p=0.093). After adjusting for commonly used covariates including age, sex, BMI and eGFR in model 4, heritability reached statistical significance (h2=0.317, p=5.75×10−4). AGE levels were highly heritable in the fully adjusted model when accounting for age, gender, BMI, eGFR, T2D affected status, and current/former smoking status (h2=0.628, p=8.96×10−10) (Table 2).
Table 2.
Heritability Analysis of Advanced Glycation End Products
| Covariates | h^2 (SE) | p-value |
|---|---|---|
| None | 0.045 (0.084) | 0.291 |
| Age, Sex | 0.084 (0.090) | 0.167 |
| Age, Sex, BMI | 0.114 (0.090) | 0.093 |
| Age, Sex, BMI, eGFR | 0.317 (0.110) | 5.75×10−4 |
| Age, Sex, BMI, eGFR, T2D, Smoking | 0.628 (0.112) | 8.96×10−10 |
Bold – indicates statistical significance; BMI: Body Mass Index, eGFR: Estimated Glomerular Filtration Rate, T2D: Type 2 Diabetes
Candidate Genes
We performed an association analysis using 121 SNPs genotyped on the GWAS and exome chips from known AGE receptor genes including, Advanced Glycosylation End Product-Specific Receptor (AGER), Dolichyl-Diphosphooligosaccharide-Protein Glycosyltransferase Subunit (non-catalytic) (DDOST), Protein Kinase C substrate 80K-H (PRKCSH), Galactoside-Binding, Soluble, 3 (LGALS3), and Macrophage Scavenger Receptor 1 (MSR1) genes. All polymorphic SNPs in the genes (± 20 kb) from the GWAS and exome chips were included in this analysis. In addition to the receptor genes, Metallothionein 1A (MT1A) was included based upon prior literature that observed an association with AGE levels [12]. We did not find significant associations of any of the candidate SNPs with AGEs after adjusting for multiple comparisons (threshold p < 0.00041) (Table 3). Thirty-three SNPs had a p < 0.25 and are shown in Table 3, including 8 variants with a p < 0.05. The top SNP was rs1035798 in the AGER gene (p=0.007). The next most significant variant was a SNP in the MT1A gene, rs7198427 (p=0.0099). DDOST contained one SNP with a p<0.05, rs113092523. Top SNPs in other candidate genes did not reach p<0.05 (MSR1, rs10503574, p=0.052; PRKCSH, rs17426435, p=0.054; and LGALS3, rs11125, p=0.22).
Table 3.
Association Results for Advanced Glycation Endproduct Receptor Candidate Variants with Advanced Glycation Endproduct Levels
| SNP | Source | Gene/Nearest Gene | MAF | CHR | Position | p-value | β±SE |
|---|---|---|---|---|---|---|---|
| rs1035798 | GWAS | AGER | 0.235 | 6 | 32,151,222 | 0.0073 | −0.089±0.033 |
| rs7198427 | GWAS | MT1A | 0.086 | 16 | 56,685,080 | 0.0099 | 0.130±0.049 |
| rs6499850 | GWAS | MT1A | 0.095 | 16 | 56,687,638 | 0.014 | −0.120±0.048 |
| rs204995 | Exome | AGER | 0.291 | 6 | 32,154,285 | 0.022 | 0.076±0.033 |
| rs7197489 | GWAS | MT1A | 0.181 | 16 | 56,683,993 | 0.031 | −0.077±0.036 |
| rs204993 | Exome | AGER | 0.001 | 6 | 32,155,581 | 0.045 | 0.064±0.032 |
| rs113092523 | Exome | DDOST | 0.001 | 1 | 20,971,057 | 0.045 | 0.850±0.420 |
| rs3130284 | Exome | AGER | 0.003 | 6 | 32,140,487 | 0.047 | 0.067±0.035 |
| rs10503574 | GWAS | MSR1 | 0.036 | 8 | 15,984,590 | 0.052 | −0.151±0.077 |
| rs1871574 | GWAS | MSR1 | 0.018 | 8 | 15,975,574 | 0.053 | 0.209±0.108 |
| rs17426435 | Exome | PRKCSH | 0.061 | 19 | 11,546,269 | 0.054 | 0.110±0.056 |
| rs3134946 | Exome | AGER | 0.209 | 6 | 32,145,993 | 0.054 | 0.067±0.035 |
| rs3134947 | Exome | AGER | 0.001 | 6 | 32,145,205 | 0.054 | 0.067±0.035 |
| rs3096697 | Exome | AGER | 0.197 | 6 | 32,134,510 | 0.054 | 0.067±0.035 |
| rs204992 | Exome | AGER | 0.002 | 6 | 32,156,908 | 0.062 | 0.063±0.034 |
| rs176095 | Exome | AGER | 0.001 | 6 | 32,158,319 | 0.070 | 0.061±0.034 |
| rs414836 | GWAS | MSR1 | 0.065 | 8 | 16,045,585 | 0.087 | −0.096±0.056 |
| rs204994 | Exome | AGER | 0.255 | 6 | 32,154,998 | 0.093 | 0.057±0.034 |
| rs17677443 | GWAS | MSR1 | 0.060 | 8 | 16,029,070 | 0.094 | 0.099±0.059 |
| rs11786960 | GWAS | MSR1 | 0.461 | 8 | 16,010,949 | 0.096 | −0.046±0.027 |
| rs142241539 | Exome | AGER | 0.001 | 6 | 32,130,655 | 0.10 | −0.690±0.420 |
| rs3130349 | Exome | AGER | 0.184 | 6 | 32,147,696 | 0.12 | 0.059±0.038 |
| rs448578 | GWAS | MSR1 | 0.066 | 8 | 16,042,900 | 0.13 | 0.085±0.056 |
| rs6530943 | GWAS | MSR1 | 0.090 | 8 | 15,973,590 | 0.14 | −0.074±0.050 |
| rs10103856 | GWAS | MSR1 | 0.065 | 8 | 16,048,551 | 0.16 | −0.079±0.056 |
| rs640742 | GWAS | DDOST | 0.412 | 1 | 20,984,554 | 0.17 | 0.041±0.029 |
| rs501257 | Exome | PRKCSH | 0.140 | 19 | 11,536,575 | 0.17 | 0.055±0.040 |
| rs149461273 | Exome | AGER | 0.001 | 6 | 32,155,917 | 0.18 | 0.560±0.420 |
| rs111334879 | Exome | DDOST | 0.001 | 1 | 20,981,976 | 0.20 | −0.360±0.280 |
| rs204989 | GWAS | AGER | 0.222 | 6 | 32,161,852 | 0.20 | 0.045±−0.035 |
| rs204990 | GWAS | AGER | 0.222 | 6 | 32,161,430 | 0.20 | 0.045±0.035 |
| rs204991 | Exome | AGER | 0.225 | 6 | 32,161,366 | 0.20 | 0.044±0.035 |
| rs11125 | Exome | LGALS3 | 0.098 | 14 | 55,611,839 | 0.22 | −0.059±0.048 |
Results listed only for associations with a p-value ≤0.25, MAF: Minor Allele Frequency, CHR: Chromosome, SE: Standard Error.
Exome-wide Analysis of Exome Chip Genotyping Data
No SNP reached conservative levels of statistical significance (threshold p<1.23×10−7). The top results are shown in Table 4. The most significant SNPs were exonic, missense variants in the Chitinase, acidic (CHIA) gene on chromosome 1 (rs41282492, p=4.1×10−5; rs41282494, p=4.1×10−5; rs41282496, p=4.1×10−5) (Table 4). Three additional SNPs had p < 1.0×10−4; rs10805470 (intergenic, p=4.9×10−5), rs139025377 (DEAD (Asp-Glu-Ala-Asp) box polypeptide 19A [DDX19A], p=7.4×10−5), and rs147902167 (myomesin 3 [MYOM3], p=8.1×10−5) (Table 4). Fig. 1 shows a Manhattan plot of the results from the exome chip analysis of AGEs.
Table 4.
Most Significant Association Results from Exome Chip Analysis for Advanced Glycation Endproduct Levels
| SNP | Gene/Nearest Gene | AA Change | MAF | CHR | Position | p-value | β±SE |
|---|---|---|---|---|---|---|---|
| rs41282492 | CHIA | N45D | 0.121 | 1 | 111854889 | 4.10×10−5 | 0.179±0.043 |
| rs41282494 | CHIA | D47N | 0.121 | 1 | 111854895 | 4.10×10−5 | 0.179±0.043 |
| rs41282496 | CHIA | R61M | 0.121 | 1 | 111854938 | 4.10×10−5 | 0.179±0.043 |
| rs10805470 | ESM1 | Intergenic | 0.384 | 5 | 54187813 | 4.90×10−5 | −0.118±0.029 |
| rs139025377 | DDX19A | K96R | 0.006 | 16 | 70398987 | 7.40×10−5 | −0.784±0.197 |
| rs147902167 | MYOM3 | I434V | 0.015 | 1 | 24417419 | 8.10×10−5 | −0.439±0.110 |
AA: Amino Acid, MAF: Minor Allele Frequency, CHR: Chromosome, SE: Standard Error.
Figure 1.
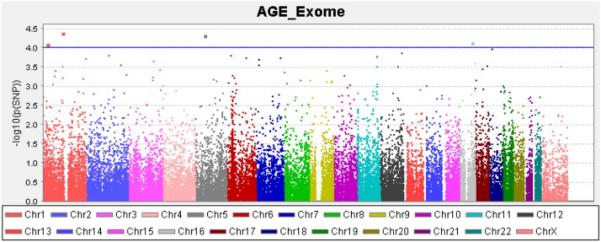
Manhattan Plot of Exome Chip Association Results for Advanced Glycation End Product Levels
Analysis of Genome-wide Association Study
There were no SNPs from the GWAS that reached statistical significance (threshold p < 1.23×10−7). The most significant SNP associated with AGE levels was rs17054480 (p=7.77×10−7), an intronic SNP in the cytoskeletal associated protein (PALLD) gene on chromosome 4 (Table 5). There were 3 SNPs with p < 1×10−5 surrounding the Niemann-Pick disease, type C2 (NCP2) and iron-sulfur cluster assembly 2(ISCA2) genes on chromosome 14 (rs11159086, p=2.70×10−6; rs4899514, p=4.18×10−6; rs1029701, p=9.11×10−6) (Table 5). Four additional SNPs showed nominal association with p < 1×10−5 including rs4454866 in the F-box Protein 33 (FBXO33) gene (p=5.07×10−6), rs716316 in the MACRO domain containing 2 (MACROD2) gene (p=7.53×10−6), rs679597 (p=8.42×10−6), and rs6765857 (p=9.32×10−6) in the zinc finger and BTB domain containing 38 (ZBTB38) gene (Table 5). There were 52 additional SNPs that were nominally associated with a p < 1×10−4 (Table S1). Fig. 2 shows a Manhattan plot of the results from the GWAS analysis of AGEs.
Table 5.
Most Significant Association Results of Genome-wide Association Study for Advanced Glycation Endproduct Levels
| SNP | Gene/Nearest Gene | AA Change | MAF | CHR | Position | p-value | β±SE |
|---|---|---|---|---|---|---|---|
| rs17054480 | PALLD | Intronic | 0.053 | 4 | 169608790 | 7.77×10−7 | −0.312±0.062 |
| rs11159086 | ISCA2/NPC2 | Intergenic | 0.141 | 14 | 74962275 | 2.70×10−6 | −0.180±0.038 |
| rs4899514 | NPC2/ISCA2 | Intergenic | 0.139 | 14 | 74940170 | 4.81×10−6 | 0.177±0.038 |
| rs4454866 | FBXO33 | Intergenic | 0.052 | 14 | 40676918 | 5.07×10−6 | 0.281±0.061 |
| rs716316 | MACROD2 | Intronic | 0.347 | 20 | 14908741 | 7.53×10−6 | 0.129±0.028 |
| rs6795197 | ZBTB38 | Intronic | 0.412 | 3 | 141047072 | 8.42×10−6 | −0.131±0.029 |
| rs1029701 | NPC2/ISCA2 | Intergenic | 0.150 | 14 | 74940815 | 9.11×10−6 | −0.161±0.036 |
| rs6765857 | ZBTB38 | Intronic | 0.420 | 3 | 141050805 | 9.32×10−6 | 0.130±0.029 |
AA: Amino Acid, MAF: Minor Allele Frequency, CHR: Chromosome, SE: Standard Error.
Figure 2.
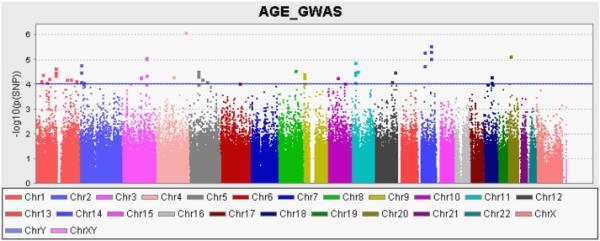
Manhattan Plot of Genome-Wide Association Results For Advanced Glycation End Product Levels
Discussion
In this study we evaluated heritability and genetic association between polymorphisms in specific genes and genome wide data with AGEs in a sample with a high prevalence of diabetes. Only one other study has calculated heritability for AGE levels and that for a specific type of AGE, Nε-carboxymethyl lysine (CML) which was highly heritable (h2 = 0.74) [10]. In the study, Leslie et al. observed a higher correlation of CML levels in monozygotic twins compared to dizygotic twins. Importantly, heritability of fasting glucose and HbA1c did not explain the CML heritability. The results of the current study are consistent with this; in the fully adjusted model we showed that circulating AGE levels are highly heritable (h2 = 0.628), suggesting that there is a substantial genetic component to not just CML levels, but also total AGEs quantitated by ELISA.
Most of the prior genetic association studies for AGE levels have focused on a few candidate genes, most notably the receptor for advanced glycation end products (RAGE) gene (AGER) on chromosome 6. Several previous studies have shown evidence of an association between polymorphisms in AGER and circulating AGE levels [11, 13]. Bansal et al. found that the serine allele of rs2070600 (Gly82Ser) and rs1800625 (C allele) were associated with increased AGEs [11]. Jang et al., on the other hand, found that the individuals with the G/G genotype at rs2070600 had increased levels of AGEs [13]. There are additional AGE receptors including AGE-R1 or Oligosaccharyl Transferase-4 coded by the DDOST (Dolichyl- Diphosphooligosaccharide-Protein Glycosyltransferase Subunit (non-catalytic)) gene, AGE-R2 (80 K-H phosphoprotein (Protein kinase C substrate)) which is coded by the Protein Kinase C substrate 80K-H (PRKCSH) gene, AGE-R3 (Galectin-3) coded by the Galactoside-Binding, Soluble, 3 (LGALS3) gene, and macrophage scavenger receptors type I and type II (SR-A) coded by the Macrophage Scavenger Receptor 1 (MSR1) gene, none of which, to our knowledge, have shown genetic associations with circulating AGE levels. In addition to AGER, Metallothionein 1A (MT1A) contains a SNP (rs8052394) that was previously found to be associated with AGE levels [12]; individuals with the G allele had increased levels of AGEs compared to individuals with the AA genotype. Significantly, to date, there are no GWAS or exome chip studies investigating circulating AGE levels.
In the present study we investigated potential associations with SNPs in and near multiple AGE receptor protein genes and genes suspected of influencing AGE levels. Several nominal associations were observed. Of particular interest is the association (p = 0.0073) of AGE levels with SNP rs1035798 located in AGER. Although the function of this SNP is unknown [20], recent studies observed rs1035798 to be associated with small-vessel disease [21] and cardiovascular death [22]. RAGE is a member of the immunoglobulin superfamily and is the best characterized of the known and suspected AGE receptors [23]. AGE interactions with RAGE mediate diverse cellular functions, possibly functioning as a promotion factor for pathologic conditions [24]. Significantly less is known of the other AGE receptors, but it is suggested that they function moreso in AGE clearance and detoxification versus transduction of intracellular signaling [25-27].
With an appreciation of the limited power due to a small sample size, we further expanded our investigation to a full GWAS and exome chip study. The top hit SNP was an intronic SNP in the PALLD gene (rs17054480, p-7.77×10−7) (Table 4). PALLD codes for an actin associated protein, palladin, involved in cytoskeleton morphology [28]. Specifically, palladin is involved in the control of the cell shape, adhesion, and contraction [29] and may play a role in cardiovascular pathology [30]. Other nominal associations highlighted the NPC2 and ISCA2 genes, involved in cholesterol transport [31, 32] and the maturation of mitochondrial sulfur proteins [33], respectively.
Further studies need to be performed to investigate AGEs in more depth. This study investigated global AGE levels; however, AGEs are a diverse set of molecules. Directly measuring these individual molecules may lead to an increased understanding of AGE genetics. In conclusion, we found that AGEs are a highly heritable trait, but were unable to identify any specific genetic components of circulating AGE levels. We saw nominal associations between SNPs in candidate genes and AGE levels, but no SNP from either the GWAS or exome chip reached statistical significance. Additional studies with increased sample size and using a more sophisticated measure of AGEs may yield stronger results.
Supplementary Material
Highlights.
We performed genetic associations with advanced glycation end products.
Advanced glycation end products were found to be highly heritable.
Genetic analysis of candidate genes showed evidence of genetic influences on AGEs.
GWAS and exome chip analysis showed nominal association with AGEs.
Acknowledgements
This study was supported in part by the National Institutes of Health through R01 HL67348, R01 HL092301, R01 NS058700 (to Donald W. Bowden), and F31 AG044879 (to Laura M. Raffield).
Abbreviations
- DHS
Diabetes heart study
- AGE
Advanced Glycation end product
- ELISA
Enzyme linked immunosorbant assay
- T2D
Type 2 diabetes
- SNP
Single nucleotide polymorphism
- GWAS
Genome Wide Association Study
- eGFR
Estimated glomerular filtration rate
- HbA1c
Glycosylated hemogobin
- SOLAR
Sequential Oligogenic Linkage Analysis
- BMI
Body mass index
- CVD
Cardiovascular disease
- RAGE
Receptor of advanced glycation end products
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. 2014;18:1–14. doi: 10.4196/kjpp.2014.18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowotny K, Jung T, Hohn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194–222. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanssen NM, Beulens JW, van Dieren S, Scheijen JL, van der AD, Spijkerman AM, van der Schouw YT, Stehouwer CD, Schalkwijk CG. Plasma advanced glycation end products are associated with incident cardiovascular events in individuals with type 2 diabetes: a case-cohort study with a median follow-up of 10 years (EPIC-NL) Diabetes. 2015;64:257–265. doi: 10.2337/db13-1864. [DOI] [PubMed] [Google Scholar]
- 4.Beisswenger PJ, Howell SK, Russell GB, Miller ME, Rich SS, Mauer M. Early progression of diabetic nephropathy correlates with methylglyoxal-derived advanced glycation end products. Diabetes Care. 2013;36:3234–3239. doi: 10.2337/dc12-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhuri S, Dutta D, Sen A, Chowdhury IH, Mitra B, Mondal LK, Saha A, Bhadhuri G, Bhattacharya B. Role of N-epsilon-carboxy methyl lysine, advanced glycation end products and reactive oxygen species for the development of nonproliferative and proliferative retinopathy in type 2 diabetes mellitus. Mol Vis. 2013;19:100–113. [PMC free article] [PubMed] [Google Scholar]
- 6.Juranek JK, Kothary P, Mehra A, Hays A, Brannagan TH, 3rd, Schmidt AM. Increased expression of the receptor for advanced glycation end-products in human peripheral neuropathies. Brain Behav. 2013;3:701–709. doi: 10.1002/brb3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goh SY, Cooper ME. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–1152. doi: 10.1210/jc.2007-1817. [DOI] [PubMed] [Google Scholar]
- 8.Nin JW, Jorsal A, Ferreira I, Schalkwijk CG, Prins MH, Parving HH, Tarnow L, Rossing P, Stehouwer CD. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: a 12-year follow-up study. Diabetes Care. 2011;34:442–447. doi: 10.2337/dc10-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka K, Nakayama M, Kanno M, Kimura H, Watanabe K, Tani Y, Kusano Y, Suzuki H, Hayashi Y, Asahi K, Sato K, Miyata T, Watanabe T. Skin autofluorescence is associated with the progression of chronic kidney disease: a prospective observational study. PLoS One. 2013;8:e83799. doi: 10.1371/journal.pone.0083799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leslie RD, Beyan H, Sawtell P, Boehm BO, Spector TD, Snieder H. Level of an advanced glycated end product is genetically determined: a study of normal twins. Diabetes. 2003;52:2441–2444. doi: 10.2337/diabetes.52.9.2441. [DOI] [PubMed] [Google Scholar]
- 11.Bansal S, Chawla D, Banerjee BD, Madhu SV, Tripathi AK. Association of RAGE gene polymorphism with circulating AGEs level and paraoxonase activity in relation to macro-vascular complications in Indian type 2 diabetes mellitus patients. Gene. 2013;526:325–330. doi: 10.1016/j.gene.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Giacconi R, Simm A, Santos AN, Costarelli L, Malavolta M, Mecocci P, Piacenza F, Basso A, Fulop T, Rink L, Dedoussis G, Kanoni S, Herbein G, Jajte J, Mocchegiani E. Influence of +1245 A/G MT1A polymorphism on advanced glycation end-products (AGEs) in elderly: effect of zinc supplementation. Genes Nutr. 2014;9:426. doi: 10.1007/s12263-014-0426-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang Y, Kim JY, Kang SM, Kim JS, Chae JS, Kim OY, Koh SJ, Lee HC, Ahn CW, Song YD, Lee JH. Association of the Gly82Ser polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating levels of soluble RAGE and inflammatory markers in nondiabetic and nonobese Koreans. Metabolism. 2007;56:199–205. doi: 10.1016/j.metabol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Bowden DW, Cox AJ, Freedman BI, Hugenschimdt CE, Wagenknecht LE, Herrington D, Agarwal S, Register TC, Maldjian JA, Ng MC, Hsu FC, Langefeld CD, Williamson JD, Carr JJ. Review of the Diabetes Heart Study (DHS) family of studies: a comprehensively examined sample for genetic and epidemiological studies of type 2 diabetes and its complications. Rev Diabet Stud. 2010;7:188–201. doi: 10.1900/RDS.2010.7.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowden DW, Lehtinen AB, Ziegler JT, Rudock ME, Xu J, Wagenknecht LE, Herrington DM, Rich SS, Freedman BI, Carr JJ, Langefeld CD. Genetic epidemiology of subclinical cardiovascular disease in the diabetes heart study. Ann Hum Genet. 2008;72:598–610. doi: 10.1111/j.1469-1809.2008.00446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox AJ, Hugenschmidt CE, Raffield LM, Langefeld CD, Freedman BI, Williamson JD, Hsu FC, Bowden DW. Heritability and genetic association analysis of cognition in the Diabetes Heart Study. Neurobiol Aging. 2014;35:1958, e1953–1958, e1912. doi: 10.1016/j.neurobiolaging.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox AJ, Ng MC, Xu J, Langefeld CD, Koch KL, Dawson PA, Carr JJ, Freedman BI, Hsu FC, Bowden DW. Association of SNPs in the UGT1A gene cluster with total bilirubin and mortality in the Diabetes Heart Study. Atherosclerosis. 2013;229:155–160. doi: 10.1016/j.atherosclerosis.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu FC, Lenchik L, Nicklas BJ, Lohman K, Register TC, Mychaleckyj J, Langefeld CD, Freedman BI, Bowden DW, Carr JJ. Heritability of body composition measured by DXA in the diabetes heart study. Obes Res. 2005;13:312–319. doi: 10.1038/oby.2005.42. [DOI] [PubMed] [Google Scholar]
- 19.Lange LA, Burdon K, Langefeld CD, Liu Y, Beck SR, Rich SS, Freedman BI, Brosnihan KB, Herrington DM, Wagenknecht LE, Bowden DW. Heritability and expression of C-reactive protein in type 2 diabetes in the Diabetes Heart Study. Ann Hum Genet. 2006;70:717–725. doi: 10.1111/j.1469-1809.2006.00280.x. [DOI] [PubMed] [Google Scholar]
- 20.Duan Z, Chen G, Chen L, Stolzenberg-Solomon R, Weinstein SJ, Mannisto S, White DL, Albanes D, Jiao L. Determinants of concentrations of N(epsilon)-carboxymethyl-lysine and soluble receptor for advanced glycation end products and their associations with risk of pancreatic cancer. Int J Mol Epidemiol Genet. 2014;5:152–163. [PMC free article] [PubMed] [Google Scholar]
- 21.Olsson S, Jood K. Genetic variation in the receptor for advanced glycation end-products (RAGE) gene and ischaemic stroke. Eur J Neurol. 2013;20:991–993. doi: 10.1111/ene.12041. [DOI] [PubMed] [Google Scholar]
- 22.Biros E, Moran CS, Norman PE, Hankey GJ, Yeap BB, Almeida OP, Flicker L, White R, Jones R, Golledge J. Association between the Advanced Glycosylation End Product-Specific Receptor Gene and Cardiovascular Death in Older Men. PLoS One. 2015;10:e0134475. doi: 10.1371/journal.pone.0134475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt AM, Hori O, Cao R, Yan SD, Brett J, Wautier JL, Ogawa S, Kuwabara K, Matsumoto M, Stern D. RAGE: a novel cellular receptor for advanced glycation end products. Diabetes. 1996;45(Suppl 3):S77–80. doi: 10.2337/diab.45.3.s77. [DOI] [PubMed] [Google Scholar]
- 24.Stern DM, Yan SD, Yan SF, Schmidt AM. Receptor for advanced glycation endproducts (RAGE) and the complications of diabetes. Ageing Res Rev. 2002;1:1–15. doi: 10.1016/s0047-6374(01)00366-9. [DOI] [PubMed] [Google Scholar]
- 25.Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, Taguchi A, Yan SF, Yan SD, Stern DM, Schmidt AM. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci. 2002;59:1117–1128. doi: 10.1007/s00018-002-8491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411–429. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stitt AW, Bucala R, Vlassara H. Atherogenesis and advanced glycation: promotion, progression, and prevention. Ann N Y Acad Sci. 1997;811:115–127. doi: 10.1111/j.1749-6632.1997.tb51994.x. discussion 127-119. [DOI] [PubMed] [Google Scholar]
- 28.Mykkanen OM, Gronholm M, Ronty M, Lalowski M, Salmikangas P, Suila H, Carpen O. Characterization of human palladin, a microfilament-associated protein. Mol Biol Cell. 2001;12:3060–3073. doi: 10.1091/mbc.12.10.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otey CA, Dixon R, Stack C, Goicoechea SM. Cytoplasmic Ig-domain proteins: cytoskeletal regulators with a role in human disease. Cell Motil Cytoskeleton. 2009;66:618–634. doi: 10.1002/cm.20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin L. The actin associated protein palladin in smooth muscle and in the development of diseases of the cardiovasculature and in cancer. J Muscle Res Cell Motil. 2011;32:7–17. doi: 10.1007/s10974-011-9246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frolov A, Srivastava K, Daphna-Iken D, Traub LM, Schaffer JE, Ory DS. Cholesterol overload promotes morphogenesis of a Niemann-Pick C (NPC)-like compartment independent of inhibition of NPC1 or HE1/NPC2 function. J Biol Chem. 2001;276:46414–46421. doi: 10.1074/jbc.M108099200. [DOI] [PubMed] [Google Scholar]
- 32.McCauliff LA, Xu Z, Li R, Kodukula S, Ko DC, Scott MP, Kahn PC, Storch J. Multiple Surface Regions on the Niemann-Pick C2 Protein Facilitate Intracellular Cholesterol Transport. J Biol Chem. 2015 doi: 10.1074/jbc.M115.667469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brancaccio D, Gallo A, Mikolajczyk M, Zovo K, Palumaa P, Novellino E, Piccioli M, Ciofi- Baffoni S, Banci L. Formation of [4Fe-4S] clusters in the mitochondrial iron-sulfur cluster assembly machinery. J Am Chem Soc. 2014;136:16240–16250. doi: 10.1021/ja507822j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.