

Abstract
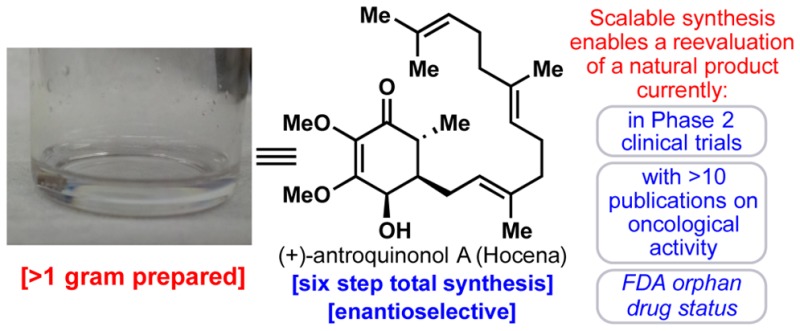
The fungal-derived Taiwanese natural product antroquinonol A has attracted both academic and commercial interest due to its reported exciting biological properties. This reduced quinone is currently in phase II trials (USA and Taiwan) for the treatment of non-small-cell lung carcinoma (NSCLC) and was recently granted orphan drug status by the FDA for the treatment of pancreatic cancer and acute myeloid leukemia. Pending successful completion of human clinical trials, antroquinonol is expected to be commercialized under the trade name Hocena. A synthesis-enabled biological re-examination of this promising natural product, however, reveals minimal in vitro and in vivo antitumor activity in preclinical models.
Antroquinonol A (1, Figure 1A) is a quinone-containing natural product reported to have remarkable medicinal potential in the areas of oncology, immunology, and even diabetes.1 More than ten publications have delineated the exciting activities of 1, and as a consequence, an investigational new drug (IND) application was filed by Golden Biotech Corp. in 2010.2 It is currently in phase II clinical trials (in the USA and Taiwan)3 for the treatment of non-small-cell lung cancer (NSCLC; phase II began in January 2014)3 and has been granted orphan drug status by the FDA for the treatment of pancreatic cancer and acute myeloid leukemia. Given the excitement surrounding this natural product4 coupled with its relative structural simplicity, an effort was launched to pursue its synthesis and biological evaluation as part of the academic–industrial symbiosis between Scripps and Bristol-Myers Squibb (BMS).5 In this communication, an enantioselective, scalable, and modular synthesis of 1 is reported. Access to copious amounts of pure 1 enabled a detailed biological reevaluation that is in contrast to the reported preclinical efficacy of the compound in a nude mouse model harboring human hepatoma xenografts and may temper the enthusiasm surrounding this natural product.
Figure 1.
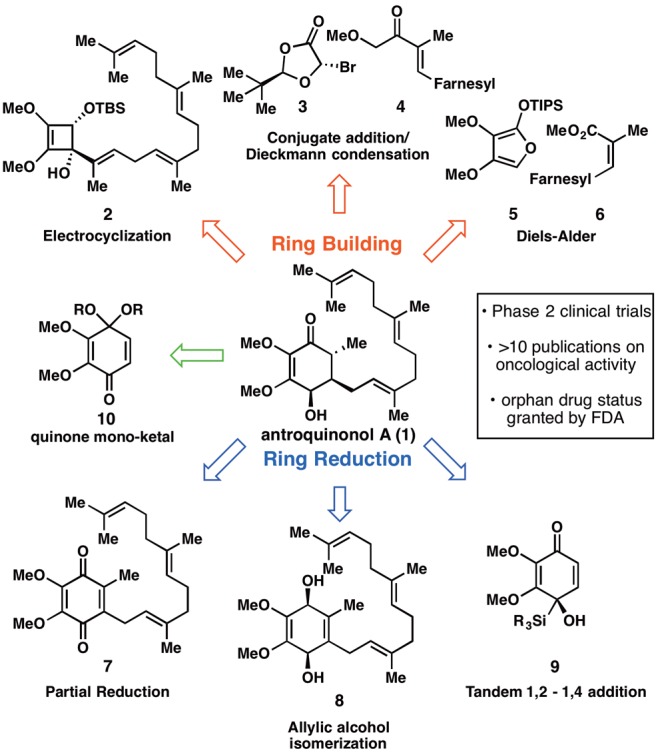
Graphical summary of retrosynthetic analyses explored for the synthesis of antroquinonol A (1).
At the outset of this project, no synthesis of 1 existed and its isolation from the rare Taiwanese fungus Antrodia camphorata was not a practical means for studying pure antroquinonol at BMS.6 A set of plans, ranging in level of ambition and precedent, were evaluated (Figure 1) before settling on the substituted quinone 10 as our final starting point. Six representative blueprints are illustrated and can be divided conceptually into approaches that build the hydroxy enone of 1 via cyclization strategies or via the semireduction of a quinone system. Among the ring-building approaches, Danheiser-type annulation (2),7 stepwise conjugate addition/annulation (3 + 4),8 and Diels–Alder (5 + 6)9 strategies were all pursued, leading either to lengthy sequences, low-yielding key steps, or unstable starting materials (4 and 6 rapidly isomerize). Inspired by the related structure of Coenzyme Q3 (7), extensive efforts to controllably reduce such systems were explored, to no avail.10 Attempted allylic alcohol isomerization11 by utilizing 1,4-diol 8 was not possible due to the inherent instability of such systems (spontaneous aromatization). Finally, efforts centering on a tandem 1,2-/1,4-addition12 to silyl-dienone 9(13) failed due to the tendency of silyl lithium reagents to reduce quinones rather than add to them. Collectively, these failures led us to the simplest possible approach: a conjugate addition to a substituted quinone. Reimagining this quinone as quinone–monoketal 10 gives the starting material “directionality”, allowing selective introduction of the nucleophile and electrophile through a Michael addition and subsequent 1,2-alkylation.14 This strategy inherently makes the synthesis modular, allowing a host of nucleophiles and electrophiles to be quickly appended to any desired quinone starting material.
Scheme 1 depicts the fully optimized six-step, enantioselective, scalable synthesis of antroquinonol A (1). Each step was meticulously studied (see Supporting Information for tables of screened conditions), and as a result, gram quantities of 1 were generated for biological screening. The synthesis commences with the formation of quinon–monoketal 12 from commercial benzaldehyde 11. Baeyer–Villiger and dearomative oxidations followed by a trans-ketalization gave the desired quinone–monoacetal in an overall yield of 64%.15 Extensive screening of the identity of the ketal protecting group (see Scheme 1B) and also of enantioselective conjugate addition conditions16 led to a one-pot procedure that produced the vicinally difunctionalized product 14 with moderate yield and high stereoselectivity. Conjugate addition product 13 was also prepared in order to confirm absolute stereochemistry via X-ray crystallography. L-Selectride proved best in producing the 4,5-cis stereochemistry of the natural product with a dr of 3:1. Hydrolysis turned out to be a significant challenge, with most acidic conditions leading to a complex mixture of elimination products. Montmorillonite K10 clay was a sufficiently mild proton source for hydrolysis of ketal 14, producing enantioenriched (+)-antroquinonol A (1) in an overall yield of 13% and with 96% ee. Over one gram of the natural product has been prepared to date.
Scheme 1. Total Synthesis of (+)-Antroquinonol A (1).

Reagents and conditions: (a) H2O2 (1.0 equiv), H2SO4 (0.1 equiv), MeOH, 0 °C, 2 h (98%); (b) PIDA (2.0 equiv), MeOH, 23 °C, (89%); (c) 2,2-dimethyl-1,3-propanediol (2.5 equiv), PPTS (0.15 equiv), toluene, 60 °C, 1 h (75%); (d) Cu(OTf)2 (0.05 equiv), phosphoramidite ligand SI2 (0.1 equiv), Me2Zn (2.5 equiv), toluene, −30 °C, 18 h; LiHMDS (1.5 equiv), HMPA (2.5 equiv), farnesyl bromide (2.0 equiv), 0 °C, 3 h; (e) L-Selectride (2.5 equiv), toluene, −78 °C to −20 °C, 6 h; (f) Montmorillonite K10 Clay (30 wt %), DCM, 23 °C, 2 h. PIDA = (diacetoxyiodo)benzene, PPTS = pyridinium p-toluenesulfonate, LiHMDS = lithium bis(trimethylsilyl)amide, HMPA = hexamethylphosphoramide, L-Selectride = lithium tri-sec-butylborohydride solution, DCM = dichloromethane.
With synthetic (+)-1 in hand, we began testing the compound against a panel of human tumor cell lines in vitro, previously reported in the literature for the natural product. As outlined in Table 1, natural antroquinonol (1) is reported to have low micromolar activity against the MDA-MB-231 breast, HepG2 hepatocellular, and LNCaP prostate tumor cell lines and low nanomolar activity versus the Hep 3B hepatocellular carcinoma (HCC) cell line.1,17 The reported IC50 value in the lung tumor cell line (A549) for the natural product is 25 μM.2c In our hands, 1 is ∼3–70-fold less cytotoxic against these cell lines except for the A549 cell line, where it is ∼4-fold more potent than previously reported. The activity of 1 in the Hep 3B cell line was particularly striking, where it is significantly less potent when compared to the natural product. To be thorough, the enantiomer of 1, (−)-antroquinonol A, was tested in a similar panel, and, much like the natural product, high micromolar activity was seen for the Hep 3B and A549 cell lines (Table S6). The observed and reported high micromolar activity of 1 against a NSCLC cell line (A549) is somewhat puzzling since the natural product is in phase II trials for the treatment of this tumor type. It was indicated in the package submitted to the FDA for phase I studies that “In vivo study in NOD/SCID mice with A549 subcutaneous xenografts consistently showed tumor growth suppression after 2 weeks of oral 30 and 60 mg/kg antroquinonol treatment.”18 In addition, the natural product is reported to have in vivo activity in a Hep 3B tumor model (vide infra).19 Because of the promising efficacy in this hepatocellular model and the limited in vitro activity against the A549 cell line, we reasoned that an active metabolite(s) could potentially be contributing to the observed in vivo activity seen with 1.
Table 1. Oncology Panel in Vitro Data.
| entry | cell line | IC50 μM reported in the lit.a | IC50 μM for 1b (72 h)c,d | IC50 μM for 15 (72 h)c,e |
|---|---|---|---|---|
| 1 | MDA-MB-231 | 2.6 ± 0.05f | 19 ± 1.6 | >25 |
| 2 | HepG2 | 4.3 ± 0.03f | >25 | >25 |
| 3 | LNCaP | 6.1 ± 0.07f | 22 ± 5.4 | >25 |
| 4 | Hep 3B | 0.13 ± 0.02f | 8.9 ± 2.1 | >25 |
| 5 | PANC-1 (48 h)c | 19g,h | >25 | >25 |
| 6 | AsPC-1 (48 h)c | 20g,h | >25 | >25 |
| 7 | A549 (12 h)c | 25i | 6.7 ± 2.5 | 10.8 ± 5.8 |
| 8 | H441 | 25i | >25e | >25 |
Source of cell line reported in the literature. MDA-MB-231: CCRC-60425. HepG2: BCRC-60025. LNCaP: CCRC-60088. Hep 3B: BCRC-60434. PANC-1: ATCC. AsPC-1: ATCC. A549: KMUH. H441: KMUH.
Source of cell lines reported in this paper: ATCC.
Incubation time.
Triplicate data.
Duplicate data.
Reference (1).
Reference (2f).
SRB assay.
Reference (2c).
When 1 was subjected to metabolite identification/biotransformation studies using human, rat, and mouse liver microsomes, it was rapidly converted to a major metabolite across all three species (Figure 2),20 the structure of which was tentatively assigned as the acid 15 based on MS/MS and 1H NMR data.
Figure 2.
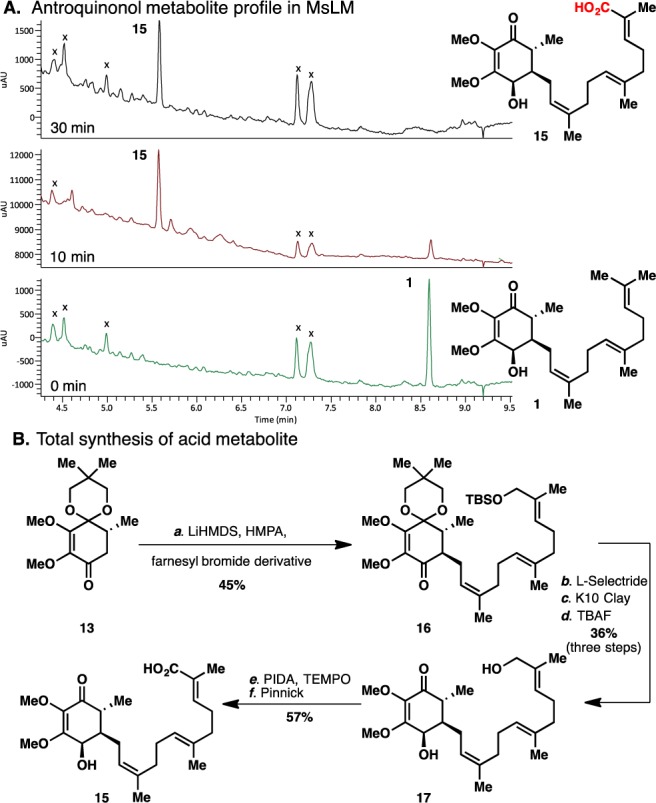
A. Antroquinonol A metabolite (15) profile in MsLM. X = sample matrix peaks, not parent related. B. Reagents and conditions: (a) LiHMDS (1.5 equiv), HMPA (2.5 equiv), allyl bromide (2.5 equiv), THF, 0 °C, 6 h (44%); (b) L-Selectride (2.0 equiv), toluene, −78 °C to −20 °C, 6 h (74%); (c) Montmorillonite K10 clay (30 wt %), DCM, 23 °C, 2 h (49%); (d) TBAF (2.0 equiv), THF, 23 °C, 2 h (99%); (e) PIDA (1.1 equiv), TEMPO (0.2 equiv), THF, 23 °C, 4 h; (f) NaClO2 (7 equiv), NaH2PO4 (7 equiv), 2-methyl-2-butene (20 equiv), t-BuOH, H2O, 23 °C, 12 h (58%). MsLM = mouse liver microsomes.
In order to confirm this structure and produce enough material for biological evaluation, a synthesis of 15 was undertaken. As a testament to the modularity of the synthetic route to 1, acid 15 was easily accessed through an identical strategy. Simply replacing farnesyl bromide with an oxidized derivative, followed by alkylation, reduction, and hydrolysis conditions, furnished antroquinonol analogue 17. Not surprisingly, chemoselective oxidation of the primary alcohol in the presence of the partially reduced quinone core of antroquinonol proved challenging. Employing TEMPO as a catalytic oxidant proved successful.21 Subsequent oxidation of the sensitive aldehyde under Pinnick conditions gave the desired acid metabolite 15, which spectroscopically matched the isolated material from liver microsomes (vide supra).
Compound 15 was subjected to the same tumor cell line panel as 1; however, little to no cytotoxicity was observed.22 Further metabolism studies where 1 was incubated with human, rat, and mouse hepatocytes showed that compound 15 is most likely rapidly oxidized and degraded to a known inactive metabolite (Met2) most likely via mitochondrial β-oxidation (Figure S2).23a−23c
Although the source of the Hep 3B tumor cell lines used in the literature is different from the one described herein (see Table 1 footnote), we decided to conduct an in vivo study with synthetic antroquinonol (1) based on the reported efficacy of the natural product.19 Before embarking on such a study, a pharmacokinetic (PK) study of 1 in mouse was performed to determine the exposure of the compound when dosed either orally (po) or intraperitoneally (ip). A single 50 mg/kg dose of 1 provided the PK profile depicted in Table 2, in which exposures following oral administration were significantly lower compared to those from ip dosing.
Table 2. Mouse Pharmacokinetic Data.
| PK parameters | ip dosinga | po dosinga |
|---|---|---|
| Cmax (nM) | 457 ± 45 | 79 ± 52 |
| Tmax (h) | 4 ± 3 | 2 ± 1 |
| AUClast (nM·h) | 2290 ± 142 | 266 ± 88 |
Corn oil was used as the dosing vehicle.
An in vivo efficacy study was conducted with 1 using Hep 3B HCC tumor xenografts implanted subcutaneously in female NSG mice. Despite literature reports of statistically significant antitumor activity observed with the natural product,2a,19 compound 1 was inactive (<70% tumor growth inhibition, TGI) in our hands when administered daily ip for 14 consecutive days (Figure 3). Paclitaxel served as a positive control in this study and was clearly efficacious (79% TGI) when administered on its optimal preclinical dosing regimen (24 mg/kg, qd×5, iv). Compound 1 was generally well-tolerated with no overt signs of toxicity and minimal weight loss (∼4%).
Figure 3.
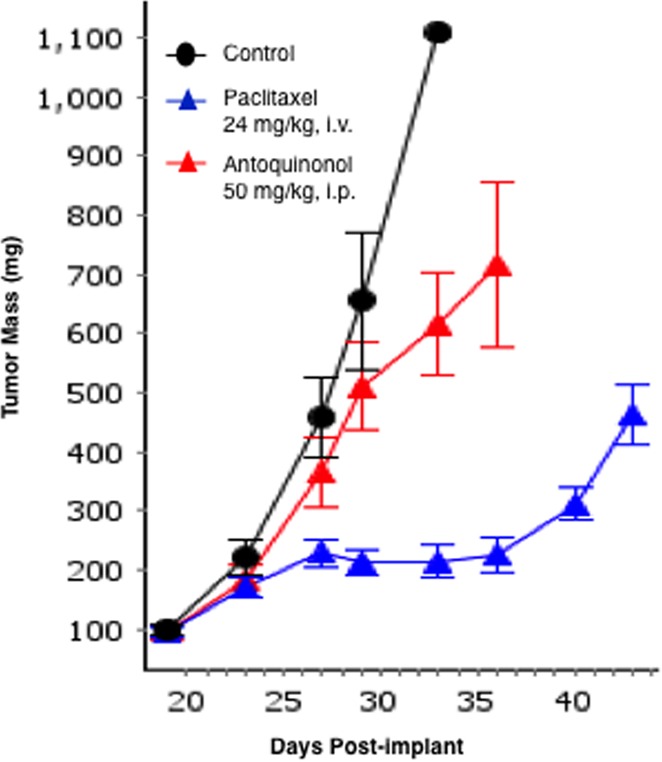
In vivo antitumor activity of synthetic antroquinonol in the Hep 3B HCC xenograft model. Last measurement in the control group includes only one animal, since all other mice in this group had to be euthanized due to severe tumor necrosis. Standard error of the mean (SE) is calculated using the average deviation built-in formula in Quantrix spreadsheets as part of IDBS’s EWorkBook Suite.
In summary, a concise, six-step, scalable, enantioselective synthesis of (+)-antroquinonol (1) has enabled an extensive reinvestigation of some of its reported preclinical biological properties. As outlined in this manuscript, compound 1 demonstrated only micromolar activity in a panel of select tumor cell lines. While the reported Hep 3B cytotoxicity data was intriguing, it was significantly less potent in our panel, therefore it was not unexpected that the compound was inactive in the xenograft model derived from this cell line. The modular synthetic route enabled us to prepare and evaluate a major metabolite identified from biotransformation studies; this compound was inactive and therefore unlikely contributing to the reported in vivo activity. Despite differences in the cell lines used for the in vitro assays24 and the manner in which the in vivo studies were conducted (e.g., the different initial tumor sizes), the lack of efficacy in an in vivo preclinical model would be a cause for concern if the compound were to be advanced into the clinic for treating hepatocellular carcinoma. As indicated earlier, antroquinonol is currently in phase II clinical trials for the treatment of NSCLC. However, the reported in vitro IC50 for antroquinonol A (1) as well as the data with synthetic antroquinonol (Table 1) suggest that the activity of the parent compound 1 for this tumor cell line is in the high micromolar range. Based on the in vitro cytotoxicity data for the A549 cell line, metabolic stability data in microsomes, and mouse PK profile, very high exposures25 or the presence of a yet unknown active metabolite may be necessary for 1 to show efficacy in treating NSCLC in the clinic.
Acknowledgments
Financial support for this work was provided by NSF (predoctoral fellowship to M.T.V.) and George E. Hewitt Foundation (postdoctoral fellowship to E.S.). The authors thank Prof. Arnold L. Rheingold and Dr. Curtis E. Moore for X-ray crystallographic analysis. We thank Dr. Joel C. Barrish for useful discussions, Sarah Traeger for NMR analysis of the metabolite, Robin Moore, Paul Elzinga, and Georgia Cornelius for PK support, and Christopher Mulligan for in vivo support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00345.
Author Contributions
‡ M.T.V. and E.S. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Isolation and structural determination of antroquinonol A (1):Lee T.; Lee C.; Tsou W.; Liu S.; Kuo M.; Wen W. Planta Med. 2007, 73, 1412. 10.1055/s-2007-990232. [DOI] [PubMed] [Google Scholar]
- Biological activity studies of 1:; a Chiang P. C.; Lin S. C.; Pan S. L.; Kuo C. H.; Tsai I. L.; Kuo M. T.; Wen W. C.; Chen P.; Guh J. H. Biochem. Pharmacol. 2010, 79, 162. 10.1016/j.bcp.2009.08.022. [DOI] [PubMed] [Google Scholar]; b Kumar K. J.; Chu F. H.; Hsieh H. W.; Liao J. W.; Li W. H.; Lin J. C.; Shaw J. F.; Wang S. Y. J. Ethnopharmacol. 2011, 136, 168. 10.1016/j.jep.2011.04.030. [DOI] [PubMed] [Google Scholar]; c Kumar V. B.; Yuan T. C.; Liou J. W.; Yang C. J.; Sung P. J.; Weng C. F. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2011, 707, 42. 10.1016/j.mrfmmm.2010.12.009. [DOI] [PubMed] [Google Scholar]; d Tsai P. Y.; Ka S. M.; Chao T. K.; Chang J. M.; Lin S. H.; Li C. Y.; Kuo M. T.; Chen P.; Chen A. Free Radical Biol. Med. 2011, 50, 1503. 10.1016/j.freeradbiomed.2011.02.029. [DOI] [PubMed] [Google Scholar]; e Tsai P. Y.; Ka S. M.; Chang J. M.; Lai J. H.; Dai M. S.; Jheng H. L.; Kuo M. T.; Chen P.; Chen A. Arthritis Rheum. 2012, 64, 232. 10.1002/art.33328. [DOI] [PubMed] [Google Scholar]; f Yu C. C.; Chiang P. C.; Lu P. H.; Kuo M. T.; Wen W. C.; Chen P.; Guh J. H. J. Nutr. Biochem. 2012, 23, 900. 10.1016/j.jnutbio.2011.04.015. [DOI] [PubMed] [Google Scholar]; g Yang S. M.; Ka S. M.; Hua K. F.; Wu T. H.; Chuang Y. P.; Lin Y. W.; Yang F. L.; Wu S. H.; Yang S. S.; Lin S. H.; Chang J. M.; Chen A. Free Radical Biol. Med. 2013, 61, 285. 10.1016/j.freeradbiomed.2013.03.024. [DOI] [PubMed] [Google Scholar]; h Chen C. K.; Kang J. J.; Wen W. C.; Chiang H. F.; Lee S. S. J. Nat. Prod. 2014, 77, 1061. 10.1021/np400670a. [DOI] [PubMed] [Google Scholar]; i Ho C. L.; Wang J. L.; Lee C. C.; Cheng H. Y.; Wen W. C.; Cheng H. H.; Chen M. C. Biomed. Pharmacother. 2014, 68, 1007. 10.1016/j.biopha.2014.09.008. [DOI] [PubMed] [Google Scholar]; j Hsu C. Y.; Sulake R. S.; Huang P. K.; Shih H. Y.; Sie H. W.; Lai Y. K.; Chen C.; Weng C. F. Br. J. Pharmacol. 2015, 172, 38. 10.1111/bph.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Lee W. T.; Lee T. H.; Cheng C. H.; Chen K. C.; Chen Y. C.; Lin C. W. Food Chem. Toxicol. 2015, 78, 33. 10.1016/j.fct.2015.01.012. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT02047344.
- GoldenBiotech’s new cancer drug Hocena has been awarded the “Research and Development Innovation Award” in Taipei Biotech Awards 2014. http://www.goldenbiotech.com.tw/en/newslist.html, accessed on July 18, 2015.
- Michaudel Q.; Ishihara Y.; Baran P. S. Acc. Chem. Res. 2015, 48, 712. 10.1021/ar500424a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Three total syntheses of 1 were reported while this manuscript was in preparation:; a Hsu C. S.; Chou H. H.; Fang J. M. Org. Biomol. Chem. 2015, 13, 5510. 10.1039/C5OB00411J. [DOI] [PubMed] [Google Scholar]; b Sulake R. S.; Chen C. Org. Lett. 2015, 17, 1138. 10.1021/acs.orglett.5b00046. [DOI] [PubMed] [Google Scholar]; c Sulake R. S.; Lin H. H.; Hsu C. Y.; Weng C. F.; Chen C. J. Org. Chem. 2015, 80, 6044. 10.1021/acs.joc.5b00345. [DOI] [PubMed] [Google Scholar]
- a Magomedov N. A.; Ruggiero P. L.; Tang Y. J. Am. Chem. Soc. 2004, 126, 1624. 10.1021/ja0399066. [DOI] [PubMed] [Google Scholar]; b Matsumoto T.; Hamura T.; Miyamoto M.; Suzuki K. Tetrahedron Lett. 1998, 39, 4853. 10.1016/S0040-4039(98)00920-4. [DOI] [Google Scholar]
- Beckwith A. L. J.; Chai C. L. L. Tetrahedron 1993, 49, 7871. 10.1016/S0040-4020(01)88012-1. [DOI] [Google Scholar]
- Brailsford J. A.; Lauchli R.; Shea K. J. Org. Lett. 2009, 11, 5330. 10.1021/ol902173g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kündig E. P.; Enriquez-Garcia A. Beilstein J. Org. Chem. 2008, 4, 37. 10.3762/bjoc.4.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilli L.; Gerard D.; Torche S.; Besnard C.; Mazet C. Angew. Chem., Int. Ed. 2009, 48, 5143. 10.1002/anie.200901863. [DOI] [PubMed] [Google Scholar]
- Solomon M.; Jamison C. L.; McCormick M.; Liotta D. J. Am. Chem. Soc. 1988, 110, 3702. 10.1021/ja00219a079. [DOI] [Google Scholar]
- Koreeda M.; Koo S. Tetrahedron Lett. 1990, 31, 831. 10.1016/S0040-4039(00)94639-2. [DOI] [Google Scholar]
- Meister A. C.; Sauter P. F.; Brase S. Eur. J. Org. Chem. 2013, 2013, 7110. 10.1002/ejoc.201300752. [DOI] [Google Scholar]
- Pirrung M. C.; Nunn D. S. Tetrahedron Lett. 1992, 33, 6591. 10.1016/S0040-4039(00)60993-0. [DOI] [Google Scholar]
- Imbos R.; Brilman M. H. G.; Pineschi M.; Feringa B. L. Org. Lett. 1999, 1, 623. 10.1021/ol990707u. [DOI] [Google Scholar]
- The IC50 values for the natural product against the Hep3B and HepG2 carcinoma cell lines are reported as 0.13 ± 0.02 μM and 4.3 ± 0.03 μM respectively in ref (1), as indicated in Table 1. The GI50′ values for 1 against the same cell lines are listed as >30 μM and 0.22 μM respectively in ref (2a). The cytotoxicity assay protocol used in this paper and ref (1) employs 10% FBS and a 72 h incubation time with compound (antroquinonol) using MTS or MTT stain. The protocol outlined in ref (2a) employs 5% FBS, 48 h incubation time, and an SRB stain. It is clear from the discussion (vide supra) that the protocol employed in this paper to determine cytotoxicity closely mirrors the conditions used in ref (1).
- ClinicalTrials.gov Identifier: NCT01134016.
- For discussion regarding the in vivo activity see ref (2a), page 170, left column, third paragraph. See supplementary material of ref (2a) for in vivo data. Initial tumor sizes were not identical: 50–70 mm3 (lit.)2a vs 75–100 mm3 (BMS).
- Data shown for mouse liver microsomes.
- De Mico A.; Margarita R.; Parlanti L.; Vescovi A.; Piancatelli G. J. Org. Chem. 1997, 62, 6974. 10.1021/jo971046m. [DOI] [Google Scholar]
- The Caco-2 value for 15 (Pc A → B, 62 nm/s, and B → A, 63 nm/s) suggests that cell permeability may not be an issue with 15.
- a Met2 (Figure S2; structure based on MS–MS data) is a reported metabolite of antroquinonol; b Met2 is inactive in the H838 tumor cell line (IC50 > 100 μM vs antroquinonol IC50 of ∼ 3 μM).Ho C.-L.; Wang J.-L.; Lee C.-C.; Cheng H.-Y.; Wen W.-C.; Cheng H. H-Y.; Chen M. C-M. Biomed. Pharmacother. 2014, 68, 1007. 10.1016/j.biopha.2014.09.008. [DOI] [PubMed] [Google Scholar]; c Synthetic antroquinonol appears to have a relatively better in vitro metabolic stability in human hepatocytes compared to human liver microsomes.
- A thorough investigation of the antiproliferative activity against a large panel of human tumor cell lines was not performed.
- Antroquinonol is being administered to patients at 200 mg tid in the ongoing phase II clinical trial (ref (3)).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.