

Abstract
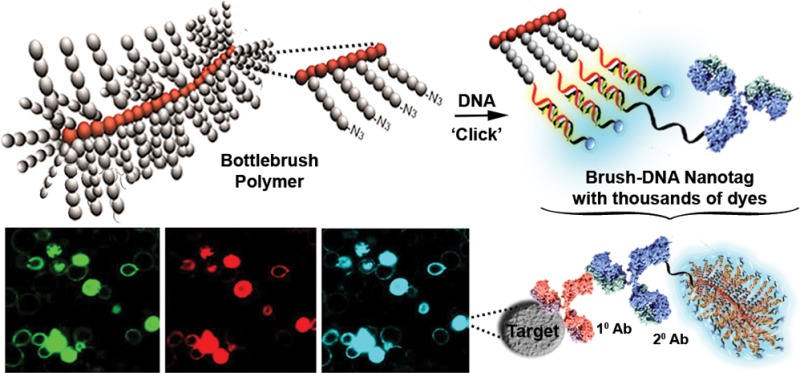
Bright signal outputs are needed for fluorescence detection of biomolecules at their native expression levels. Increasing the number of labels on a probe often results in crowding-induced self-quenching of chromophores, and maintaining the function of the targeting moiety (e.g., an antibody) is a concern. Here we demonstrate a simple method to accommodate thousands of fluorescent dye molecules on a single antibody probe while avoiding the negative effects of self-quenching. We use a bottlebrush polymer from which extend hundreds of duplex DNA strands that can accommodate hundreds of covalently attached and/or thousands of noncovalently intercalated fluorescent dyes. This polymer–DNA assembly sequesters the intercalated fluorophores against dissociation and can be tethered through DNA hybridization to an IgG antibody. The resulting fluorescent nanotag can detect protein targets in flow cytometry, confocal fluorescence microscopy, and dot blots with an exceptionally bright signal that compares favorably to commercially available antibodies labeled with organic dyes or quantum dots.
Short abstract
Here we demonstrate a simple method to accommodate thousands of fluorescent dye molecules on a single antibody probe while avoiding the negative effects of self-quenching. We use a bottlebrush polymer from which extend hundreds of duplex DNA strands that can accommodate hundreds of covalently attached and/or thousands of noncovalently intercalated fluorescent dyes. The fluorescent bottlebrush polymer is then attached to an antibody via DNA hybridization, resulting in an exceptionally bright immunofluorescent label.
Introduction
Virtually every imaginable aspect of biological systems has succumbed to labeling through fluorescent probes that have been developed over the years.1a,1b Fluorescent dyes coupled to affinity binders such as antibodies are common reporters in fluorescence microscopy, flow cytometry, and microplate assays as well as in protein and nucleic acid blots.2a−2d Despite the advent of competing approaches such as recombinant peptide tagging and mass spectrometry, antibody-based detection remains the most broadly applicable means of localizing and quantitating specific components in a complex sample.3a−3c Labeled secondary antibodies make stable and specific complexes with unlabeled primary antibodies, providing the foundation for most immunofluorescence protocols.
The number of target molecules per surface area or volume unit is a key variable in biological detection applications. To detect functionally important proteins with a natural low expression level, there remains a need to enhance the detectable signal.4a,4b The most straightforward way to enhance fluorescence signals is to increase the number of fluorophores available for detection.5 In one approach, signal amplification methods can be used to get brighter signals. For example, in catalytic reporter deposition (CARD) technology, the high turnover rate of enzymes such as horseradish peroxidase and alkaline phosphatase generate high density, in situ labeling of a target protein or nucleic acid.6a−6f Hence in both immunohistochemical and immunoassay applications, careful control of timing is essential to obtaining quantitative and reproducible results. To avoid these potential limitations of amplification methods, an alternative is to increase the number of labels directly attached to affinity binders. A typical IgG antibody molecule has about 90 lysine residues, of which at most 30 can be modified under forcing conditions.7 However, maintenance of functional properties typically requires a degree of labeling of less than 10 dyes per IgG, representing a low fraction of modification with individual fluorescent dyes. For example, antibodies labeled with more than four to six fluorophores per protein can exhibit reduced specificity and binding affinity.8 Furthermore, with higher degrees of substitution, the fluorescence obtained per added fluorophore is typically much lower than expected, due to self-quenching by nearby fluorophores.9
The use of soluble and relatively stable fluorescent proteins such as the phycobiliproteins, conjugated to antibodies, could overcome the limitations arising from the high loading of low molecular weight dyes.10 On a molar basis, the fluorescence yield of a phycobiliprotein is equivalent to at least 30 unquenched fluorescein or 100 rhodamine molecules at comparable wavelengths. On the other hand, fluorescent polystyrene microspheres heavily loaded with fluorescent dyes have been used as immunofluorescent reagents to deliver strong signals.11a−11c
Here we show that limitations in a high loading of fluorescent molecules in a label can be overcome through a simple DNA–polymer macromolecular scaffold. This DNA–polymer scaffold can incorporate thousands of fluorescent dyes and can be attached to a single antibody to give an intense fluorescent signal that compares favorably with current immunofluorescence technology. The macromolecular scaffold is based on a polymeric core with bottlebrush architecture and is functionalized with hundreds of DNA duplexes attached to the tips of the bottlebrush “bristles” on which can be assembled both covalent and intercalated fluorescent dyes. While synthetic tools that make use of polymeric structures with compact but flexible brush-like architectures are gaining wide use,12a−12h we further enhance the bottlebrush polymer functionality by readily grafting nucleic acids to the bottlebrush side chains. These nucleic acids can host thousands of fluorescent dyes leading to exceptionally bright labels with highly tunable colors for biological imaging and detection.13a−13c
Results and Discussion
The methods of atom transfer radical polymerization (ATRP) have yielded rich and diverse polymer architectures.14a−14d For a scaffold that could display a large and functionalizable array, we chose a bottlebrush polymer (BBP) with reactive azide groups at the tips of the bristles (Figure 1a). Full synthetic details and characterization data are provided in Supporting Information. To obtain the BBP core, a poly[2-(2-bromoisobutyryloxy)ethyl methacrylate] (PBiBEM) macroinitiator with a degree of polymerization of 400 (m′ + m″) was used as the backbone.15 From this PBiBEM core, approximately half of the bromoisobutyrate groups initiated the side chains, leading to grafting of 200 (m″) side chains with 2-(2-(2-methoxyethoxy)ethoxy)ethyl methacrylate (MEO3MA) monomeric units. The 50% initiation efficiency was calculated based on the content of the azide groups; the lack of complete initiation can be attributed to the large size of the monomer, particularly the long PEO substituent. The degree of polymerization of side chains was calculated based on absolute molecular weight of the bottlebrush measured by light scattering (Mn = 8.6 × 106) yielding n = 180.16 The bromides at the side chain termini were then substituted with azides to provide the BBP-N3. Clickable bottlebrush polymers prepared through other methods have been described,17 though here there is a greater density of side chains for functionalization with a 5′-hexynyl modified DNA oligonucleotide (hexynyl-A; red strand in Figure 1a), which was grafted to the side chain termini via a Cu(I)-catalyzed azide alkyne cycloaddition (“click”) reaction. Our pseudoligandless click conditions are optimized for nucleic acid conjugations in a buffered solution (phosphate buffered saline (PBS), pH 7.5) with acetonitrile as a minor cosolvent.18a,18b Three hours of reaction time was sufficient for near quantitative reaction of the DNA with the BBP, as followed by the disappearance of the azide peak at 2110 cm–1 in aqueous phase FTIR (e.g., Figure S2). Simple purification using molecular weight cutoff filters provided the DNA-conjugated polymer, A-BBP, with about 200 DNA strands per brush (see Supporting Information). Annealing of the complementary strand, Acomp (black strand in Figure 1a) completed the formation of the full DNA-BBP (DBBP) scaffold, A/Acomp-BBP.
Figure 1.

A bottlebrush polymer with DNA bristles as a scaffold for fluorescent dyes. (a) Straightforward assembly from a bottlebrush polymer (BBP; m′ = m″ = 200, n = 180) with azide side chain terminii by CuAAC or click reaction with hexynyl-DNA (red strand) results in single-stranded A-BBP. Annealing of the complementary strands (Acomp; black) gives hundreds of base pairs for the intercalation of YOYO-1 (yellow) in double-stranded DNA bottlebrush polymer (DBBP). Inset shows the structure of the BBP-azide. (b) Graph of increasing fluorescence intensities (λex = 488 nm) with increasing YOYO-1 up to 5 equiv relative to DNA (c) Graph of the fluorescence emission at 510 nm versus YOYO-1 equivalents relative to DNA concentration shows the saturation of the DBBP with YOYO-1 occurs around 5 equiv as expected (18 bp duplex, 4 bp per bis intercalator. See Supporting Information for synthesis and characterization of the BBP and sequences of DNAs used).
With about 200 double-stranded DNAs (dsDNA) per brush and 18 base pairs per dsDNA, this DBBP can accommodate ca. 1800 intercalating fluorescent dyes. For the DNA intercalating fluorescent dye, we chose YOYO-1, a commercially available bis-intercalating dimer of oxazole yellow19 that has been useful in our previous nanotag designs.13a,13b Titrating YOYO-1 into the A/Acomp-BBP scaffold and monitoring the fluorescence intensity at 510 nm revealed a monotonic increase (Figure 1b,c) with saturation at approximately five YOYO-1 dimers per 18 bp dsDNA (arrow in Figure 1c), as expected for a bis-intercalator having a four base-pair binding site. When bound to dsDNA, YOYO-1 has an extinction coefficient of 98 900 M–1 cm–1 at 491 nm. Given that there are, on average, 200 dsDNAs per brush and five YOYO-1 dimers per dsDNA, the extinction coefficient of the YOYO-1 loaded DBBP scaffold (A/Acomp[YOYO-1]-BBP) reaches ∼108 M–1 cm–1.
It is often desirable to red shift the emission wavelength away from the excitation wavelength in order to minimize interference from background autofluorescence, particularly in biological samples. Wavelength shifting can be achieved by allowing the light-absorbing chromophores (donors) to transfer the excitation energy to lower energy acceptor chromophores that can fluoresce at a longer wavelength. To incorporate a wavelength shifting property to our DBBP scaffold, we designed the complementary strand with a terminal acceptor Cy5 dye (Cy5-Acomp). Hybridization of this strand to the covalently attached complement introduces ca. 200 Cy5 acceptor dyes to the DBBP (Figure 2a).
Figure 2.

Wavelength shifting via energy transfer (ET). (a) Schematic of energy transfer pathways from intercalated YOYO-1 to terminal Cy5 acceptors. Both intra (blue) and inter (gray) duplex pathways are possible in a DBBP system, whereas only intraduplex ET is possible in nonbrush DNA. (b) Brush-immobilized DNA shows higher ET (left spectra) compared to nonbrush DNA in solution (right spectra). The red and blue traces correspond to DBBPs that include or exclude, respectively, covalent terminal Cy5 dyes on all the hybridized DNA strands. The ET is calculated based on the decrease in YOYO-1 fluorescence at 510 nm (cyan arrow); see Methods. (c) Comparison of brush (black curves) and nonbrush (red curves) DNA at different excitation wavelengths. Left spectra: Excitation at 633 nm (direct excitation of C5). Similar intensities indicate lack of self-quenching of Cy5 in brush. Right spectra: Excitation at 450 nm (excitation of YOYO-1). Higher Cy5 emission in brush is due to higher ET efficiency.
Prior work on nanostructured DNA systems demonstrated efficient energy transfer (ET) between YOYO-1 and Cy5.13a,13b In the DBBP context, ET is clearly observed based on a decrease in YOYO-1 emission at 510 nm and the appearance of sensitized Cy5 emission at 665 nm (Figure 2b, left spectra). On the basis of the quenching of YOYO-1 emission in the presence of Cy5, we estimate the ET efficiency to be 52%. The emission spectrum of a control sample with the same concentration of the A/Cy5-Acomp dsDNA free in the buffered solution (i.e., nonbrush DNA) shows ET but at a somewhat lower efficiency compared to the DBBP (Figure 2b, right spectra). Although the dsDNA sequences are identical, their three-dimensional arrangement and localization around the polymeric core of the DPPB likely lead to the enhanced ET efficiency through an interduplex mechanism, which is not possible for the dsDNA in dilute solution.
It should be noted that even though the DBBP contains about 200 Cy5 dyes at the tips of the brush bristles, comparison of direct excitation of Cy5 in the DBBP versus free DNA duplex revealed no significant difference in Cy5 fluorescence intensity, indicating a lack of self-quenching even within the relatively dense environment of the DBBP (Figure 2c, left). This could result from stacking of Cy5 dyes on the ends of their respective duplexes,21 which would suppress quenching due to interaction between Cy5 dyes on adjacent duplexes. In contrast, when the samples are excited in the YOYO-1 absorption band (Figure 2c, right), significantly stronger Cy5 emission is observed for the DBBP, due to the more efficient energy transfer in the polymeric context.
While the ease of assembly of intercalating dyes in a DNA-based scaffold is appealing, the dissociation of dyes from the intercalating sites can lead to reduced brightness and nonspecific staining. For example, Benvin et al. reported that bisintercalating YOYO-1 readily dissociates from a DNA three-way junction in the presence of excess calf thymus (CT) DNA.13a We therefore tested the stability of our DBBP nanotag by adding a 4-fold excess of CT DNA (in base pairs). In this experiment, DBBP-bound YOYO-1 can undergo ET to nearby Cy5 dyes, so dissociation of the intercalator with subsequent binding to unlabeled CT DNA should result in a decrease in ET. This is observed for a control experiment, in which YOYO-1 was prebound to a free (i.e., nonbrush) Cy5-labeled DNA duplex and then mixed with excess CT DNA. An immediate drop in ET upon addition of CT DNA was observed, consistent with rapid dissociation of the intercalator from the free DNA duplex (Figure 3, left spectra).
Figure 3.

High density of DNA on the DBBP retains assembled intercalated dyes. Fluorescence and ET before and after addition of calf thymus (CT) DNA (black and red traces, respectively) in four times excess of A/Cy5-Acomp/YOYO-1/DNA either free in solution (left spectra), on brush (center spectra) or in a brush with lower (40%) DNA loading and PEO chains (right spectra). While intercalated YOYO-1 in free or nonbrush DNA dissociates and migrates to CT DNA resulting in an immediate drop in fluorescence due to ET (green arrow), the fluorescence from brush-DNA shows no such drop even after 15 h as the high density of DNA on the brush prevents dissociation of positively charged YOYO-1 into the CT DNA. In the brushes with lower DNA density, the dyes can dissociate and migrate to CT DNA resulting in an immediate drop in ET.
In stark contrast, when CT DNA was added to the YOYO-1 loaded DBBP, the ET signal remains unchanged, even after 15 h, indicating that the polymeric scaffold with dsDNA retains the intercalators (Figure 3, middle spectra). We attribute the strong retention of YOYO-1 by the DBBP to the electrostatic attraction between the tetracationic intercalating dye and the relatively dense polyanionic DNA assembly, as opposed to free DNA, where the negative charge density of the Cy5-labeled duplex is equivalent to that of the unlabeled CT-DNA competitor. To provide support for this hypothesis, we designed a scaffold with a reduced loading of DNA (40%, relative to the original DBBP). To mimic the density of strands extending from the polymeric core, we also used an alkyne-terminated poly(ethylene oxide) (PEO) to react with the remaining azide groups (60%) of the BBP during the click conjugation reaction (Figure S11). The completion of the reaction of the azide groups was verified by aqueous phase FTIR (Figure S3). After preloading with YOYO-1 and mixing with CT-DNA, an immediate drop in ET was observed (Figure 3, right spectra). This is consistent with our hypothesis that the greater negative charge density on the brush that is fully modified with DNA plays a significant role in sequestering YOYO-1 against dissociation. More broadly, the ability of the DBBP to noncovalently trap a DNA-binding small molecule suggests other applications of these materials as, e.g., drug delivery vehicles.
We sought to exploit dye-loaded DBBPs as fluorescent nanotags for labeling applications. We therefore conjugated a DBBP to a secondary IgG antibody for specific targeting. Functionalization of secondary antibodies is appealing because a single secondary antibody can be used with many different primary antibodies generated against diverse antigens. In prior work we have shown that DNA hybridization can be used to generate protein-polymer hybrids.20 Furthermore, we demonstrated that an antibody–DNA nanotag conjugate could be constructed through hybridization of dye-labeled DNA strands to a secondary antibody labeled with the complementary DNA strand.13c We used this approach so that a DNA strand on the DBBP could be used to hybridize to a complementary strand covalently conjugated to an antibody. The antibody was functionalized with an average of one DNA strand per antibody through a bisaryl hydrazone linkage (Figures S4 and S5). To favor hybridization of a single antibody to each brush, we synthesized a DBBP in which two different DNA sequences were clicked onto the bristles. Using proportionally 0.5% of one DNA sequence (“B”) along with the previous sequence A during the click reaction gave, on average, a single copy of sequence B on each brush (as each brush has 200 azides available for functionalization). This separate sequence B is fully complementary to the antibody-conjugated DNA sequence (B′). The remaining (199) DNA sequences on the brush were hybridized as previously described to the Cy5 bearing complementary strand, Cy5-Acomp, to give the final DBBP-nanotag (Figure 4a).
Figure 4.

A nanotag based on antibody tethered to the DBBP (a) A DBBP with a single specific sequence B can be hybridized to Cy5-Acomp and antibody bearing fully complementary B′comp strand and loaded with YOYO-1. The single sequence B per brush is to ensure one antibody per brush. (b) The c-myc protein detection system using primary and secondary antibody interactions where the tag (X) is on the secondary antibody. (c) Flow cytometry data shows high fluorescent brightness of the brush nanotag compared to other commercially available antibody tags. (d) Confocal microscopic images show the exceptional brightness of the nanotag antibodies compared to Alexa 647 and QD655 tagged antibodies. (e) Detection of maltose binding protein (MBP) using the primary and secondary antibody system (left) and the visual confirmation of the specificity and brightness of DBBP nanotags via dot blots. The nanotag antibodies allow visualization with at least an order of magnitude greater sensitivity. See Supporting Information for additional imaging and quantitation.
This DBBP nanotag was tested on yeast cells that expressed on their surface a single chain variable fragment (scFv) protein bearing a c-Myc binding epitope. After incubation with a primary antibody specific for the c-Myc epitope, the cells were stained with the corresponding secondary IgG antibody hybridized to the fluorescent DBBP (Figure 4b) for analysis by flow cytometry and microscopy. Commercially available Alexa 647 and Quantum Dot (QD) 655 tagged secondary IgGs were used as controls.
The ability of the antibody-tagged DBBP to stain yeast cells is clearly demonstrated by the flow cytometry histograms shown in Figure 4c. Data shown in the left and right panels were obtained from excitation at 488 nm (i.e., YOYO-1) or 633 nm (Cy5). For excitation at 488 nm, the bottom histogram corresponds to unlabeled yeast and sets the background signal. Samples stained with Alexa 647 or QD 655 (top two histograms) also represent negative controls because they do not absorb/emit at 488/530 nm. Yeast stained with the DBBP-conjugated antibody gave strong signals (green and orange histograms; the multimodal shape of the histograms arises from populations of yeast that express varying levels of the Myc epitope-tagged scFv even after induction). Comparing the two histograms reveals a slight shift to lower intensity for the DBBP labeled with Cy5 acceptor dyes, reflecting the lower YOYO-1 emission due to energy transfer. Meanwhile, excitation/emission at 633/695 nm shows background-only signal from yeast alone (red), DBBP without Cy5 (which lacks absorbance in the red) and, surprisingly, Alexa 647, which absorbs strongly at 633 nm. We hypothesize that the lack of signal from the Alexa 647-labeled antibody is due to our use of this reagent at 10-fold lower concentration than is recommended by the manufacturer. The QD 655-labeled antibody (blue histogram) clearly stains the yeast cells, although staining by the DBBP+Cy5 antibody is more than 10-fold stronger, based on mean intensity. The superior performance of the DBBP+Cy5 antibody is most likely due to the ca. 10-fold higher extinction coefficient of 199 Cy5 dyes versus 1–2 QDs per antibody. (We note that 633 nm is not the optimal wavelength for exciting QD 655; i.e. the difference in performance for the two types of antibody conjugates will depend on the excitation wavelength.) In addition to demonstrating the significantly higher brightness of DBBP-conjugated antibodies, these results demonstrate that, although a rather large polymer brush DNA conjugate (120 × 20 nm) is tethered to the antibody, antigen binding is not hindered, perhaps because of the low loading (one DBBP per antibody). Furthermore, while control over the extent of functionalization of the antibody was optimized, the conjugation was not directed to a specific residue on the protein. With emerging methods for greater control and specific attachment of DNA to antibodies,22 the hybridization method could be even more powerful for the development of probes.
We also performed confocal microscopy on the yeast (Figure 4d). Samples were analyzed at three excitation/emission wavelengths: (i) 488/530 (YOYO-1), (ii) 633/695 (Cy5), and (iii) 488/695 (ET). The images shown in Figure 4d reinforce the flow cytometry results. 488/530 gives strong signal for the DBBP-tagged antibody due to residual YOYO-1 fluorescence that was not quenched by ET to Cy5. 633/695 gives strong signal for both QD655 and the DBBP, whereas the Alexa 647-labeled antibody fails to stain the cells. We also observe labeling by both the QD and DBBP-labeled antibodies in the energy transfer channel. Although ET is not occurring in the quantum dot, the broad absorption spectrum of QDs allows excitation in the blue. Two additional control experiments were performed to confirm the specificity of labeling. First, yeast cells that were not induced to express the scFv antigen exhibited no detectable staining (Figure S6). Second, DBBPs lacking the single copy of sequence B; i.e., the complement to the oligonucleotide attached to the secondary antibody also failed to stain yeast cells (Figure S7). These control experiments demonstrate that the DBBP nanotags do not give rise to nonspecific staining of the cells. Finally, as shown in Figures S8–S10, similar labeling experiments were also performed using biotin-coated polystyrene beads. In both flow cytometric and confocal imaging experiments, compared to the commercially available probes, the DBBP nanotags again were an order of magnitude brighter.
We extended these experiments to dot blots, a widely used format for screening and detection of specific biomolecules. This represents a simplification of the Northern blot, Southern blot, or Western blot methods. To evaluate the efficiency of the DBBP nanotag in this method, we used maltose binding protein (MBP) as a target. MBP is part of the maltose/maltodextrin system of Escherichia coli, which is responsible for the uptake and efficient catabolism of maltodextrins. We chose an anti-MBP monoclonal primary antibody sandwiched between the MBP target blotted on a nitrocellulose paper, with the DBBP nanotag as the detection system (Figure 4e). Increasing amounts of MBP blotted on the paper could be detected with the DBBP nanotag, as shown by increasing density of the spots in Figure 4e. The sample was excited at either 473 nm (YOYO-1) or 650 nm (Cy5 or Alexa 647), and the emission was recorded at 695 nm. At both excitation wavelengths, as little as 0.5 ng of MBP could be detected by eye for the DBBP-conjugated antibody. The observation of discrete spots for excitation of YOYO-1 indicates that the dye remains sequestered by the DNA within the polymer brush, instead of nonspecifically staining the filter paper, since YOYO-1 that released from the DNA scaffold would be incapable of transferring energy to Cy5 dyes that remained attached to the DBBP through hybridization. In separate experiments using an antibody conjugated to a DNA tetrahedron nanotag, YOYO-1 was not retained by the antibody, leading to strong nonspecific background fluorescence from the paper (data not shown). An experiment performed under similar conditions with an Alexa 647-tagged secondary antibody gave a detection limit of 5 ng of MBP protein, i.e., at least 10-fold lower sensitivity. The DBBP nanotag is thus a versatile and bright multichromophore system for labeling applications.
Conclusions
We demonstrate here the use of a DNA-conjugated bottlebrush polymer as a brightly fluorescent nanotag that is useful in applications for sensitive detection of proteins both in blots and directly on cell surfaces. In particular we show that the polymer brush architecture provides the means to graft DNA side chains at a high local density. The conjugation of DNA to the polymeric scaffold is readily achieved via the high efficiency Cu(I) promoted azide–alkyne cycloaddition reaction with a simple filtration to purify the brush-DNA conjugate. These brush-localized DNA bristles provide a dense scaffold for intercalating fluorescent dyes such as YOYO-1, generating extinction coefficients as high as ∼108 M–1 cm–1. Importantly, the intercalated YOYO-1 dyes are retained on the DBBP scaffold without dissociation, avoiding complications from nonspecific fluorescence. This property eliminates the need for obligatory covalent conjugation of dyes and tremendously eases the assembly of >1000 fluorescent dyes via simple mixing. The approach of using DNA bristles on the polymer brush also provides access to scaffolds with the same donor chromophore but with additional and different terminal acceptor dyes at the end of the DNA strands. The ability to excite each fluorescent scaffold at a single wavelength while monitoring emission in different channels would enable multiplex detection schemes involving antibodies labeled with unique DBBPs, similar to what is already possible with quantum dot technology, although with substantially brighter signal.
The high degree of control over the DNA sequences on the brush allows us to tailor specific sequences for attaching targeting agents through simple hybridization and without any further modification to the generic scaffold design. In the example reported here, conjugation of the relatively large DBBP to a secondary antibody through DNA hybridization does not hinder the antibody’s binding. We show that these versatile DBBP nanotags on antibodies provide probes that are at least 10 times brighter than commercially available QD or Alexa fluor-tagged IgG antibodies under identical conditions. By simply lengthening the DNA sequences (to provide more intercalation sites) or the number of DNA strands on the brushes, it should be possible to increase the chromophore content of the antibody nanotags even further. We envision that using other targeting agents such as an internalizing antibody or aptamer will potentially expand the technology to detect targets in intracellular compartments or for other types of in vitro detection assays. Furthermore, the design of the scaffold not only allows attachment of fluorescent dyes but also provides an attractive compartment in which to transport and deliver cargoes such as DNA intercalating cancer therapeutics and short interfering RNAs. These concepts are currently under further investigation.
Methods
See Supporting Information for detailed methods.
Click Conjugation of 5′-Hexynyl DNA or PEO-Alkyne to Azide Functionalized Brush Polymers
A reaction with 200 μM final concentrations of azide from BBP-N3 and equivalent concentration of hexynyl from the required oligonucleotide/oligonucleotide mixture or oligonucleotide and PEO-alkyne mixture (see Supporting Information) was mixed to give a final volume of 50.0 μL in PBS buffer (100 mM Na+, pH = 7.5). The final concentrations of acetonitrile (ACN), CuSO4, and sodium ascorbate were 0.6%, 2 mM and 16 mM, respectively. The reaction was allowed to run for 3 h at 25 °C with shaking and was purified with a 30 K nanosep filters (Millipore, 14000 rcs, 30 min).
Hybridization of DNA Duplexes
Mixtures of BBP conjugated DNA or unconjugated DNA with an equivalent amount of their respective complementary DNA strands were heated in PBS buffer to 90 °C for 2 min and then maintained at 65 °C for 15 min in a water bath and allowed to slowly cool to 25 °C within an hour.
Fluorescence Measurements
Fluorescence measurements were taken in 0.1 μM final concentrations of duplex DNA. YOYO-1 was purchased from Life Technologies. YOYO-1 was added to give 0.5 μM final concentration to the preannealed DNA duplexes and mixed for 1 min before taking fluorescence measurements. Energy transfer signals were monitored by exciting the donor YOYO-1 at 450 nm, and the resulting emission of Cy5 was measured at 670 nm. The band passes for both excitation and emission monochromators were 5 nm. The energy transfer (ET) efficiency was calculated on the basis of the fluorescence intensity of the donor in the presence (FDA) and absence (FD) of the acceptor dye based to the following equation: ET = 1 – (FDA/FD).
YOYO-1 Intercalation Competition Experiments with Calf Thymus DNA
A DBBP sample with 0.1 μM duplex DNA and a control sample of free duplex DNA of equivalent concentration were prepared in buffered solution (100 mM Na+, pH 7.5, PBS buffer). YOYO-1 (0.5 μM; ca. 1 YOYO-1 per 4 bp of DNA) was added, and the fluorescence emission was recorded. Type XV activated CT DNA (0.4 μM; Sigma-Aldrich) was added to each sample and mixed for 1 min, and the fluorescence emission was recorded immediately and at the indicated times following the initial measurement.
Acknowledgments
The authors are grateful to Matthew Saunders for providing the yeast cells expressing c-Myc-tagged scFv. K.M. acknowledges NSF Grants DMR 1501324 and DMR 1407645, and B.A.A. and S.R.D. gratefully acknowledge financial support from the David Scaife Family Charitable Foundation (Award 141RA01).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00259.
Additional data and extended methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Lichtman J. W.; Conchello J. Nat. Methods 2005, 2, 910–919. 10.1038/nmeth817. [DOI] [PubMed] [Google Scholar]; b Evanko D. Nat. Methods 2005, 2, 901–901. 10.1038/nmeth1205-901. [DOI] [PubMed] [Google Scholar]
- a Kherlopian A. R.; Song T.; Duan Q.; Neimark M. A.; Po M. J.; Gohagan J. K.; Laine A. F. BMC Syst. Biol. 2008, 2, 74. 10.1186/1752-0509-2-74. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lesaicherre M. L.; Uttamchandani M.; Chen G. Y. J.; Yao S. Q. Bioorg. Med. Chem. Lett. 2002, 12, 2085–2088. 10.1016/S0960-894X(02)00378-5. [DOI] [PubMed] [Google Scholar]; c Gong H.; Urlacher T. Anal. Biochem. 2015, 469, 1–3. 10.1016/j.ab.2014.09.008. [DOI] [PubMed] [Google Scholar]; d Korb M. L.; Hartman Y. E.; Kovar J.; Zinn K. R.; Rosenthal E. L.; Bland K. I. J. Surg. Res. 2014, 188, 119–128. 10.1016/j.jss.2013.11.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Giesen C.; Wang H. A. O.; Schapiro D.; Zivanovic N.; Jacobs A.; Hattendorf B.; Schüffler P. J.; Grolimund D.; Buhmann J. M.; Brandt S.; Varga Z.; Wild P. J.; Günther D.; Bodenmiller B. Nat. Methods 2014, 11, 417–22. 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]; b Bendall S. C.; Simonds E. F.; Qiu P.; Amir E. D.; Krutzik P. O.; Finck R.; Bruggner R. V.; Melamed R.; Trejo A.; Ornatsky O. I.; Balderas R. S.; Plevritis S. K.; Sachs K.; Pe’er D.; Tanner S. D.; Nolan G. P. Science 2011, 332, 687–696. 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bandura D. R.; Baranov V. I.; Ornatsky O. I.; Antonov A.; Kinach R.; Lou X.; Pavlov S.; Vorobiev S.; Dick J. E.; Tanner S. D. Anal. Chem. 2009, 81, 6813–6822. 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- a Ohya T.; Hayashi T.; Kiyama E.; Nishii H.; Miki H.; Kobayashi K.; Honda K.; Omasa T.; Ohtake H. Biotechnol. Bioeng. 2008, 100, 317–324. 10.1002/bit.21758. [DOI] [PubMed] [Google Scholar]; b Lobbestael E.; Reumers V.; Ibrahimi A.; Paesen K.; Thiry I.; Gijsbers R.; Van den Haute C.; Debyser Z.; Baekelandt V.; Taymans J. BMC Biotechnol. 2010, 10, 16. 10.1186/1472-6750-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora G. J.; Meador C. E.; Anderson G. P.; Taitt C. R. Mol. Cell. Probes 2008, 22, 294–300. 10.1016/j.mcp.2008.07.002. [DOI] [PubMed] [Google Scholar]
- a Bobrow M. N.; Moen P. T. Curr. Protoc. Cytom. 2001, 11:8.9, 8.9.1–8.9.16. 10.1002/0471142956.cy0809s11. [DOI] [PubMed] [Google Scholar]; b Speel E. J.; Hopman A. H.; Komminoth P. J. Histochem. Cytochem. 1999, 47, 281–288. 10.1177/002215549904700302. [DOI] [PubMed] [Google Scholar]; c Mayer G.; Bendayan M. J. Histochem. Cytochem. 1999, 47, 421–430. 10.1177/002215549904700401. [DOI] [PubMed] [Google Scholar]; d Clutter M. R.; Heffner G. C.; Krutzik P. O.; Sachen K. L.; Nolan G. P. Cytometry, Part A 2010, 77A, 1020–1031. 10.1002/cyto.a.20970. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Karkmann U.; Radbruch A.; Hölzel V.; Scheffold A. J. Immunol. Methods 1999, 230, 113–120. 10.1016/S0022-1759(99)00130-1. [DOI] [PubMed] [Google Scholar]; f Wang N.; Gibbons C. H.; Freeman R. J. Histochem. Cytochem. 2011, 59, 382–390. 10.1369/0022155410396931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bernardes G. J. L.; Steiner M.; Hartmann I.; Neri D.; Casi G. Nat. Protoc. 2013, 8, 2079–2089. 10.1038/nprot.2013.121. [DOI] [PubMed] [Google Scholar]
- Lefevre C.; Kang H. C.; Haugland R. P.; Malekzadeh N.; Arttamangkul S.; Haugland R. P. Bioconjug. Chem. 1996, 7, 482–489. 10.1021/bc960034p. [DOI] [PubMed] [Google Scholar]
- Ogawa M.; Kosaka N.; Choyke P. L.; Kobayashi H. ACS Chem. Biol. 2009, 4, 535–546. 10.1021/cb900089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siiman O.; Wilkinson J.; Burshteyn A.; Roth P.; Ledis S. Bioconjugate Chem. 1999, 10, 1090–1106. 10.1021/bc990077g. [DOI] [PubMed] [Google Scholar]
- a Yan X.; Schielke E. G.; Grace K. M.; Hassell C.; Marrone B. L.; Nolan J. P. J. Immunol. Methods 2004, 284, 27–38. 10.1016/j.jim.2003.09.016. [DOI] [PubMed] [Google Scholar]; b Dasso J.; Lee J.; Bach H.; Mage R. G. J. Immunol. Methods 2002, 263, 23–33. 10.1016/S0022-1759(02)00028-5. [DOI] [PubMed] [Google Scholar]; c Yan X.; Tang A.; Schielke E. G.; Hang W.; Nolan J. P. Int. Congr. Ser. 2004, 1263, 342–345. 10.1016/j.ics.2004.02.075. [DOI] [Google Scholar]
- a Sheiko S. S.; Sumerlin B. S.; Matyjaszewski K. Prog. Polym. Sci. 2008, 33, 759–785. 10.1016/j.progpolymsci.2008.05.001. [DOI] [Google Scholar]; b Lee H. il; Pietrasik J.; Sheiko S. S.; Matyjaszewski Prog. Polym. Sci. 2010, 35, 24–44. 10.1016/j.progpolymsci.2009.11.002. [DOI] [Google Scholar]; c Kwak M.; Herrmann Angew. Chem., Int. Ed. 2010, 49, 8574–8587. 10.1002/anie.200906820. [DOI] [PubMed] [Google Scholar]; d Jiang H.; Xu F.-J. Chem. Soc. Rev. 2013, 42, 3394. 10.1039/c2cs35453e. [DOI] [PubMed] [Google Scholar]; e Bracha D.; Bar-Ziv R. H. J. Am. Chem. Soc. 2014, 136, 4945–4953. 10.1021/ja410960w. [DOI] [PubMed] [Google Scholar]; f Liu K.; Zheng L.; Liu Q.; de Vries J. W.; Gerasimov J. Y.; Herrmann A. J. Am. Chem. Soc. 2014, 136, 14255. 10.1021/ja5080486. [DOI] [PubMed] [Google Scholar]; g Krishnamoorthy M.; Hakobyan S.; Ramstedt M.; Gautrot J. E. Chem. Rev. 2014, 114, 10976–11026. 10.1021/cr500252u. [DOI] [PubMed] [Google Scholar]; h Barbey R.; Lavanant L.; Paripovic D.; Schüwer N.; Sugnaux C.; Tugulu S.; Klok H. A. Chem. Rev. 2009, 109, 5437–5527. 10.1021/cr900045a. [DOI] [PubMed] [Google Scholar]
- a Benvin A. L.; Creeger Y.; Fisher G. W.; Ballou B.; Waggoner A. S.; Armitage B. A. J. Am. Chem. Soc. 2007, 129, 2025–2034. 10.1021/ja066354t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Özhalici-Ünal H.; Armitage B. A. ACS Nano 2009, 3, 425–433. 10.1021/nn800727x. [DOI] [PubMed] [Google Scholar]; 15; c Stadler A. L.; Delos Santos J. O.; Stensrud E. S.; Dembska A.; Silva G. L.; Liu S.; Shank N. I.; Kunttas-Tatli E.; Sobers C. J.; Gramlich P. M. E.; Carell T.; Peteanu L. A.; McCartney B. M.; Armitage B. A. Bioconjugate Chem. 2011, 22, 1491–1502. 10.1021/bc100485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Matyjaszewski K.; Xia J. Chem. Rev. 2001, 101, 2921–2990. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]; b Matyjaszewski K. Macromolecules 2012, 45, 4015–4039. 10.1021/ma3001719. [DOI] [Google Scholar]; c Matyjaszewski K.; Tsarevsky N. V. J. Am. Chem. Soc. 2014, 136, 6513–6533. 10.1021/ja408069v. [DOI] [PubMed] [Google Scholar]; d Averick S.; Mehl R. A.; Das S. R.; Matyjaszewski K. J. Controlled Release 2015, 205, 45–57. 10.1016/j.jconrel.2014.11.030. [DOI] [PubMed] [Google Scholar]
- Beers K. L.; Gaynor S. G.; Matyjaszewski K.; Sheiko S. S.; Möller M. Macromolecules 1998, 31, 9413–9415. 10.1021/ma981402i. [DOI] [Google Scholar]
- Mukumoto K.; Li Y.; Nese A.; Sheiko S. S.; Matyjaszewski K. Macromolecules 2012, 45, 9243–9219. 10.1021/ma3020867. [DOI] [Google Scholar]
- Johnson J. a.; Lu Y. Y.; Burts A. O.; Lim Y. H.; Finn M. G.; Koberstein J. T.; Turro N. J.; Tirrell D. A.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 559–566. 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Paredes E.; Das S. R. Bioorg. Med. Chem. Lett. 2012, 22, 5313–5316. 10.1016/j.bmcl.2012.06.027. [DOI] [PubMed] [Google Scholar]; b Averick S. E.; Paredes E.; Dey S. K.; Snyder K. M.; Tapinos N.; Matyjaszewski K.; Das S. R. J. Am. Chem. Soc. 2013, 135, 12508–12511. 10.1021/ja404520j. [DOI] [PubMed] [Google Scholar]
- Glazer A. N.; Rye H. S. Nature 1992, 359, 859–861. 10.1038/359859a0. [DOI] [PubMed] [Google Scholar]
- Averick S. E.; Paredes E.; Grahacharya D.; Woodman B. F.; Miyake-Stoner S. J.; Mehl R. A.; Matyjaszewski K.; Das S. R. Langmuir 2012, 28, 1954–1958. 10.1021/la204077v. [DOI] [PubMed] [Google Scholar]
- Ouellet J.; Schorr S.; Iqbal A.; Wilson T. J.; Lilley D. M. J. Biophys. J. 2011, 101, 1148–1154. 10.1016/j.bpj.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen C. B.; Kodal A. L. B.; Nielsen J. S.; Schaffert D. H.; Scavenius C.; Okholm A. H.; Voigt N. V.; Enghild J. J.; Kjems J.; Tørring T.; Gothelf K. V. Nat. Chem. 2014, 6, 804–9. 10.1038/nchem.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.