

Abstract
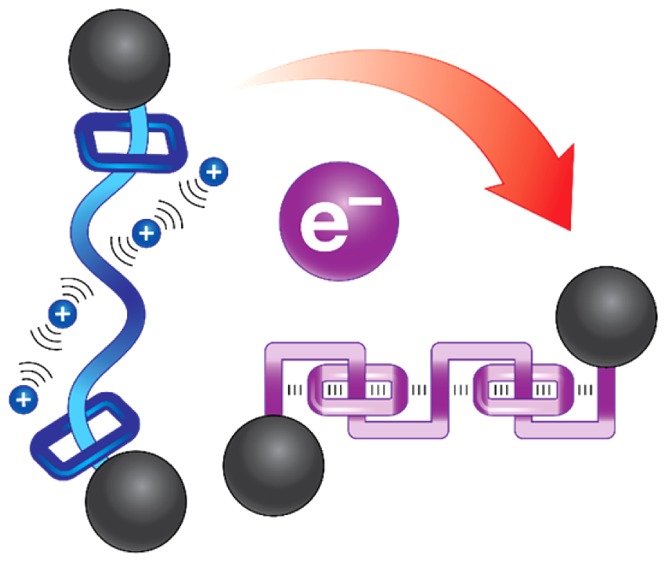
A strategy for creating foldameric oligorotaxanes composed of only positively charged components is reported. Threadlike components—namely oligoviologens—in which different numbers of 4,4′-bipyridinium (BIPY2+) subunits are linked by p-xylylene bridges, are shown to be capable of being threaded by cyclobis(paraquat-p-phenylene) (CBPQT4+) rings following the introduction of radical-pairing interactions under reducing conditions. UV/vis/NIR spectroscopic and electrochemical investigations suggest that the reduced oligopseudorotaxanes fold into highly ordered secondary structures as a result of the formation of BIPY•+ radical cation pairs. Furthermore, by installing bulky stoppers at each end of the oligopseudorotaxanes by means of Cu-free alkyne–azide cycloadditions, their analogous oligorotaxanes, which retain the same stoichiometries as their progenitors, can be prepared. Solution-state studies of the oligorotaxanes indicate that their mechanically interlocked structures lead to the enforced interactions between the dumbbell and ring components, allowing them to fold (contract) in their reduced states and unfold (expand) in their fully oxidized states as a result of Coulombic repulsions. This electrochemically controlled reversible folding and unfolding process, during which the oligorotaxanes experience length contractions and expansions, is reminiscent of the mechanisms of actuation associated with muscle fibers.
Short abstract
The secondary extended−contracted structures of oligorotaxanes are redox-switchable between equilibrium and far-from-equilibrium states as prototypes for artificial molecular muscles.
Introduction
Motivated by the desire to understand the structure–property relationships of biomolecules including DNA, RNA, and membranes and the roles they play in life processes, chemists have striven to manipulate molecular-scale phenomena, resulting from noncovalent bonding interactions, in ever-increasingly complex and organized situations.1−4 By employing noncovalent bonding interactions, synthetic foldamers,5−8 which are promising candidates for mimicking the behavior of biomacromolecules under different kinds of stimuli—and mechanical interlocked molecules9−11 (MIMs), which are the result of the formation of mechanical bonds and have already found applications in drug delivery12,13 and molecular electronics14−17—have been developed and investigated in some detail. Foldamers and MIMs, both utilizing intra- and intermolecular interactions in order to regulate the shapes of molecules, however, seldom result in their paths’ crossing.
Foldameric rotaxanes,18−21 which lie at the intersection between synthetic foldamers and MIMs, have made their ways into chemists’ sights recently. Usually expressed in the context of oligorotaxanes, in which the dumbbell component is threaded by multiple ring components in order to regulate the folded secondary structure, they can exhibit remarkable physicochemical as well as mechanochemical properties22−26 in response to external stimuli. For example, it has already been22,23,25 demonstrated that mechanical responses of oligorotaxanes toward external forces can be controlled by the mobile rings trapped along their one-dimensional dumbbell components, representing a new class of entropy-dominated molecules and materials. It follows that, combining the features of both foldamers and MIMs, foldameric oligorotaxanes not only help us to understand the nature of the folding mechanisms from a fundamental point of view but also make it possible to explore functional materials by scaling11,27 the concerted mechanical actuation of MIMs into the macroscopic regime where applications can be sought and witnessed. To date, we have reported28 the syntheses and properties of a family of foldameric oligorotaxanes which rely on the presence of donor–acceptor recognition between electron-rich 1,5-dioxynaphthalene (DNP) units and electron-deficient cyclobis(paraquat-p-phenylene) (CBPQT4+) rings. Recently, we have shown that the radical-pairing interactions29,30 associated with BIPY(•+) radical cations—the monoreduced state of dicationic BIPY2+ units—can be utilized in the preparation31−33 of MIMs based on a template-directing strategy. Taking advantage of this powerful new molecular recognition motif, herein we describe (Scheme 1) a new class of functional foldameric oligorotaxanes composed of only positively charged components whose construction relies on the interactions between the oligoviologen threads and the CBPQT4+ ring under reducing conditions. This design is based on the consideration that, unlike the donor–acceptor-based examples wherein the folded secondary structures are “permanent” aside from the influence of solvent and temperature, the radical-pairing interactions enable the coconformations of the resulting oligorotaxanes to be switched reversibly between folded and unfolded states by altering the external redox potential. Specifically, in their oxidized states, the positively charged dumbbells apparently become extended and the CBPQT4+ rings are repelled from each other and also from the dumbbells as a result of Coulombic repulsion. Upon reduction back to their radical states, however, solution studies indicate the formation of folded structures driven by radical-pairing interactions. This reversible process, which switches the interactions of bipyridinium units between being repulsive and attractive and giving rise to the extension and contraction of the oligorotaxane chains, can lead to drastic changes in their lengths. This property makes it possible for us to control the operation of artificial molecular motors. The relative movements of the components in these oligorotaxanes, at the behest of external stimuli, are reminiscent of the actions of macroscopic springs.34 In addition, these molecular-level movements, resembling those of the workings of muscle tissue, can potentially be developed further in the context of artificial molecular muscles35−39 that respond to electrochemical stimuli.
Scheme 1. Structural Formulas of the Oligorotaxanes 3R|4BP·16PF6 and 3R|5BP·18PF6 Composed of Only Positively Charged Components.

Note that in the acronyms used to identify compounds, R corresponds to Rotaxane while BP stands for BIPY units.
Results and Discussion
Precise designs of molecular components are necessary in order to optimize noncovalent bonding interactions required for the efficient production of MIMs employing template-directed strategies. Recently, we have demonstrated40 that strong intra- and intermolecular radical-pairing interactions come into play upon reduction of linear oligoviologen chains in which the dicationic BIPY2+ units are separated periodically by xylylene linkers, rendering them to fold both in solution and in the solid state. It should be emphasized, however, that the nature of the folded (super)structures of these oligoviologens either (i) are susceptible to changes in concentration or (ii) lack imposed linear geometries, i.e., they can form loops, which limits their potential applications at least as far as serving as a prototype for artificial molecular muscles is concerned. As a consequence, it is of paramount importance to introduce ring components onto the oligoviologens in order to arrest the chains self-entangling and further regulate the folded secondary superstructures so that they are (i) less influenced by changes in concentration since they are MIMs and (ii) obliged to adopt linear geometries. On the basis of these considerations, we have chosen (Scheme 2) oligoviologens with four and five BIPY2+ units—namely, 4V8+ and 5V10+—to serve as the linear components of the oligorotaxanes, since (iii) their self-folding tendencies40 under reducing conditions are less pronounced, when compared with their longer analogues, making it possible for them to interact with the rings to form the desired oligorotaxanes, while (iv) compared with their shorter analogues, they can potentially bind more CBPQT2(•+) rings under reducing conditions, a situation which is expected to provide additional (co)conformational control during the folding and unfolding processes by (v) generating more BIPY•+ recognition sites to stabilize their radical-state superstructures, and (vi) providing stronger Coulombic repulsion so as to force the secondary structures to become extended upon oxidation.
Scheme 2. Structural Formulas and Graphical Represent-ations of the Oligopseudorotaxanes 4V4(•+)⊂2CBPQT2(•+) and 5V5(•+)⊂2CBPQT2(•+) Formed as a Result of Radical-Pairing Interactions.
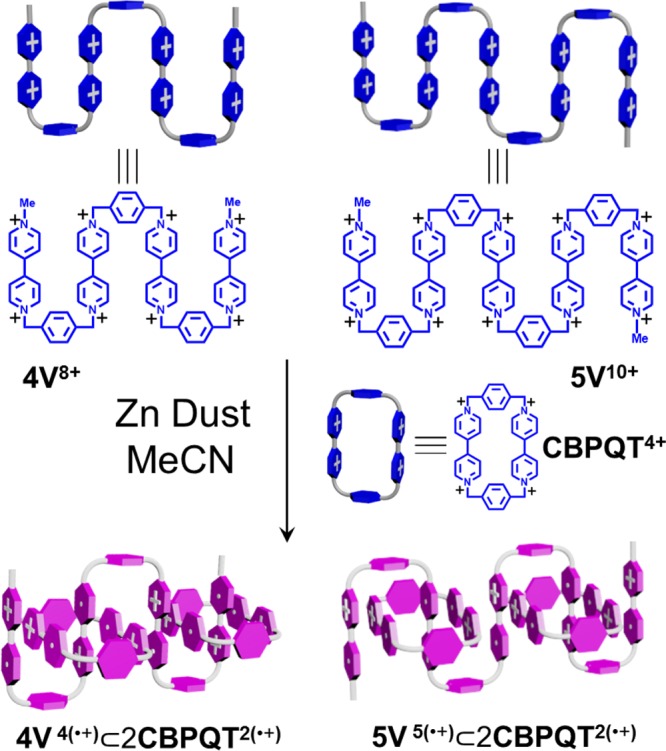
As the key intermediates in the construction of these oligorotaxanes, the formation (Scheme 2) of the oligopseudorotaxanes between the reduced oligoviologens—namely, 4V4(•+) and 5V5(•+)—and the CBPQT2(•+) ring was first of all investigated (Figure 1a,b) by performing UV/vis/NIR titrations. Following the reduction of the oligoviologen 4V8+ to its radical cationic state by Zn dust, the absorption spectrum of an MeCN solution of 4V4(•+) (10 μM) was recorded at room temperature. Next, an increasing amount of CBPQT2(•+) from 1 to 10 equiv was titrated into this MeCN solution, and the UV/vis/NIR spectra were recorded. The results reveal (Figure 1, black trace) that, when only 4V4(•+) is present in the solution, an absorption band around 900 nm is observed, indicating40 the formation of BIPY•+ dimers induced by intramolecular radical-pairing interactions. Upon the addition of CBPQT2(•+), however, a new absorption band emerges (Figure 1, red–purple traces) centered on 1110 nm, which clearly indicates41 the formation of trisradical complexes. As the concentration of CBPQT2(•+) in the solution increases, the intensity of the trisradical band grows with a gradual decrease in its intensity increment until finally a saturation point is reached, a situation which suggests that the maximum number of BIPY•+ units on the 4V4(•+) have been encircled by the CBPQT2(•+) rings. It is also noteworthy that this absorption band is significantly red-shifted, compared (1066 nm) with the example32 of the inclusion complex MV•+⊂CBPQT2(•+) between reduced methyl viologen (MV•+) and CBPQT2(•+). This observation possibly comes about because of the fact that 4V4(•+) binds multiple CBPQT2(•+) rings in solution, such that the resulting radical pairs interact with each other intermolecularly through space to form (Scheme 2) a continuous π-stack, giving rise to a narrower electron-migrating energy gap—in other words, a red-shifted absorption.
Figure 1.

(a) UV/vis/NIR absorption spectrophotometric titration of 4V4(•+) by CBPQT2(•+). Solvent: MeCN; black, [4V4(•+)] = 10 μM; purple, (CBPQT2(•+))/(4V4(•+)) = 10. (b) Enlargement of the spectra from 800 to 1500 nm. The rising peak intensity observed at 1120 nm upon titration indicates the formation of trisradical inclusion complexes.
A similar phenomenon was observed in the case of 5V5(•+), where an absorption band, centered on 1140 nm, emerges (see Figure S11a) immediately after the addition of CBPQT2(•+), indicating rapid formation of trisradical inclusion complexes. It is worth noting that the absorption band in the case of 5V5(•+) is further red-shifted with respect to that observed in the case of 4V4(•+), presumably because of the participation of an additional BIPY•+ subunit in the π-stack results (Scheme 2) in a stacked superstructure of even greater length. All these observations suggest that the CBPQT2(•+) rings interact strongly with both 4V4(•+) and 5V5(•+), despite the existence of competitive intramolecular radical-pairing interactions within 4V4(•+) and 5V5(•+) themselves. This situation possibly pertains because BIPY•+ units prefer to stack in a face-to-face manner in solution, and the CBPQT2(•+) rings, whose rigid geometry already dictates that two BIPY•+ units be parallel, facilitates this type of stacking fashion.
In order to determine the binding stoichiometry between both the reduced oligoviologens 4V4(•+) and 5V5(•+) and CBPQT2(•+), Job plots were obtained. See the Supporting Information, section 5. The titrations reveal that CBPQT2(•+) forms 2:1 complexes with both 4V4(•+) and 5V5(•+) in MeCN solutions, confirming the formation of the oligopseudorotaxanes.42 More importantly, these 2:1 binding stoichiometries support the formation of favorable radical-pairing interactions between all of the BIPY•+ units in both the oligoviologens and the CBPQT2(•+) rings—a coconformation which is in good agreement with the red-shifted band observed in the UV/vis/NIR spectra—as a consequence of the assembly (Scheme 2) of well-defined secondary structures. Furthermore, the binding constants between reduced oligoviologens and CBPQT2(•+) were calculated (see the Supporting Information, section 6), demonstrating that both 4V4(•+) and 5V5(•+) bind strongly (Ka ∼ 109 M–2) with two CBPQT2(•+) rings in solution.
In order to elucidate the binding mechanism between the reduced oligoviologens and CBPQT2(•+), cyclic voltammetry (CV) was also performed. Upon scanning the potential to negative values up to −600 mV, an equimolar mixture of 4V8+ and CBPQT4+ is reduced to radical species, revealing (Figure 2a) a single reduction peak at −300 mV. Indeed, six electrons are involved in this reduction process: two electrons go into the CBPQT4+ ring, forming the diradical dication CBPQT2(•+), and four electrons go to 4V8+, forming the tetraradical tetracation 4V4(•+). As a consequence of this simultaneous six-electron process, the formation of the 4V4(•+)⊂CBPQT2(•+) inclusion complex occurs spontaneously. It is noteworthy that the reduction potential at −300 mV is cathodically shifted significantly, compared with those for the individual 4V8+ oligomer (at −330 mV) and the CBPQT4+ rings (at −360 mV),29 i.e., the mixture is easier to reduce, indicating that the formation of the inclusion complex stabilizes the radical species. On reoxidation, the result is that one of the BIPY•+ radical cations of the complexed CBPQT2(•+) associates more weakly with the 4V4(•+) than the other BIPY•+, leading to the conclusion that the oxidation of this inclusion complex occurs in a stepwise manner, with the more weakly interacting BIPY•+ in the CBPQT2(•+) ring and the unpaired BIPY•+ in 4V4(•+) being oxidized first of all at −209 mV, leaving the strongly interacting BIPY•+ subunits to become oxidized at more positive potentials, i.e., +32 mV.
Figure 2.

Cyclic voltammograms of (a) 4V8+ (black) and an equimolar mixture of 4V8+ and CBPQT4+ (red) and (b) 5V10+ (black) and an equimolar mixture of 5V10+ and CBPQT4+ (red). A glassy carbon working electrode, a platinum counter electrode, and a Ag/AgCl reference electrode were used in the characterization of 0.1 mM MeCN solutions of the oligoviologens at 298 K with 0.1 M TBAPF6 serving as the electrolyte. A scan rate of 200 mV s–1 was used in all analyses.
In the case of 5V10+ and CBPQT4+, an equimolar mixture also gives (Figure 2b) a more positive reduction potential at −264 mV, compared with those of their individual components, indicating the formation of the inclusion complex. More significantly, when the inclusion complex is undergoing oxidation, it registers the first potential at −205 mV, a value which is close to that of the inclusion complex between 4V8+ and CBPQT4+, indicating that the unpaired BIPY•+ radical cations have a similar tendency to become oxidized. By contrast, the second potential is shifted slightly to +45 mV, presumably because the presence of an additional BIPY•+ radical cation makes the dissociation between 5V5(•+) and CBPQT2(•+) even more difficult.
Computational studies were carried out in order to demonstrate how the superstructures of the oligopseudorotaxanes are regulated by radical-pairing interactions. In the case of 4V4(•+)⊂2CBPQT2(•+), we examined four possible co-conformations and discovered that the one (Figure 3a) incorporating two CBPQT2(•+) rings which are centered on the first and the third BIPY•+ subunits has the highest stability. This co-conformation allows all the BIPY•+ radical cations, both in 4V4(•+) and in the CBPQT2(•+) rings, to stack employing a total of seven (BIPY•+)2 radical pairs. The open superstructures with the middle BIPY•+ subunit in 4V4(•+) twisted away (Figure 3b–d), which releases some strain at the angle of BIPY•+–p-xylyene–BIPY•+ in 4V4(•+), is not sufficient to compensate for the loss of one radical pair—leaving six (BIPY•+)2 radical pairs in total—between the BIPY•+ radical cations, rendering them much higher energy (2–7 kcal mol–1) coconformations.
Figure 3.

Simulated coconformations of the oligopseudorotaxane 4V4(•+)⊂2CBPQT2(•+) (a–d) and 5V5(•+)⊂2CBPQT2(•+) (e–h) in different binding modes stabilized by radical-pairing interactions. The numbers (black, in kcal mol–1) show their relative energies, demonstrating that the coconformations with the greatest number (red) of (BIPY•+)2 pairs have the highest stability.
Four coconformations of 5V5(•+)⊂2CBPQT2(•+), where the one with the largest number of (BIPY•+)2 pairs is (Figure 3e) the most stable coconformation, constitutes a result which is in good agreement with 4V4(•+)⊂2CBPQT2(•+). It is also noteworthy that, compared with the 4V4(•+)⊂2CBPQT2(•+) superstructure, once the continuous BIPY•+ stacking is interrupted in the case of 5V5(•+)⊂2CBPQT2(•+), the resulting coconformations (Figure 3f–h) are significantly more destabilized (10.0, 14.1, and 20.2 kcal mol–1), indicating that the π–π stacking contributes to the stabilization energy. These observations can be rationalized by the presence of a continuous π–π stack, in which all the orbitals can interact with each other, leading to a lower orbital binding energy. In the case of the longer π–π stack, 5V5(•+)⊂2CBPQT2(•+), this effect is even more pronounced. The computational investigations also reveal how the number of BIPY•+ subunits affects the secondary structures of the possible coconformations, providing a unique example where longer oligoviologens have a greater tendency to fold.
Having shown that both oligopseudorotaxanes prefer highly ordered secondary structures in solution, we decided to investigate whether this behavior can be promoted in the case of the oligorotaxanes and so facilitate potential applications. Therefore, we carried out the syntheses of the oligorotaxanes, which rely on the templation present in their oligopseudorotaxane progenitors. In the beginning, an azide group is attached by means of hexamethylene chain linkers to each end of the oligoviologens. These linkers are expected to be long enough to act as collecting zones for the CBPQT4+ rings in their fully oxidized states. The azide-functionalized oligoviologens are then mixed with a gross excess (10 equiv) of CBPQT4+ in MeCN under an Ar atmosphere. Upon reduction to their radical cationic states, the solutions turn, first of all, to dark blue and then, after a few minutes, to an intense purple color, indicating the formation of the inclusion complexes. After stirring the solutions overnight to allow the formation of the inclusion complexes to reach thermodynamic equilibrium, a bulky alkyne 4, which acts as the stopper precursor, is added and the solutions are stirred for a further 20 days. The highly charged oligorotaxanes, 3R|4BP·16PF6 and 3R|5BP·18PF6, were isolated (Scheme 3) from the corresponding reaction mixtures by preparative-HPLC in yields43 of 10 and 6%, respectively. 1H NMR and 1H–1H COSY spectra show (see the Supporting Information, section 3) that the CBPQT4+ rings become located, after oxidation, on the hexamethylene chains as a result of Coulombic repulsions, as evidenced by the significantly lower resonating frequencies (<0 ppm) of protons on the hexamethylene chains. Therefore, it is apparent that 3R|4BP16+ and 3R|5BP18+ are fully stretched in their oxidized states. It is also noteworthy that both the oligorotaxanes 3R|4BP16+ and 3R|5BP18+ are composed of one oligoviologen dumbbell and two CBPQT4+ rings, as confirmed by the 1H NMR integration and high resolution mass spectrometry (HR-MS). The outcome is also consistent with the solution-state experiments performed on the oligopseudorotaxanes, demonstrating that the binding stoichiometries are retained during the production of the oligorotaxanes, in spite of the fact that the constitutions of oligoviologens are slightly different.
Scheme 3. Syntheses of Oligorotaxanes 3R|4BP16+ and 3R|5BP18+ by Radical Templation Using Cu-Free Alkyne–Azide Cycloadditions.

With the two oligorotaxanes 3R|4BP16+ and 3R|5BP18+ in hand, we then set out to investigate the behavior of their radical cationic states—namely, 3R|4BP8(•+) and 3R|5BP9(•+)—in MeCN solutions. The comparison of their UV/vis/NIR spectra (Figure 4) with those of the oligopseudorotaxanes shows that, while the peaks around 600 nm still remain (Figure 4a,c) a feature characteristic of the free CBPQT2(•+) rings in the case of oligopseudorotaxane, blue-shifted absorption bands centered on 550 nm emerge in the case of 3R|4BP8(•+) and 3R|5BP9(•+), an observation which is typical of strong BIPY•+ radical pimerization.44 This absorption peak assignment is further confirmed by a variable-temperature UV/vis/NIR experiment (see the Supporting Information, section 8). Moreover, the absorption intensities of the trisradical bands of 3R|4BP8(•+) and 3R|5BP9(•+) are significantly higher (Figure 4b,d) than those of the 1:2 molar mixtures of (i) 4V4(•+) and (ii) 5V5(•+) with CBPQT2(•+), despite their almost identical chemical compositions. Indeed, we found that the absorption intensities are close to those of the saturated situations in the cases of oligopseudorotaxanes. These observations suggest that the molecular recognition between 4V4(•+), 5V5(•+), and CBPQT2(•+) and the strengths of the radical-pairing interactions are enhanced on account of the interlaced superstructures, which restrict the motions of the CBPQT2(•+) rings so that they rest exclusively along the oligoviologen chains, facilitating the folding process. In addition, this enhancement was also observed45 when performing electron paramagnetic resonance (EPR) spectroscopic investigations.
Figure 4.

(a) Partial UV/vis/NIR absorption spectra of MeCN solution of 4V4(•+) (c = 10 μM) with 2 equiv (red) and 10 equiv (blue) of CBPQT2(•+) and oligorotaxane 3R|4BP8(•+) (green). (c) Partial UV/vis/NIR absorption spectra of MeCN solution of 5V5(•+) (c = 10 μM) with 2 equiv (red) and 12 equiv (blue) of CBPQT2(•+) and oligorotaxane 3R|5BP9(•+) (green). (b, d) Enlargement of the corresponding spectra recorded in panels a and c from 800 to 1400 nm, indicating that mechanical bonds enhance molecular recognition.
In order to gain a deeper insight into the mechanically interlocked structures and understand the properties of the radical–radical pairing recognition between the interlocked dumbbells and ring components, we performed (Figure 5a,b) CV experiments on the oligorotaxanes 3R|4BP16+ and 3R|5BP18+ and compared the results with those obtained (Figure 5c–f) using the oligopseudorotaxanes. It transpires (Figure 5a,b) that the CV profiles of the oligorotaxanes display three reduction peaks with potentials at −60, –190, and −271 mV for 3R|4BP16+ and at 0, −174, and −273 mV for 3R|5BP18+. The two additional reduction peaks in both cases, whose potentials are shifted toward positive values compared with those of their oligopseudorotaxane progenitors, can be interpreted in terms of a stepwise formation of the (BIPY•+)n pairs in 3R|4BP16+ and 3R|5BP18+ upon reduction. In the case of 3R|4BP16+, all the BIPY2+ units experience repulsion in their fully oxidized state. Upon reduction, a two-electron process is observed at a potential of −60 mV. Considering that the 4V8+ dumbbell has a higher reduction potential than the CBPQT4+ rings, we believe that both these electrons go preferentially into the dumbbell components in order to relieve the repulsion between the BIPY2+ units. Subsequently the oligorotaxane accepts another two electrons at a potential of −190 mV, whereupon both rings become reduced to CBPQT2+(•+), leading to their translation from the hexamethylene chains to the BIPY•+ radical cations of the dumbbell so as to form (BIPY•+)2 dimeric units. The reduction of the remaining four BIPY2+ dications in both the dumbbell and the rings gives rise to the formation of trisradicals. Differential pulse voltammetry (DPV) experiments (see the Supporting Information, section 9) confirm the numbers of electrons involved in each step of the reduction process. Upon reoxidation, these reduction processes are fully reversible, allowing the partially oxidized intermediates to be observed at −115 mV for 3R|4BP16+ and at −135 mV for 3R|5BP18+. These reduction processes are not observed in the corresponding oligopseudorotaxanes. These results suggest that the radical cationic forms of the oligorotaxanes are more difficult to oxidize than their oligopseudorotaxane progenitors, demonstrating their increased stabilities as a consequence of their mechanically interloscked structures, enforcing the BIPY•+ radical cations to come into close proximity with one another. Taken together, the results illustrate the switching of the oligorotaxanes between (i) the extended forms in their fully oxidized states—as indicated by 1H NMR spectroscopy as well as by computational analysis46—and (ii) the contracted–folded forms adopted under reducing conditions, as revealed by UV/vis/NIR spectroscopy and electrochemistry.
Figure 5.

Cyclic voltammograms of oligorotaxanes 3R|4BP16+ and 3R|5BP18+ and their oligopseudorotaxane progenitors. Voltammograms of (a) 3R|4BP16+, (c) 4V8+ with 2 equiv of CBPQT4+, and (e) 4V8+ with 7 equiv of CBPQT4+. Voltammograms of (b) 3R|5BP18+, (d) 5V10+ with 2 equiv of CBPQT4+, and (f) 5V10+ with 7 equiv of CBPQT4+. A glassy carbon working electrode, a platinum counter electrode, and a Ag/AgCl reference electrode were used in the characterization of 0.1 mM MeCN solutions of the oligoviologens at 298 K with 0.1 M TBAPF6 serving as the electrolyte. A scan rate of 200 mV s–1 was used in all analyses.
In summary, we have reported a new class of oligorotaxanes, 3R|4BP·16PF6 and 3R|5BP·18PF6, which combine the advantages of both foldamers and mechanically interlocked molecules under reducing conditions. Composed of positively charged components, it is only possible to access them by a template-directed approach that takes advantage of radical-pairing interactions, followed by a stoppering protocol employing Cu-free alkyne–azide cycloadditions. The formation of the key intermediates, oligopseudorotaxanes 4V4(•+)⊂2CBPQT2(•+) and 5V5(•+)⊂2CBPQT2(•+), is confirmed by both spectroscopic and electrochemical studies in solution. Computational studies reveal that these oligopseudorotaxanes preferentially form highly ordered secondary structures, wherein the CBPQT2(•+) ring components play an important role in promoting all the BIPY•+ radical cations to stack in extended arrays, in order to maximize the stabilizing effect resulting from radical-pairing interactions. Comparison of the properties of the oligopseudorotaxanes with those of the oligorotaxanes shows that the secondary structures are further regulated in the oligorotaxanes since the components are obliged to remain in close proximity. More importantly, the redox-controlled actuation processes present (Figure 6b) in these oligorotaxanes, which allow their secondary structures to be switched between folded and unfolded states, differentiate them from donor–acceptor,20,28 interactions-based systems (Figure 6a). Moreover, these actuation processes lead to contractions and extensions of the oligorotaxanes, rendering them ideal prototypes of artificial molecular muscles. This research sheds light not only on how to perform chemistry away-from-equilibrium4,47−50 but also on the behavior of foldameric oligorotaxanes so that their structural and mechanical properties can be harnessed in the future in device settings.
Figure 6.
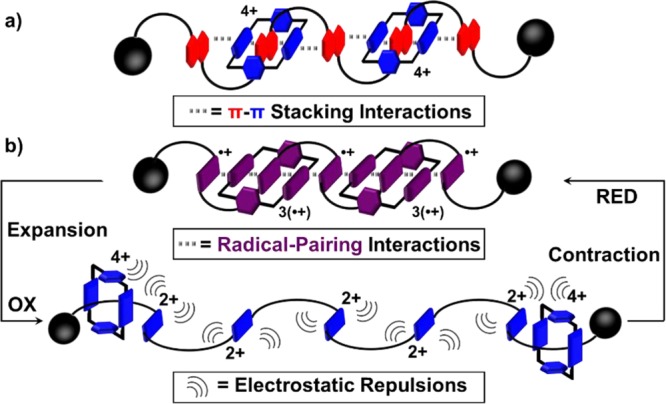
Contrasting (a) an oligorotaxane with donor–acceptor interactions20,28 with (b) an oligorotaxane where radical-pairing interactions stabilize the contracted form under reducing conditions and electrostatic repulsions favor the expanded form under oxidizing conditions.
Methods
See Supporting Information for detailed methods.
Computational Studies of the Oligoviologen 4V8+ and Oligopseudorotaxanes 4V4(•+)⊂2CBPQT2(•+) and 5V5(•+)⊂ 2CBPQT2(•+)
The extended conformation of 4V8+ and the folded coconformations of the two oligopseudorotaxanes, 4V4(•+)⊂2CBPQT2(•+) and 5V5(•+)⊂2CBPQT2(•+), were investigated using the M0651 suite of density functionals. In addition to the general gradient approximation and kinetic energy functionals, M06 includes hybrid exact exchange to account for the localization needed to give good energies and has been optimized to account for van der Waals interactions important in supramolecular complexes. The superstructures were optimized at the M06L using the 6-31G* basis set while more accurate energies were obtained with single-point calculations at the M06 level using the 6-311++G** basis set. All calculations included solvation based on the Poisson–Boltzmann solvation model52 for MeCN (ε = 37.5 and R0 = 2.18 Å) implemented53 in Jaguar 7.7.
Acknowledgments
This research is part (Project 34-945) of the Joint Center of Excellence in Integrated Nano-Systems (JCIN) at King Abdul-Aziz City for Science and Technology (KACST) and Northwestern University (NU). The authors would like to thank both KACST and NU for their continued support of this research. This work was also supported by the National Science Foundation (NSF) under grant no. CHE-1266201. Y.W. thanks the Fulbright Scholar Program for a fellowship and the NU International Institute of Nanotechnology (IIN) for a Ryan Fellowship. The computational studies (W.-G.L., W.A.G.) were supported by NSF (EFRI-ODISSEI 1332411).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00377.
Instrumentation, synthetic procedures, NMR investigations of oligorotaxanes, UV/vis/NIR data, cyclic voltammetry, and titration and EPR experiments (PDF)
Author Present Address
‡ M.F.: Department of Chemical Sciences, University of Padova, Via Marzolo 1, Padova 35131, Italy.
The authors declare no competing financial interest.
Supplementary Material
References
- Aida T.; Meijer E. W.; Stupp S. I. Functional supramolecular polymers. Science 2012, 335, 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans N. H.; Beer P. D. Advances in anion supramolecular chemistry: From recognition to chemical applications. Angew. Chem., Int. Ed. 2014, 53, 11716–11754. 10.1002/anie.201309937. [DOI] [PubMed] [Google Scholar]
- Schneider H.-J. Dispersive interactions in solution complexes. Acc. Chem. Res. 2015, 48, 1815–1822. 10.1021/acs.accounts.5b00111. [DOI] [PubMed] [Google Scholar]
- Mattia E.; Otto S. Supramolecular systems chemistry. Nat. Nanotechnol. 2015, 10, 111–119. 10.1038/nnano.2014.337. [DOI] [PubMed] [Google Scholar]
- Hill D. J.; Mio M. J.; Prince R. B.; Hughes T. S.; Moore J. S. A field guide to foldamers. Chem. Rev. 2001, 101, 3893–4011. 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- Sessler J. L.; Jayawickramarajah J. Functionalized base-pairs: Versatile scaffolds for self-assembly. Chem. Commun. 2005, 1939–1949. 10.1039/b418526a. [DOI] [PubMed] [Google Scholar]
- Guichard G.; Huc I. Synthetic foldamers. Chem. Commun. 2011, 47, 5933–5941. 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]
- Zhang D.-W.; Zhao X.; Li Z.-T. Aromatic amide and hydrazide foldamer-based responsive host-guest systems. Acc. Chem. Res. 2014, 47, 1961–1970. 10.1021/ar5000242. [DOI] [PubMed] [Google Scholar]
- Kay E. R.; Leigh D. A.; Zerbetto F. Synthetic molecular motors and mechanical machines. Angew. Chem., Int. Ed. 2007, 46, 72–191. 10.1002/anie.200504313. [DOI] [PubMed] [Google Scholar]
- Bruns C. J.; Stoddart J. F. The mechanical bond: A work of art. Top. Curr. Chem. 2011, 323, 19–72. 10.1007/128_2011_296. [DOI] [PubMed] [Google Scholar]
- Coskun A.; Banaszak M.; Astumian R. D.; Stoddart J. F.; Grzybowski B. A. Great expectations: Can artificial molecular machines deliver on their promise?. Chem. Soc. Rev. 2012, 41, 19–30. 10.1039/C1CS15262A. [DOI] [PubMed] [Google Scholar]
- Luo Z.; Ding X.; Hu Y.; Wu S.; Xiang Y.; Zeng Y.; Zhang B.; Yan H.; Zhang H.; Zhu L.; Liu J.; Li J.; Cai K.; Zhao Y. Engineering a hollow nanocontainer platform with multifunctional molecular machines for tumor-targeted therapy in vitro and in vivo. ACS Nano 2013, 7, 10271–10284. 10.1021/nn404676w. [DOI] [PubMed] [Google Scholar]
- Barat R.; Legigan T.; Tranoy-Opalinski I.; Renoux B.; Péraudeau E.; Clarhaut J.; Poinot P.; Fernandes A. E.; Aucagne V.; Leigh D. A.; Papot S. A mechanically interlocked molecular system programmed for the delivery of an anticancer drug. Chem. Sci. 2015, 6, 2608–2613. 10.1039/C5SC00648A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood A. H.; Stoddart J. F.; Steuerman D. W.; Heath J. R. Whence molecular electronics?. Science 2004, 306, 2055–2056. 10.1126/science.1106195. [DOI] [PubMed] [Google Scholar]
- Coskun A.; Spruell J. M.; Barin G.; Dichtel W. R.; Flood A. H.; Botros Y. Y.; Stoddart J. F. High hopes: Can molecular electronics realise its potential?. Chem. Soc. Rev. 2012, 41, 4827–4859. 10.1039/c2cs35053j. [DOI] [PubMed] [Google Scholar]
- Venturi M.; Credi A. Electroactive [2]catenanes. Electrochim. Acta 2014, 140, 467–475. 10.1016/j.electacta.2014.02.061. [DOI] [Google Scholar]
- Götz G.; Zhu X.; Mishra A.; Segura J.-L.; Mena-Osteritz E.; Bäuerle P. π-Conjugated [2]catenanes based on oligothiophenes and phenanthrolines: Efficient synthesis and electronic properties. Chem. - Eur. J. 2015, 21, 7193–7210. 10.1002/chem.201406039. [DOI] [PubMed] [Google Scholar]
- Gan Q.; Ferrand Y.; Bao C.; Kauffmann B.; Grélard A.; Jiang H.; Huc I. Helix-rod host-guest complexes with shuttling rates much faster than disassembly. Science 2011, 331, 1172–1175. 10.1126/science.1200143. [DOI] [PubMed] [Google Scholar]
- Zhang K.-D.; Zhao X.; Wang G.-T.; Liu Y.; Zhang Y.; Lu H.-J.; Jiang X.-K.; Li Z.-T. Foldamer-tuned switching kinetics and metastability of [2]rotaxanes. Angew. Chem., Int. Ed. 2011, 50, 9866–9870. 10.1002/anie.201104099. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Bruns C. J.; Li H.; Lei J.; Ke C.; Liu Z.; Shafaie S.; Colquhoun H. M.; Stoddart J. F. Synthesis and solution-state dynamics of donor-acceptor oligorotaxane foldamers. Chem. Sci. 2013, 4, 1470–1483. 10.1039/c3sc00015j. [DOI] [Google Scholar]
- Woźny M.; Pawl̷owska J.; Osior A.; Świder P.; Bilewicz R.; Korybut-Daszkiewicz B. An electrochemically switchable foldamer—A surprising feature of a rotaxane with equivalent stations. Chem. Sci. 2014, 5, 2836–2842. 10.1039/c4sc00449c. [DOI] [Google Scholar]
- Pinson M. B.; Sevick E. M.; Williams D. R. M. Mobile rings on a polyrotaxane lead to a yield force. Macromolecules 2013, 46, 4191–4197. 10.1021/ma4000094. [DOI] [Google Scholar]
- Katsuno C.; Konda A.; Urayama K.; Takigawa T.; Kidowaki M.; Ito K. Pressure-responsive polymer membranes of slide-ring gels with movable cross-links. Adv. Mater. 2013, 25, 4636–4640. 10.1002/adma.201301252. [DOI] [PubMed] [Google Scholar]
- Van Quaethem A.; Lussis P.; Leigh D. A.; Duwez A.-S.; Fustin C.-A. Probing the mobility of catenane rings in single molecules. Chem. Sci. 2014, 5, 1449–1452. 10.1039/c3sc53113a. [DOI] [Google Scholar]
- Bin Imran A.; Esaki K.; Gotoh H.; Seki T.; Ito K.; Sakai Y.; Takeoka Y. Extremely stretchable thermosensitive hydrogels by introducing slide-ring polyrotaxane cross-linkers and ionic groups into the polymer network. Nat. Commun. 2014, 5, Article 5124. 10.1038/ncomms6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inutsuka M.; Inoue K.; Hayashi Y.; Inomata A.; Sakai Y.; Yokoyama H.; Ito K. Highly dielectric and flexible polyrotaxane elastomer by introduction of cyano groups. Polymer 2015, 59, 10–15. 10.1016/j.polymer.2014.12.055. [DOI] [Google Scholar]
- Du G.; Moulin E.; Jouault N.; Buhler E.; Giuseppone N. Muscle-like supramolecular polymers: Integrated motion from thousands of molecular machines. Angew. Chem., Int. Ed. 2012, 51, 12504–12508. 10.1002/anie.201206571. [DOI] [PubMed] [Google Scholar]
- Bruns C. J.; Stoddart J. F. Mechanically interlaced and interlocked donor-acceptor foldamers. Adv. Polym. Sci. 2013, 261, 271–294. 10.1007/12_2013_245. [DOI] [Google Scholar]
- Trabolsi A.; Khashab N.; Fahrenbach A. C.; Friedman D. C.; Colvin M. T.; Coti K. K.; Benitez D.; Tkatchouk E.; Olsen J. C.; Belowich M. E.; Carmielli R.; Khatib H. A.; Goddard W. A. III; Wasielewski M. R.; Stoddart J. F. Radically enhanced molecular recognition. Nat. Chem. 2010, 2, 42–49. 10.1038/nchem.479. [DOI] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Zhu Z.; Cao D.; Liu W.-G.; Li H.; Dey S. K.; Basu S.; Trabolsi A.; Botros Y. Y.; Goddard W. A. III; Stoddart J. F. Radically enhanced molecular switches. J. Am. Chem. Soc. 2012, 134, 16275–16288. 10.1021/ja306044r. [DOI] [PubMed] [Google Scholar]
- Barnes J. C.; Fahrenbach A. C.; Cao D.; Dyar S. M.; Frasconi M.; Giesener M. A.; Benítez D.; Tkatchouk E.; Chernyashevskyy O.; Shin W. H.; Li H.; Sampath S.; Stern C. L.; Sarjeant A. A.; Hartlieb K. J.; Liu Z.; Carmieli R.; Botros Y. Y.; Choi J. W.; Slawin A. M. Z.; Ketterson J. B.; Wasielewski M. R.; Goddard W. A. III; Stoddart J. F. A radically configurable six-state compound. Science 2013, 339, 429–433. 10.1126/science.1228429. [DOI] [PubMed] [Google Scholar]
- Li H.; Zhu Z.; Fahrenbach A. C.; Savoie B. M.; Ke C.; Barnes J. C.; Lei J.; Zhao Y.-L.; Lilley L. M.; Marks T. J.; Ratner M. A.; Stoddart J. F. Mechanical bond-induced radical stabilization. J. Am. Chem. Soc. 2013, 135, 456–467. 10.1021/ja310060n. [DOI] [PubMed] [Google Scholar]
- Cheng C.; McGonigal P. R.; Schneebeli S. T.; Li H.; Vermeulen N. A.; Ke C.; Stoddart J. F. An artificial molecular pump. Nat. Nanotechnol. 2015, 10, 547–553. 10.1038/nnano.2015.96. [DOI] [PubMed] [Google Scholar]
- Feringa B. L. Molecular machines springing into action. Nat. Chem. 2010, 2, 429–430. 10.1038/nchem.676. [DOI] [PubMed] [Google Scholar]
- Dawson R. E.; Lincoln S. F.; Easton C. J. The foundation of a light driven molecular muscle based on stilbene and α-cyclodextrin. Chem. Commun. 2008, 34, 3980–3982. 10.1039/b809014a. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Credi A.; Venturi M. Light powered molecular machines. Chem. Soc. Rev. 2009, 38, 1542–1550. 10.1039/b806328c. [DOI] [PubMed] [Google Scholar]
- Takashima Y.; Hatanaka S.; Otsubo M.; Nakahata M.; Kakuta T.; Hashidzume A.; Yamaguchi H.; Harada A. Expansion-contraction of photoresponsive artificial muscle regulated by host-guest interactions. Nat. Commun. 2012, 3, 1270–1277. 10.1038/ncomms2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns C. J.; Stoddart J. F. Rotaxane-based molecular muscles. Acc. Chem. Res. 2014, 47, 2186–2199. 10.1021/ar500138u. [DOI] [PubMed] [Google Scholar]
- Durot S.; Heitz V.; Sour A.; Sauvage J.-P. Transition-metal-complexed catenanes and rotaxanes: From dynamic systems to functional molecular machines. Top. Curr. Chem. 2014, 354, 35–70. 10.1007/128_2013_514. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Frasconi M.; Liu W.-G.; Liu Z.; Sarjeant A. A.; Nassar M. S.; Botros Y. Y.; Goddard W. A. III; Stoddart J. F. Folding of oligoviologens induced by radical-radical interactions. J. Am. Chem. Soc. 2015, 137, 876–885. 10.1021/ja5111305. [DOI] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Barnes J. C.; Lanfranchi D. A.; Li H.; Coskun A.; Gassensmith J. J.; Liu Z.; Benítez D.; Trabolsi A.; Goddard W. A. III; Elhabiri M.; Stoddart J. F. Solution-phase mechanistic study and solid-state structure of a tris(bipyridinium radical cation) inclusion complex. J. Am. Chem. Soc. 2012, 134, 3061–3072. 10.1021/ja2089603. [DOI] [PubMed] [Google Scholar]
- According to this binding stoichiometry, we found that the stronger interactions between CBPQT2(•+) and 4V4(•+), as well as between CBPQT2(•+) and 5V5(•+), compared to the self-dimerization of 4V4(•+) and 5V5(•+), are also supported by the results of DFT calculations. The formation enthalpies (ΔH) of the inclusion complexes 4V4(•+)⊂2CBPQT2(•+) and 5V5(•+)⊂2CBPQT2(•+) are –66.1 and −73.6 kcal mol–1, whereas the ΔH values for the 4V4(•+) and 5V5(•+) dimers are only –42.7 and −48.4 kcal mol–1, respectively, indicating that the organized geometry provided by the CBPQT2(•+) rings amount to approximately 24 kcal mol–1 stabilizing energy.
- The low yield can be ascribed to (i) thermodynamic phenomena, the competing self-radical-pairing interactions within oligoviologens and the binding constants between oligoviologens and CBPQT2(•+) which determine that a certain portion of oligoviolgens will remain uncomplexed, lowering the reaction efficiency; and (ii) kinetical phenomena, e.g., the slow reaction rate of the copper-free click reaction which prevents the complete conversion of azide groups to the triazole rings within the limited time of the reaction.
- Geuder W.; Hünig S.; Suchy A. Single and double bridged viologenes and intramolecular pimerization of their cation radicals. Tetrahedron 1986, 42, 1665–1677. 10.1016/S0040-4020(01)87583-9. [DOI] [Google Scholar]
- The EPR signal for the purple-colored 3R|4BP8(•+), obtained under the same experimental conditions (0.2 mM in MeCN at RT) as those used to generate the benzyl viologen radical cation BnV•+, is 4-fold weaker, despite the fact that it contains eight viologen units per molecule. The relatively weak intensity of the EPR signal for 3R|4BP8(•+) is indicative of a pronounced spin-pairing as a result of intramolecular diamagnetic π-dimerization. The presence of an EPR signal can be attributed to a small thermal population of paramagnetic coconformations.
- The change in the length of the oligorotaxane 3R|4BP16+ on reduction to 3R|4BP8(•+) is supported by computational analysis. See the Supporting Information, section 10. Since 3R|4BP16+ is too large to be simulated by DFT calculations, we sought an approximation by comparing the “central regions” of 3R|4BP16+ and 3R|4BP8(•+). We measured the centroid–centroid distance between the two terminal BIPY2+ units in the simulated conformation (Figure S18a) of 4V8+ and the centroid–centroid distance between the two terminal BIPY•+ units—one on the 4V4(•+) component and the other on the distant CBPQT2(•+) ring—in the coconformation (Figure S18b) of the oligopseudorotaxane 4V4(•+)⊂2CBPQT2(•+). It turns out that, upon reduction, the molecular length contracts by 6 Å from 29.8 to 23.8 Å.
- Debnath S.; Roy S.; Ulijn R. V. Peptide nanofibers with dynamic instability through nonequilibrium biocatalytic assembly. J. Am. Chem. Soc. 2013, 135, 16789–16792. 10.1021/ja4086353. [DOI] [PubMed] [Google Scholar]
- Li H.; Cheng C.; McGonigal P. R.; Fahrenbach A. C.; Frasconi M.; Liu W.-G.; Zhu Z.; Zhao Y.; Ke C.; Lei J.; Young R. M.; Dyar S. M.; Co D. T.; Yang Y.-W.; Botros Y. Y.; Goddard W. A. III; Wasielewski M. R.; Astumian R. D.; Stoddart J. F. Relative unidirectional translation in an artificial molecular assembly fueled by light. J. Am. Chem. Soc. 2013, 135, 18609–18620. 10.1021/ja4094204. [DOI] [PubMed] [Google Scholar]
- Cheng C.; McGonigal P. R.; Liu W.-G.; Li H.; Vermeulen N. A.; Ke C.; Frasconi M.; Stern C. L.; Goddard W. A. III; Stoddart J. F. Energetically demanding transport in a supramolecular assembly. J. Am. Chem. Soc. 2014, 136, 14702–14705. 10.1021/ja508615f. [DOI] [PubMed] [Google Scholar]
- Li Q.; Fuks G.; Moulin E.; Maaloum M.; Rawiso M.; Kulic I.; Foy J. T.; Giuseppone N. Macroscopic contraction of a gel induced by the integrated motion of light-driven molecular motors. Nat. Nanotechnol. 2015, 10, 161–165. 10.1038/nnano.2014.315. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Tannor D. J.; Marten B.; Murphy R.; Friesner R. A.; Sitkoff D.; Nicholls A.; Ringnalda M.; Goddard W. A. III; Honig B. J. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. 10.1021/ja00105a030. [DOI] [Google Scholar]
- Jaguar; Schrodinger, LLC: New York, NY, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.