

Abstract
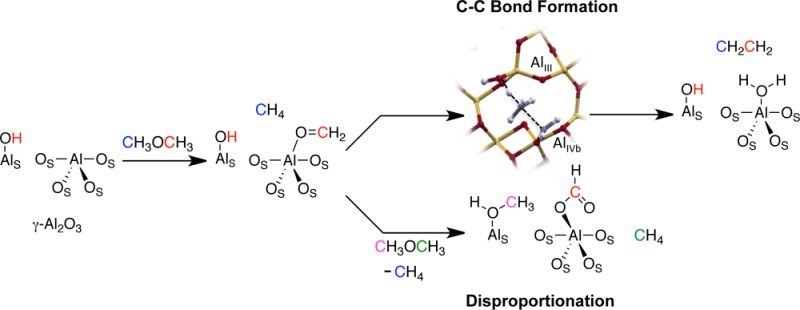
The methanol-to-olefin (MTO) process allows the conversion of methanol/dimethyl ether into olefins on acidic zeolites via the so-called hydrocarbon pool mechanism. However, the site and mechanism of formation of the first carbon–carbon bond are still a matter of debate. Here, we show that the Lewis acidic Al sites on the 110 facet of γ-Al2O3 can readily activate dimethyl ether to yield CH4, alkenes, and surface formate species according to spectroscopic studies combined with a computational approach. The carbon–carbon forming step as well as the formation of methane and surface formate involves a transient oxonium ion intermediate, generated by a hydrogen transfer between surface methoxy species and coordinated methanol on adjacent Al sites. These results indicate that extra framework Al centers in acidic zeolites, which are associated with alumina, can play a key role in the formation of the first carbon–carbon bond, the initiation step of the industrial MTO process.
Short abstract
Adjacent Lewis acid sites on alumina play in concert to activate dimethyl ether via H-transfer yielding CH4 and surface formate or olefins and H2O.
Forming carbon–carbon bonds from C1 species is a long-standing scientific and industrial challenge.1−15 An industrial breakthrough stemmed from the discovery of the methanol to olefin (MTO) process in the 1970s, which allowed the catalytic conversion of methanol to ethylene and propylene by zeolites (Scheme 1). This process constitutes an alternative route to light alkenes not relying on crude oil. Several industrial plants are being opened, in particular in Asia, in view of the increasing demand for alkene feedstocks.16,17
Scheme 1. Methanol to Olefin Process Catalyzed by Zeolites.
The mechanism of this process is a matter of intense debate and investigation both in industry and academia.17−31 Initially, direct pathways from methanol to olefin were suggested (Scheme 2b–e), but in the 1990s Dahl and Kolboe proposed an indirect pathway that proceeds via a hydrocarbon pool as shown in Scheme 2a.32−34 A dual operating cycle,30,31 one for alkenes (Scheme 2a, left) and another one for aromatics (Scheme 2a, right), forms the currently accepted mechanism operating under industrial MTO conditions. Carbenium species have been proposed as the active species of the hydrocarbon pool cycle,18 and a complete catalytic cycle combining theory and experiment was proposed for the HZSM-5 zeolite.20,35
Scheme 2. A Selection out of the More than 20 Proposed Mechanisms for the Carbon–Carbon Bond Formation Step Is Shown: (a) Hydrocarbon Pool, (b) Methane-formaldehyde Route, (c) Oxonium-ylide, (d) Carbine-carbenoid Mechanisms, (e) Alkoxy Mechanism.

Nevertheless, the question related to the site and mechanism of formation of the first olefins and aromatics, requiring carbon–carbon bond forming processes from methanol/dimethyl ether, has remained a matter of debate. More than 20 mechanisms have been proposed for this step,17,28 involving intermediates such as methane–formaldehyde,23 oxonium ylide,27 and carbenoid or alkoxy species (Scheme 2b–e), and some proposals also include the presence of adventitious organic compounds such as aromatics. Extensive attention was devoted to the oxonium ylide mechanism (Scheme 2c), which by an intramolecular Steven’s rearrangement would produce methylethyl ether, which could then form ethene by β-hydride elimination. This mechanism and parent ones were discarded because of the need for formation of highly unstable oxonium ylide species.22,36 Other direct C–C bond formation routes, via methane-formaldehyde (Scheme 2b) or carbenoid species (Scheme 2d), were found unfavorable on the basis of an extensive ab initio study.25 It is worth noting that all the zeolites active in the MTO process contain Al sites. This element provides acidic sites that play a significant role in the zeolite catalytic activity, and they could participate in the initial carbon–carbon bond formation step. Moreover, there is always a debate whether or not extraframework aluminum, which is in the form of alumina, could be responsible for side-reactions in zeolite catalysis.37−39 Hence, one important aspect that has not been considered is the participation of multiple Al centers, possibly belonging to alumina, in the promotion of carbon–carbon bonds in the MTO process. Though the formation of hydrocarbons by reaction of methanol on alumina or silica–alumina was reported,40 most studies have concentrated on the hydrolysis of dimethyl ether (DME) into methanol and the reverse reaction and also on the alcohol dehydration to the corresponding ether and olefins.41−54 Recent works have shown that γ-Al2O3 and δ-Al2O3 contain on their 110 facet55 highly reactive tricoordinated AlIII and four-coordinated AlIVb sites, which are able to activate the C–H bond of methane at low temperatures (<150 °C)56,57 and to convert CH3F into branched olefinic products isobutene and 2-methylbutene at relatively mild temperatures (200 °C) showing that the key carbon–carbon formation step occurs on alumina surfaces via the growth of surface alkoxy chains.58 In such processes, adsorbed water plays a key role to facilitate the activation of hydrocarbons, through the formation of basic O sites and the stabilization of the otherwise unstable 110 facet.57,59
Here, we show that alumina converts DME at 300 °C into methane along with smaller amounts of higher olefin products, such as ethene, propene, butenes, and pentenes, analogously to the MTO process. In addition, combined IR and solid-state NMR show that this process is accompanied by the formation of methoxy and formate surface species besides methane and other hydrocarbons detected in the gas phase. Ab initio simulations suggest a step involving a hydride transfer from a methoxy aluminum surface species and an activated DME, adsorbed on adjacent Al sites. This process involves a transition state structure with an oxonium (AlO=CH2+)/methane adduct, which evolves either to the formation of methane and adsorbed formate species or olefins through a carbon–carbon bond forming process, consistent with experimental observation. This indicates that two Al sites can play in concert to generate AlO=CH2+ species as key transient species, able to activate C–H bonds and to promote a C–C bond formation step.
First, the reaction of DME (0.05 mmol) with alumina (Evonik aluC, 130 m2·g–1 or SBA, 200 m2·g–1) partially dehydroxylated at 700 °C was monitored by gas chromatography as a function of temperature (Table 1a and Table S1). Methane evolved at 300 °C,60 0.029 mmol as a sole gaseous product, which corresponds to 2.7 CH4 molecules per nm2. This amount of methane approximately corresponds to one CH4 per two Al sites considering the 110 surface of γ-Al2O3.61,62 Treatment of the solids under high vacuum (10–5 mBar) at 100 °C led to desorption of ethene, propene, butenes, and pentenes as major products along with traces of hexenes according to GC and GC/MS (Table 1b). In contrast, no DME conversion was observed under the same reaction conditions in the absence of alumina, indicating that alumina is critical to promote the formation of these hydrocarbon products from DME.
Table 1. (a) Composition of the Gas Phase during the Reaction of Dimethyl Ether with Pyrogenic Evonik–Degussa AluC Dehydroxylated at 700 °Ca and (b) Desorption of the Surface.
| (a) Composition of the Gas Phase during the Reaction of Dimethyl Ether with Pyrogenic Evonik–Degussa AluC Dehydroxylated at 700 °C | ||
|---|---|---|
| Al2O3 with dimethyl ether | composition of the gas phase | |
| temperature | CH3OCH3 | CH4 |
| room temperature | 100% | 0% |
| 100 °C | 100% | 0% |
| 200 °C | 100% | 0% |
| 300 °C | 42% | 58% |
| (b) Desorption of the Surface (molecules/nm2) | |
|---|---|
| hydrocarbon removed from the surface | molecules per nm2 |
| ethylene | 3.1 × 10–2 |
| propylene | 1.6 × 10–2 |
| Z-2-butene | 0.3 × 10–2 |
| E-2-butene | 0.7 × 10–2 |
| isobutene | 0.2 × 10–2 |
| pentene | 1.0 × 10–2 |
Similar data are found for a boehmite-derived pure γ-Al2O3 provided by Sasol (SBA-200, see Table S1).
To further understand the reaction, an in situ IR study was carried out. Figure 1a–d shows a series of IR spectra taken at different stages of the reaction (full IR spectrum are available in Figures S1 and S2, Supporting Information). Addition of DME to Al2O3 at room temperature (40 mbar, 4.6 molecules of DME per nm2) led to the decrease of the intensity of the alumina OH bands at 3840 and 3600 cm–1 (Figure 1a,b).
Figure 1.

On the right: in situ FT-IR transmission spectra of (a) Al2O3 dehydroxylated at 700 °C, then reacted with dimethyl ether at room temperature (b), at 200 °C (c), and 300 °C (d) (for full spectra, see Figure S1). All the spectra were recorded with the gas phase condensed at −190 °C. In situ IR was used to determine the main changes of the surface species upon heating. We evidenced the activation of the C–O bond of DME as well as the formation of new surface species including formate. The same IR study can be found in Figure S2 for pure γ-Al2O3. On the left: 1H–13C CPMAS NMR, 400 MHz NMR spectrometer, spinning rate of 10 kHz. Spectrum of Al2O3 reacted with 2-13C-(CH3)2O (e) at room temperature, number of scans was set to 5k; (f) at 200 °C, number of scans was set to 100k; and (g) at 300 °C, number of scans was set to 50k. The recycling delay was set to 1 s of all the spectra. The radiofrequency field for 1H excitation was set to 100 kHz.
It also showed the presence of CH3 and CH2 groups, a C–O bond as well as a few Csp2-H species (tentatively attributed to the presence of a peak at 3040 cm–1). At 200 °C two new peaks in the region of hydroxyl appeared (3570 and 3675 cm–1), and the C–O band (1157 cm–1) disappeared, consistent with the cleavage of that bond (Figure 1c). At 300 °C, the intensity of the ν(C–H) vibration decreased while the intensity of the OH band increased (Figure 1d). This was accompanied by the formation of methane in the gas phase and the appearance of two new peaks of strong intensity at 1578 and 1321 cm–1, which can be assigned to the vibration of the C=O double bond and C–O bond of surface formate species.63 Adsorbing methylformate on Al2O3 led to the appearance of the same bands, but also an additional band at 1683 associated with the carbonyl of physisorbed methyl formate (Figure S3). Similar bands were also obtained upon adsorption of formic acid on alumina.64,65 Overall, these IR data suggest that carbon–carbon forming and carbon–oxygen cleavage reactions took place on the Al2O3 surfaces upon reaction with DME, while methane and formate species are formed.
The reaction was also monitored by solid-state NMR using 13C dilabeled DME (Figure 1e–g). The insets e–g of Figure 1 show the 13C cross polymerization magic angle spinning NMR (CP-MAS) spectra obtained on Al2O3-(700) after reaction with DME at three different temperatures: 25, 200, and 300 °C. At low temperature, only one peak was observed at 62 ppm (Figure 1e), which is assigned to adsorbed DME. The intensity of the peak at 62 ppm decreased at higher temperatures, while a new peak progressively appeared at 49 ppm, reaching a maximum of intensity at 200 °C (Figure 1f). This peak is assigned to surface methoxy species.58 In addition, a shoulder appeared at a higher field for the peak at 64 ppm that we assigned to the adsorption of DME molecule on different Al sites. Three peaks are observed in 13C NMR after treatment at 300 °C (Figure 1g): Two at 49, 62–64 ppm assigned to methoxy and DME respectively, and a third one at 169 ppm assigned to formate. The observation of formate by carbon-13 NMR is also confirmed by 2D NMR: the carbon at 169 ppm, which is typical of an ester carbonyl, correlates with a proton at 9.2 ppm in the 2D NMR, which clearly identifies its attribution to a formate species (Figures S4–S6). In addition, adsorption of methyl formate on γ-Al2O3-(700) leads to the same NMR signal consistent with its attribution to formate surface species (Figure S6) as previously discussed from the IR data. The correlation with the OCH3 group suggests that the formate species are in close proximity to the methoxy species. No direct detection of hydrocarbons could be observed, suggesting that they are present in small amounts or remain adsorbed as minor alkoxy surface species.
The formation of methane, higher hydrocarbons, methoxy, and formate species was investigated by means of DFT periodic calculations. We used the 110 termination of γ-Al2O3 because it is the most abundant one for γ-Al2O3 (75%) and δ-Al2O3 (one of the component of our sample AluC) particles. In addition, this surface when completely dehydrated contains the most reactive sites (strong Lewis acid sites, see Figure S7).61,62,66
The adsorption (coordination) of CH3OCH3 on the most acidic AlIII site of the fully dehydrated alumina surface (s0 surface, see Supporting Information) forms the species 0-III in an exothermic step by 131 kJ mol–1. This adsorbed species can further react through either the C–H or the C–O bonds of CH3OCH3. The C–O activation route with the transfer of the methoxy on the bare alumina surface is associated with a high-energy barrier equal to 179 kJ mol–1 and is endothermic by 16 kJ mol–1.67 On the more realistic monohydrated surfaces, the initial C–O activation step is lowered by more than 50 kJ mol–1 (vide infra). The alternative C–H bond activation pathway presents overall higher energy barriers and would also lead to the formation of unlikely Al-alkyl intermediates in the presence of proton sources (see Figure S8). In a previous study, we did an extensive analysis of the possible adsorption sites of water on the 110 termination of the γ-Al2O3 surface.59 Depending on the initial adsorption site of water in the unit cell, corresponding to an OH coverage equal to 3.0 OH/nm2, analogous energy profiles can be obtained for the ethylene formation route (see Supporting Information, Figure S9). Here, we will discuss ethylene formation from the most stable and probable s1a surface, in which one OH group coordinates to AlIII and one proton is bonded to the O2a center, since the minima and the transition-states of the corresponding energy profile present the lowest energies of all the different surfaces evaluated with water initially adsorbed. From the s1a surface, the coordination of DME to the AlIVb center yields a binding energy equal to 118 kJ mol–1 (s1a-IVb species in Figure 2a). In this case, the coordinated OH eases the CH3 migration of the DME to produce the CO-1 intermediate, via a barrier of 127 kJ mol–1 (hence significantly lower than for the nonwater asssisted process) and a reaction step endoenergetic by 31 kJ mol–1. These results show that water assists the DME/methanol conversion as already proposed for acidic zeolites.68,69 Hence, it is essential to generate Lewis acid sites adjacent to Lewis basic sites, so-called Frustrated Lewis acid base pairs, which display unexpected reactivities as more recently illustrated in molecular chemistry.70 Overall, the participation of (frustrated) Lewis acid–base pairs, acidic AlIII and AlIVb centers, and the basic oxygen atoms of both dimethyleter molecule and the OH group coordinated to such centers acts in a synergistic way providing a low energy pathway for the activation of DME.
Figure 2.

(a) C-OCH3 activation process assisted by an OH group of the CH3OCH3 molecule on γ-Al2O3. (b) Formation of methane and oxonium, carbon–carbon bond formation step (from CO-1 to CO-3) and subsequent ethylene formation along with s2a surface (c) Formate route from the CO-2 species. (d) Transition state structures corresponding to the formation of methane and oxonium (TS-CO-1-2) and (e) carbon–carbon bond formation steps (TS-CO-2-3). (f) Electronic energy profiles (in kJ mol–1) for the ethylene and formate formation. The energies refer to two CH3OCH3 and the γ-Al2O3 surface. For the ethylene route (dark blue), the second DME molecule is not depicted since it does not participate in the reaction. The formate route is depicted in brown.
After this step, the participation of two Al acid sites allows the previously transferred methyl group to abstract a hydride from the remaining methoxy group coordinated to AlIVb, generating methane and a Al–O=CH2 species with the O bound to AlIVb (CO-2a species in Figure 2b). In CO2a the −O=CH2 species is not interacting with the OH group of AlIII while it is in CO2b.71 In the corresponding transition-state (Figure 2d), a Al–O=CH2+ oxonium group is being formed along with methane. The oxonium species is characterized by a C=O distance equal to 1.328 Å at the transition-state, while the newly formed C–H bond has a distance equal to 1.452 Å and the one being broken equal to 1.240 Å (see Figure 2d). The formation of the oxonium has an energy barrier of 134 kJ mol–1 in a process endothermic by 26 kJ mol–1 when reaching CO2a. CH4 can be released or subsequently undergo a C–H bond activation by the Al–O=CH2+ oxonium group leading to the formation of ethanol coordinated to AlIVb (CO-3 in Figure 2b).58 This carbon–carbon bond forming step is exoenergetic by 146 kJ mol–1 and has an energy barrier of 86 kJ mol–1. The corresponding transition-state for the carbon–carbon bond formation step is shown as an inset in Figure 2e, in which the incoming carbon–carbon bond has a distance at the transition-state equal to 2.016 Å. Because of the structural similarity between TS-CO-12 and TS-CO-23 structures (corresponding to Figure 2d,e, respectively), there is also the possibility that from the TS-CO-12 structure a slight rotation of the methane molecule could lead directly to the TS-CO-23 structure. Finally, the formation of ethene (from the dehydration of the ethanol group in CO-3 giving the hydrated termination s2a of the alumina surface in Figure 2b) has an energy barrier equal to 159 kJ mol–1 in a process endothermic by 40 kJ mol–1. The formed water remains adsorbed on the AlIVb center. Alcohol dehydration on γ-Al2O3 has been addressed previously both experimentally44−47 and using DFT calculations on both 110 and 100 terminations.44,48−52 While the 100 facet is more active than the 110 one toward alcohol dehydration,50 both facets can allow this reaction, and the 110 facet exposes unsaturated AlIII and AlIVb sites, which are significantly more reactive toward C–H activation and able to allow for the carbon–carbon bond formation from CH3F to yield isobutene formation,58 in contrast to the Al sites present in the 100 surface.59 From the CO-2 intermediate, in the event where the formed methane departs, an alternative route can lead to the formation of the formate species. The OH group present in AlIII can interact with the Al–O=CH2+ group in the AlIVb via an interaction favorable by 57 kJ mol–1 (CO-2b). Subsequently, the OH group can decoordinate from the AlIII site, and a new DME molecule can coordinate to this Al center in a practically isoenergetic reaction (CO-4 from Figure 2c). In a subsequent step a hydrogen is transferred from the CH2 group of the AlIVb–OCH2–OH species to the DME molecule coordinated to AlIII in a process similar to that from CO-1 to CO-2a. This step gives methane as product, while a O=CHOH group remains bonded to AlIVb and a OCH3 group to AlIII (CO-5 species in Figure 2c). This step is exoenergetic (−95 kJ mol–1) and associated with an energy barrier equal to 115 kJ mol–1. Finally, the proton of the OH group can be transferred from the AlIVb-OCHOH species to the AlIII-OCH3 species via a very low energy barrier equal to 4 kJ mol–1 in a process exoergic by 76 kJ mol–1.
The formation of formate and methanol adsorbed on the surface (CO-6 species in Figure 2c) and two methane molecules is globally exoenergetic by 281 kJ mol–1 with respect to initial reactants (Al2O3 and two DME molecules), in agreement with the experimental observations. This route competes with ethylene formation in view of its similar energy barrier and more favorable thermodynamics. The whole energy profile for the formation of the ethylene from one DME molecule is shown in Figure 2f. In this energy profile, the energy barriers present values equal to 127–143 kJ mol–1, except the elimination step which produces ethylene, which is slightly higher: 159 kJ mol–1. All these barriers are accessible at 300 °C. From the partially hydrated alumina surface s1a and DME, the formation of ethylene and of the more hydrated alumina s2a surface is exothermic by 162 kJ mol–1. From this s2a surface, the water adsorbed on the AlIVb site can be exchanged by an incoming DME molecule regenerating the s1a-IVb species in an step endothermic by 44 kJ mol–1. The formate route is very favored thermodynamically, being exothermic by more than 281 kJ mol–1 with respect to initial reactants, in agreement with the experimental observation of formate on the γ-Al2O3 surface. The formate route is kinetically favored without considering entropic contributions, since at the branching point in the energy profile the barrier is lower by 28 kJ mol–1.
Overall, the formation of ethylene and water from DME is endoenergetic by 20 kJ mol–1, while when including entropic contributions the reaction is exothermic by 85 kJ mol–1 (eq 1). By comparison, the formation of formate and methanol from DME and water is exothermic by 210 kJ mol (eq 2).
| 1 |
| 2 |
In Gibbs free energy (see Supporting Information, Figure S10), the formate route is slightly more demanding than the ethylene route by 16 kJ mol–1. The energy barriers for both ethylene and formate routes are higher due to the stabilization of the gas phase species. By including the entropic terms, the formation of ethylene and the s2a surface is exothermic by 146 kJ mol–1, while the formate route leading to CO-6 and two CH4 molecules is exothermic by 209 kJ mol–1 (104 kJ mol–1 per DME molecule).
In conclusion, the reaction of DME on transition Al2O3 at 300 °C yields methane and higher olefins, which is reminiscent of the MTO process occurring on acidic zeolites. This reaction also generates methoxy and formate surface species according to IR and NMR data. These experiments and computational studies show that oxonium ions are key reaction intermediates. They form upon reaction of methoxy surface species with coordinated methanol on adjacent Al centers. The process involves C–H bond activation processes via a hydride abstraction from a surface methoxy species leading to a transition-state with methoxy, an oxonium group coordinated to AlIII and methane. The transient oxonium group can further react with methane yielding the first carbon–carbon bond or react with one additional DME molecule to form through a subsequent hydrogen transfer step a formate species, which was observed experimentally. These results show that the cooperation between adjacent aluminum sites of Al2O3 can readily participate in hydrogen transfer and C–C bond forming reaction processes. It also suggests that the “carbon pool” in the MTO process, which essentially takes place on the zeolite cavites of acidic zeolites via carbenium ions, can solely originate from methanol/DME and not from adventitious organic compounds. Higher hydrocarbons can be formed through the reaction of methanol/DME on highly acidic Al surface sites, which are also present in zeolites as part of the extraframework aluminum.
Acknowledgments
A.C.-V. and M.V. acknowledge support from the Swiss National Foundation (Grant numbers PZ00P2_148059 and 200021_143600, respectively). We thank Evonik and Sassol for providing Alu C and Sba 200 alumina samples, respectively. Dr. D. Estes is acknowledged for helping with the edition of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00226.
Details concerning preparation of the γ-Al2O3, additional NMR and IR spectra, and computational parameters (PDF)
Author Contributions
# A.C.-V. and M.V. contributed equally.
Swiss National Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Lunsford J. H. Die Katalytische Oxidative Kupplung von Methan. Angew. Chem. 1995, 107, 1059–1070. 10.1002/ange.19951070905. [DOI] [Google Scholar]
- Lunsford J. H. The Catalytic Oxidative Coupling of Methane. Angew. Chem., Int. Ed. Engl. 1995, 34, 970–980. 10.1002/anie.199509701. [DOI] [Google Scholar]
- Crabtree R. H. Aspects of Methane Chemistry. Chem. Rev. 1995, 95, 987–1007. 10.1021/cr00036a005. [DOI] [Google Scholar]
- Lunsford J. H. Catalytic Conversion of Methane to More Useful Chemicals and Fuels: a Challenge for the 21st Century. Catal. Today 2000, 63, 165–174. 10.1016/S0920-5861(00)00456-9. [DOI] [Google Scholar]
- Labinger J. A.; Bercaw J. E. Understanding and Exploiting C-H Bond Activation. Nature 2002, 417, 507–514. 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- Woertink J. S.; Smeets P. J.; Groothaert M. H.; Vance M. A.; Sels B. F.; Schoonheydt R. A.; Solomon E. I. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18908–18913. 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree R. H. Introduction to Selective Functionalization of C-H Bonds. Chem. Rev. 2010, 110, 575–575. 10.1021/cr900388d. [DOI] [PubMed] [Google Scholar]
- Copéret C. C-H Bond Activation and Organometallic Intermediates on Isolated Metal Centers on Oxide Surfaces. Chem. Rev. 2010, 110, 656–680. 10.1021/cr900122p. [DOI] [PubMed] [Google Scholar]
- Schwarz H. Chemie mit Methan: Studieren geht über Probieren!. Angew. Chem. 2011, 123, 10276–10297. 10.1002/ange.201006424. [DOI] [Google Scholar]
- Schwarz H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem., Int. Ed. 2011, 50, 10096–10115. 10.1002/anie.201006424. [DOI] [PubMed] [Google Scholar]
- Hazari N.; Iglesia E.; Labinger J. A.; Simonetti D. A. Selective Homogeneous and Heterogeneous Catalytic Conversion of Methanol/Dimethyl Ether to Triptane. Acc. Chem. Res. 2012, 45, 653–662. 10.1021/ar2002528. [DOI] [PubMed] [Google Scholar]
- Schwarz H. How and Why Do Cluster Size, Charge State, and Ligands Affect the Course of Metal-Mediated Gas-Phase Activation of Methane?. Isr. J. Chem. 2014, 54, 1413–1431. 10.1002/ijch.201300134. [DOI] [Google Scholar]
- Kwapien K.; Paier J.; Sauer J.; Geske M.; Zavyalova U.; Horn R.; Schwach P.; Trunschke A.; Schlögl R. Zentren der Methanaktivierung auf Oberflächen von Lithium-dotiertem MgO. Angew. Chem. 2014, 126, 8919–8923. 10.1002/ange.201310632. [DOI] [PubMed] [Google Scholar]
- Kwapien K.; Paier J.; Sauer J.; Geske M.; Zavyalova U.; Horn R.; Schwach P.; Trunschke A.; Schlögl R. Sites for Methane Activation on Lithium-Doped Magnesium Oxide Surfaces. Angew. Chem., Int. Ed. 2014, 53, 8774–8778. 10.1002/anie.201310632. [DOI] [PubMed] [Google Scholar]
- Tsai M.-L.; Hadt R. G.; Vanelderen P.; Sels B. F.; Schoonheydt R. A.; Solomon E. I. [Cu2O]2+ Active Site Formation in Cu-ZSM-5: Geometric and Electronic Structure Requirements for N2O Activation. J. Am. Chem. Soc. 2014, 136, 3522–3529. 10.1021/ja4113808. [DOI] [PubMed] [Google Scholar]
- Tian P.; Wei Y.; Ye M.; Liu Z. Methanol to Olefins (MTO): From Fundamentals to Commercialization. ACS Catal. 2015, 5, 1922–1938. 10.1021/acscatal.5b00007. [DOI] [Google Scholar]
- Stöcker M.Methanol to Olefins (MTO) and Methanol to Gasoline (MTG). In Zeolites and Catalysis; Cejka J., Corma A., Zones S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2010; pp 687–711. [Google Scholar]
- Haw J. F.; Song W.; Marcus D. M.; Nicholas J. B. The Mechanism of Methanol to Hydrocarbon Catalysis. Acc. Chem. Res. 2003, 36, 317–326. 10.1021/ar020006o. [DOI] [PubMed] [Google Scholar]
- Marcus D. M.; McLachlan K. A.; Wildman M. A.; Ehresmann J. O.; Kletnieks P. W.; Haw J. F. Experimental Evidence from H/D Exchange Studies for the Failure of Direct C-C Coupling Mechanisms in the Methanol-to-Olefin Process Catalyzed by HSAPO-34. Angew. Chem., Int. Ed. 2006, 45, 3133–3136. 10.1002/anie.200504372. [DOI] [PubMed] [Google Scholar]
- McCann D. M.; Lesthaeghe D.; Kletnieks P. W.; Guenther D. R.; Hayman M. J.; Van Speybroeck V.; Waroquier M.; Haw J. F. A Complete Catalytic Cycle for Supramolecular Methanol-to-Olefins Conversion by Linking Theory with Experiment. Angew. Chem., Int. Ed. 2008, 47, 5179–5182. 10.1002/anie.200705453. [DOI] [PubMed] [Google Scholar]
- Van Speybroeck V.; De Wispelaere K.; Van der Mynsbrugge J.; Vandichel M.; Hemelsoet K.; Waroquier M. First principle chemical kinetics in zeolites: the methanol-to-olefin process as a case study. Chem. Soc. Rev. 2014, 43, 7326–7357. 10.1039/C4CS00146J. [DOI] [PubMed] [Google Scholar]
- Blaszkowski S. R.; van Santen R. A. Theoretical Study of C-C Bond Formation in the Methanol-to-Gasoline Process. J. Am. Chem. Soc. 1997, 119, 5020–5027. 10.1021/ja963530x. [DOI] [Google Scholar]
- Tajima N.; Tsuneda T.; Toyama F.; Hirao K. A New Mechanism for the First Carbon-Carbon Bond Formation in the MTG Process: A Theoretical Study. J. Am. Chem. Soc. 1998, 120, 8222–8229. 10.1021/ja9741483. [DOI] [Google Scholar]
- Li J.; Wei Z.; Chen Y.; Jing B.; He Y.; Dong M.; Jiao H.; Li X.; Qin Z.; Wang J.; Fan W. A route to form initial hydrocarbon pool species in methanol conversion to olefins over zeolites. J. Catal. 2014, 317, 277–283. 10.1016/j.jcat.2014.05.015. [DOI] [Google Scholar]
- Lesthaeghe D.; Van Speybroeck V.; Marin G. B.; Waroquier M. Understanding the failure of direct C-C coupling in the zeolite-catalyzed methanol-to-olefin process. Angew. Chem., Int. Ed. 2006, 45, 1714–1719. 10.1002/anie.200503824. [DOI] [PubMed] [Google Scholar]
- Sun X.; Mueller S.; Liu Y.; Shi H.; Haller G. L.; Sanchez-Sanchez M.; van Veen A. C.; Lercher J. A. On reaction pathways in the conversion of methanol to hydrocarbons on HZSM-5. J. Catal. 2014, 317, 185–197. 10.1016/j.jcat.2014.06.017. [DOI] [Google Scholar]
- Olah G. A.; Doggweiler H.; Felberg J. D.; Frohlich S.; Grdina M. J.; Karpeles R.; Keumi T.; Inaba S.-i.; Ip W. M.; Lammertsma K.; Salem G.; Tabor D. Onium Ylide chemistry. 1. Bifunctional acid-base-catalyzed conversion of heterosubstituted methanes into ethylene and derived hydrocarbons. The onium ylide mechanism of the C1 -> C2 conversion. J. Am. Chem. Soc. 1984, 106, 2143–2149. 10.1021/ja00319a039. [DOI] [Google Scholar]
- Stöcker M. Methanol-to-hydrocarbons: catalytic materials and their behavior. Microporous Mesoporous Mater. 1999, 29, 3–48. 10.1016/S1387-1811(98)00319-9. [DOI] [Google Scholar]
- Hemelsoet K.; Van der Mynsbrugge J.; De Wispelaere K.; Waroquier M.; Van Speybroeck V. Unraveling the Reaction Mechanisms Governing Methanol-to-Olefins Catalysis by Theory and Experiment. ChemPhysChem 2013, 14, 1526–1545. 10.1002/cphc.201201023. [DOI] [PubMed] [Google Scholar]
- Olsbye U.; Svelle S.; Bjørgen M.; Beato P.; Janssens T. V. W.; Joensen F.; Bordiga S.; Lillerud K. P. Umwandlung von Methanol in Kohlenwasserstoffe: Wie Zeolith-Hohlräume und Porengröße die Produktselektivität bestimmen. Angew. Chem. 2012, 124, 5910–5933. 10.1002/ange.201103657. [DOI] [Google Scholar]
- Olsbye U.; Svelle S.; Bjørgen M.; Beato P.; Janssens T. V. W.; Joensen F.; Bordiga S.; Lillerud K. P. Conversion of Methanol to Hydrocarbons: How Zeolite Cavity and Pore Size Controls Product Selectivity. Angew. Chem., Int. Ed. 2012, 51, 5810–5831. 10.1002/anie.201103657. [DOI] [PubMed] [Google Scholar]
- Dahl I. M.; Kolboe S. On the Reaction-Mechanism for Propene Formation in the Mto Reaction over Sapo-34. Catal. Lett. 1993, 20, 329–336. 10.1007/BF00769305. [DOI] [Google Scholar]
- Dahl I. M.; Kolboe S. On the Reaction-Mechanism for Hydrocarbon Formation from Methanol over Sapo-34 0.1. Isotopic Labeling Studies of the Co-Reaction of Ethene and Methanol. J. Catal. 1994, 149, 458–464. 10.1006/jcat.1994.1312. [DOI] [Google Scholar]
- Dahl I. M.; Kolboe S. On the reaction mechanism for hydrocarbon formation from methanol over SAPO-34 0.2. Isotopic labeling studies of the co-reaction of propene and methanol. J. Catal. 1996, 161, 304–309. 10.1006/jcat.1996.0188. [DOI] [Google Scholar]
- Lesthaeghe D.; Van der Mynsbrugge J.; Vandichel M.; Waroquier M.; Van Speybroeck V. Full Theoretical Cycle for both Ethene and Propene Formation during Methanol-to-Olefin Conversion in H-ZSM-5. ChemCatChem 2011, 3, 208–212. 10.1002/cctc.201000286. [DOI] [Google Scholar]
- Lesthaeghe D.; Van Speybroeck V.; Marin G. B.; Waroquier M. What Role do Oxonium Ions and Oxonium Ylides play in the ZSM-5 Catalysed Methanol-to-Olefin Process?. Chem. Phys. Lett. 2006, 417, 309–315. 10.1016/j.cplett.2005.09.136. [DOI] [Google Scholar]
- Silaghi M.-C.; Chizallet C.; Raybaud P. Challenges on molecular aspects of dealumination and desilication of zeolites. Microporous Mesoporous Mater. 2014, 191, 82–96. 10.1016/j.micromeso.2014.02.040. [DOI] [Google Scholar]
- Corma A.; Martinez A.; Martinez C. The role of extraframework aluminum species in USY catalysts during isobutane/2-butene alkylation. Appl. Catal., A 1996, 134, 169–182. 10.1016/0926-860X(95)00228-6. [DOI] [Google Scholar]
- Cejka J., Corma A.; Zones S.. Zeolites and Catalysis: Synthesis, Reactions and Applications; Wiley-VCH: Weinheim, 2010. [Google Scholar]
- Chang C. D. Hydrocarbons from Methanol. Catal. Rev.: Sci. Eng. 1983, 25, 1–118. 10.1080/01614948308078874. [DOI] [Google Scholar]
- Xu M.; Lunsford J. H.; Goodman D. W.; Bhattacharyya A. Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts. Appl. Catal., A 1997, 149, 289–301. 10.1016/S0926-860X(96)00275-X. [DOI] [Google Scholar]
- Yaripour F.; Baghaei F.; Schmidt I.; Perregaard J. Catalytic dehydration of methanol to dimethyl ether (DME) over solid-acid catalysts. Catal. Commun. 2005, 6, 147–152. 10.1016/j.catcom.2004.11.012. [DOI] [Google Scholar]
- Schiffino R. S.; Merrill R. P. A mechanistic study of the methanol dehydration reaction on.gamma.-alumina catalyst. J. Phys. Chem. 1993, 97, 6425–6435. 10.1021/j100126a017. [DOI] [Google Scholar]
- Roy S.; Mpourmpakis G.; Hong D.-Y.; Vlachos D. G.; Bhan A.; Gorte R. J. Mechanistic Study of Alcohol Dehydration on γ-Al2O3. ACS Catal. 2012, 2, 1846–1853. 10.1021/cs300176d. [DOI] [Google Scholar]
- Kang M.; DeWilde J. F.; Bhan A. Kinetics and Mechanism of Alcohol Dehydration on γ-Al2O3: Effects of Carbon Chain Length and Substitution. ACS Catal. 2015, 5, 602–612. 10.1021/cs501471r. [DOI] [Google Scholar]
- DeWilde J. F.; Chiang H.; Hickman D. A.; Ho C. R.; Bhan A. Kinetics and Mechanism of Ethanol Dehydration on γ-Al2O3: The Critical Role of Dimer Inhibition. ACS Catal. 2013, 3, 798–807. 10.1021/cs400051k. [DOI] [Google Scholar]
- Phung T. K.; Lagazzo A.; Rivero Crespo M. Á.; Sánchez Escribano V.; Busca G. A study of commercial transition aluminas and of their catalytic activity in the dehydration of ethanol. J. Catal. 2014, 311, 102–113. 10.1016/j.jcat.2013.11.010. [DOI] [Google Scholar]
- Christiansen M. A.; Mpourmpakis G.; Vlachos D. G. Density Functional Theory-Computed Mechanisms of Ethylene and Diethyl Ether Formation from Ethanol on γ-Al2O3(100). ACS Catal. 2013, 3, 1965–1975. 10.1021/cs4002833. [DOI] [Google Scholar]
- Jenness G. R.; Christiansen M. A.; Caratzoulas S.; Vlachos D. G.; Gorte R. J. Site-Dependent Lewis Acidity of γ-Al2O3 and Its Impact on Ethanol Dehydration and Etherification. J. Phys. Chem. C 2014, 118, 12899–12907. 10.1021/jp5028349. [DOI] [Google Scholar]
- Kwak J. H.; Rousseau R.; Mei D.; Peden C. H. F.; Szanyi J. The Origin of Regioselectivity in 2-Butanol Dehydration on Solid Acid Catalysts. ChemCatChem 2011, 3, 1557–1561. 10.1002/cctc.201100173. [DOI] [Google Scholar]
- Christiansen M. A.; Mpourmpakis G.; Vlachos D. G. DFT-driven multi-site microkinetic modeling of ethanol conversion to ethylene and diethyl ether on γ-Al2O3 (111). J. Catal. 2015, 323, 121–131. 10.1016/j.jcat.2014.12.024. [DOI] [Google Scholar]
- Larmier K.; Chizallet C.; Cadran N.; Maury S.; Abboud J.; Lamic-Humblot A.-F.; Marceau E.; Lauron-Pernot H. Mechanistic investigation of isopropanol conversion on alumina catalysts: location of active sites for alkene/ether production. ACS Catal. 2015, 5, 4423–4437. 10.1021/acscatal.5b00723. [DOI] [Google Scholar]
- Chen J. G.; Basu P.; Ballinger T. H.; Yates J. T. A transmission infrared spectroscopic investigation of the reaction of dimethyl ether with alumina surfaces. Langmuir 1989, 5, 352–356. 10.1021/la00086a011. [DOI] [Google Scholar]
- Kipnis M. A.; Samokhin P. V.; Volnina E. A.; Lin G. I. Surface reactions of dimethyl ether on γ-Al2O3: 1. Adsorption and thermal effects. Kinet. Catal. 2014, 55, 456–462. 10.1134/S0023158414040107. [DOI] [Google Scholar]
- Wischert R.; Copéret C.; Delbecq F.; Sautet P. Dinitrogen: a selective probe for tri-coordinate Al ″defect″ sites on alumina. Chem. Commun. 2011, 47, 4890–4892. 10.1039/c1cc10623f. [DOI] [PubMed] [Google Scholar]
- Wischert R.; Copéret C.; Delbecq F.; Sautet P. Optimal Water Coverage on Alumina: A Key to Generate Lewis Acid–Base Pairs that are Reactive Towards the C-H Bond Activation of Methane. Angew. Chem. 2011, 123, 3260–3263. 10.1002/ange.201006794. [DOI] [PubMed] [Google Scholar]
- Wischert R.; Copéret C.; Delbecq F.; Sautet P. Optimal Water Coverage on Alumina: A Key to Generate Lewis Acid–Base Pairs that are Reactive Towards the C-H Bond Activation of Methane. Angew. Chem., Int. Ed. 2011, 50, 3202–3205. 10.1002/anie.201006794. [DOI] [PubMed] [Google Scholar]
- Comas-Vives A.; Schwarzwälder M.; Copéret C.; Sautet P. Carbon–Carbon Bond Formation by Activation of CH3F on Alumina. J. Phys. Chem. C 2015, 119, 7156–7163. 10.1021/jp512598p. [DOI] [Google Scholar]
- Wischert R.; Laurent P.; Copéret C.; Delbecq F.; Sautet P. γ-Alumina: The Essential and Unexpected Role of Water for the Structure, Stability, and Reactivity of ″Defect″ Sites. J. Am. Chem. Soc. 2012, 134, 14430–14449. 10.1021/ja3042383. [DOI] [PubMed] [Google Scholar]
- Note that above 300 °C the dimethyl ether undergoes a homolytic cleavage of the CH3–OCH3 bond leaving two radicals in the gas phase which can react with the alumina surface. For this reason, the system will not be studied above 300 °C (see ref (27)).
- Digne M.; Sautet P.; Raybaud P.; Euzen P.; Toulhoat H. Hydroxyl Groups on γ-Alumina Surfaces: A DFT Study. J. Catal. 2002, 211, 1–5. 10.1006/jcat.2002.3741. [DOI] [Google Scholar]
- Digne M.; Sautet P.; Raybaud P.; Euzen P.; Toulhoat H. Use of DFT to Achieve a Rational Understanding of Acid-Basic Properties of γ-Alumina Surfaces. J. Catal. 2004, 226, 54–68. 10.1016/j.jcat.2004.04.020. [DOI] [Google Scholar]
- Rataboul F.; Baudouin A.; Thieuleux C.; Veyre L.; Copéret C.; Thivolle-Cazat J.; Basset J.-M.; Lesage A.; Emsley L. Molecular Understanding of the Formation of Surface Zirconium Hydrides upon Thermal Treatment under Hydrogen of [(≡SiO)Zr(CH2tBu)3] by Using Advanced Solid-State NMR Techniques. J. Am. Chem. Soc. 2004, 126, 12541–12550. 10.1021/ja038486h. [DOI] [PubMed] [Google Scholar]
- Koga O.; Onishi T.; Tamaru K. Infrared emission spectra of formic acid adsorbed on alumina. J. Chem. Soc., Chem. Commun. 1974, 464–464. 10.1039/c39740000464. [DOI] [Google Scholar]
- Rubasinghege G.; Ogden S.; Baltrusaitis J.; Grassian V. H. Heterogeneous Uptake and Adsorption of Gas-Phase Formic Acid on Oxide and Clay Particle Surfaces: The Roles of Surface Hydroxyl Groups and Adsorbed Water in Formic Acid Adsorption and the Impact of Formic Acid Adsorption on Water Uptake. J. Phys. Chem. A 2013, 117, 11316–11327. 10.1021/jp408169w. [DOI] [PubMed] [Google Scholar]
- Krokidis X.; Raybaud P.; Gobichon A. E.; Rebours B.; Euzen P.; Toulhoat H. Theoretical Study of the Dehydration Process of Boehmite to γ-alumina. J. Phys. Chem. B 2001, 105, 5121–5130. 10.1021/jp0038310. [DOI] [Google Scholar]
- An energy barrier of 100 kJ·mol–1 at 25 °C is equivalent in rate to an energy barrier equal to 195 kJ·mol–1 at 300 °C.
- Jones A. J.; Iglesia E. Kinetic, Spectroscopic, and Theoretical Assessment of Associative and Dissociative Methanol Dehydration Routes in Zeolites. Angew. Chem., Int. Ed. 2014, 53, 12177–12181. 10.1002/anie.201406823. [DOI] [PubMed] [Google Scholar]
- Blaszkowski S. R.; van Santen R. A. Theoretical Study of the Mechanism of Surface Methoxy and Dimethyl Ether Formation from Methanol Catalyzed by Zeolitic Protons. J. Phys. Chem. B 1997, 101, 2292–2305. 10.1021/jp962006+. [DOI] [Google Scholar]
- Stephan D. W.; Erker G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem., Int. Ed. 2010, 49, 46–76. 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]
- In ref (58), we proposed the analogous pathway for the formation of an ethoxy intermediate taking CH3F as initial substrate.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.