

Abstract
Drug toxicity is frequently caused by electrophilic reactive metabolites that covalently bind to proteins. Epoxides comprise a large class of three-membered cyclic ethers. These molecules are electrophilic and typically highly reactive due to ring tension and polarized carbon–oxygen bonds. Epoxides are metabolites often formed by cytochromes P450 acting on aromatic or double bonds. The specific location on a molecule that undergoes epoxidation is its site of epoxidation (SOE). Identifying a molecule’s SOE can aid in interpreting adverse events related to reactive metabolites and direct modification to prevent epoxidation for safer drugs. This study utilized a database of 702 epoxidation reactions to build a model that accurately predicted sites of epoxidation. The foundation for this model was an algorithm originally designed to model sites of cytochromes P450 metabolism (called XenoSite) that was recently applied to model the intrinsic reactivity of diverse molecules with glutathione. This modeling algorithm systematically and quantitatively summarizes the knowledge from hundreds of epoxidation reactions with a deep convolution network. This network makes predictions at both an atom and molecule level. The final epoxidation model constructed with this approach identified SOEs with 94.9% area under the curve (AUC) performance and separated epoxidized and non-epoxidized molecules with 79.3% AUC. Moreover, within epoxidized molecules, the model separated aromatic or double bond SOEs from all other aromatic or double bonds with AUCs of 92.5% and 95.1%, respectively. Finally, the model separated SOEs from sites of sp2 hydroxylation with 83.2% AUC. Our model is the first of its kind and may be useful for the development of safer drugs. The epoxidation model is available at http://swami.wustl.edu/xenosite.
Short abstract
Epoxide metabolites frequently cause drug toxicity. A deep convolution network accurately predicts the epoxidation of drug-like molecules. This model may guide efforts to modify drug candidates to be less toxic.
Introduction
Drug discovery and development involve significant efforts to identify safe and efficacious drugs; nevertheless, unanticipated toxicity and adverse drug reactions do occur and cause approximately 40% of drug candidates to fail.1 Frequently, these harmful outcomes are linked to the formation of electrophilic metabolites that covalently bind to proteins or DNA and, in some cases, elicit an immune response in susceptible patients.2−6 One of the most common types of reactive metabolites are epoxides, the subject of this study.
Epoxides are three membered cyclic ethers and are often highly reactive due to ring tension and polarized carbon–oxygen bonds.7−11 Epoxides are formed by cytochromes P450 acting on aromatic or double bonds,12,13 and these epoxidation reactions comprise around 10%14 to 15%15 of all bioactivation reactions. Biological defense mechanisms to epoxides, including glutathione conjugation and cleavage by epoxide hydrolase, offer only partial protection.7,11,16,17 Glutathione can be depleted,18,19 and certain products of glutathione conjugation17 and epoxide hydrolase20,21 are themselves toxic.
Epoxide metabolites often drive toxicity for drugs, and accurate strategies for anticipating the formation of epoxides are critical in drug development. Knowledge of epoxide formation aids assessment of drug candidates. Furthermore, the identity of the specific bond in a molecule undergoing epoxidation, its site of epoxidation (SOE), could enable rational modification of the molecule to reduce risk of reactive metabolite formation. An example of how this knowledge can lead to drugs with improved safety is illustrated by carbamazepine (Figure 1). The metabolism of this anti-epileptic drug forms carbamazepine-10,11-epoxide. Carbamazepine metabolism can also form an iminoquinone,22 but the epoxide’s formation is the focus of this study and more correlated with adverse reactions.23−25 The molecular mechanism for this response involves reactions between the epoxide and proteins to form adducts.26 However, the epoxide formation can be blocked by modifying carbamazepine’s SOE. For example, oxcarbazepine23 or eslicarbazepine are analogues of carbamazepine that are no longer epoxidized.25 While oxcarbazepine and eslicarbazepine were not prospectively designed in order to reduce epoxide formation, they demonstrate how small molecular changes can significantly impact toxicity caused by epoxide metabolites. These analogues retain the same mechanism of action as carbamazepine, yet have a lower incidence of adverse effects because they prevent the formation of epoxides.25,27
Figure 1.

Adverse drug reactions are often caused by reactive metabolites. For example, carbamazepine is metabolized by cytochromes P450 to carbamazepine-10,11-epoxide. Carbamazepine metabolism can also form an iminoquinone,22 but the epoxide’s formation is the focus of this study and more correlated with adverse reactions.23−25 The epoxide is electrophilically reactive and covalently binds to nucleophilic sites within proteins. The resulting adduct serves as a hapten complex and elicits an immune response. This mechanism is thought to be responsible for many carbamazepine adverse reactions.35,36 This site of epoxidation is circled on carbamazepine.
A number of studies, including those by our group, have established that computational methods can predict the sites at which molecules are metabolized.28−33 A shortcoming of those approaches has been the lack of predictions for the actual metabolites generated by those reactions. Cytochromes P450 catalyze many different types of oxidative reactions, including commonly observed hydroxylations.12,30,34 While several cytochromes P450 site of metabolism models are reported in the literature, to the best of our knowledge, none of those models specifically identify SOEs in molecules. Instead, all existing methods only report which atoms undergo oxidation, without distinguishing the specific type of reaction—such as epoxidation or hydroxylation—or the resulting modification to the structure.
In this study, we construct an epoxidation model—based on the structural data of several hundred diverse molecules—that is successful at three key objectives. First, the model accurately predicts SOE within epoxidized molecules; these SOE predictions can be used to direct structural modifications to drug candidates. Second, the model distinguishes SOE from sites of sp2 hydroxylation (SOH), a key negative control. Both SOEs and SOHs are oxidized by P450s, and we expect a useful model to correctly identify which of these oxidations give rise to epoxides. In contrast, commonly reported P450 site of metabolism models will not distinguish these two cases and report both as sites of metabolism. Third, the model identifies which molecules are metabolized into epoxides, separating these molecules from closely related molecules that are not epoxidized. This enables rapid screening of drug candidates for molecules that are potentially toxic due to epoxidation.
Methods
Epoxidation Training Data
We mined a large, chemically diverse training data set from the Accelrys Metabolite Database (AMD), which includes a collection of metabolic reactions drawn from the literature. A total of 702 reactions were extracted, each of which takes place in humans, human cells, or human microsomes and is classified as epoxidation. Because of the short half-life of many epoxides, however, some product molecules do not explicitly contain an epoxide. Instead, an epoxidation product may be a dihydrodiol or a DNA, glutathione, or protein conjugate (Figure 2).37,38 An automated labeling algorithm used these motifs to label SOEs on the starting molecule of each reaction.
Figure 2.

In the database, each epoxidation reaction acting on a site of epoxidation (abbreviated SOE and circled) forms an epoxide, dihydrodiol, or a conjugate adjacent to a hydroxylation. For example, the epoxidation reaction of nevirapine forms an epoxide (top),40 of N-desmethyl triflubazam forms a dihydrodiol (middle),41 and of benzo(a)pyrene forms a DNA conjugate adjacent to a hydroxylation (bottom).38 The first case explicitly records the epoxide, while the other two record a tell-tale signature of a transient, reactive epoxide that is not directly observed.37,38 A total of 702 human epoxidation reactions were identified in the Accelrys Metabolite Database. An automated labeling algorithm labeled SOEs on the starting molecule of each reaction based on these motifs.
In this study, we defined an SOE as the bond between the two carbons to which an epoxide forms and identified these bonds in depictions with circles. When bonds were topologically equivalent to observed SOEs, as identified using the Pybel python library, they were themselves labeled as SOE.39 Duplicate starting molecules were identified by canonical SMILES and merged into a single training example with all observed SOEs labeled. The final data set included 389 epoxidized molecules, each with its SOEs labeled. These epoxidized molecules included 411 aromatic bond SOEs and 168 double bond SOE. Additionally, 20 single bond SOEs were included; the labeling of single bonds as SOEs is likely due to rearrangements or intermediates—absent from the database—allowing epoxidation to occur at an aromatic or double bond.
We also identified structurally similar but non-epoxidized molecules. These target compounds were mined from the reaction network for each previously identified epoxidized molecule. This strategy ensured the inclusion of the metabolic parent and sibling molecules so that a robust distinction between molecules undergoing epoxidation and those that are not became possible. After excluding molecules already classified as epoxidized, the remaining 135 molecules were marked non-epoxidized. Each one was metabolically studied and chemically similar to an epoxidized molecule in the data set.
Our license for the AMD data did not allow us to disclose the structures of the full data set. However, all molecule registry numbers are included in the Supporting Information, and this is sufficient data to rebuild the database and reproduce our results.
Hydroxylation Negative Control Data
As discussed in the Introduction, sp2 sites can be either epoxidized or hydroxylated. An epoxidation model must be validated using hydroxylation data as a negative control to distinguish the epoxidation model from a general oxidation model. An epoxidation model should rank SOEs above SOHs, whereas an oxidation model would rank them approximately equally. For use as negative controls, we also extracted SOHs from the AMD. Both SOHs and SOEs are acted on by cytochromes P450, but the epoxides formed from SOEs are more likely to be toxic. To build a hydroxylation test data set, 3000 human hydroxylation reactions were randomly sampled from the AMD. We filtered out sp3 hydroxylations and any SOHs that included non-carbon atoms, both of which are easily distinguishable from epoxidations. After these filtrations, 1105 hydroxylations remained. Duplicate starting molecules were identified by canonical SMILES and merged by labeling all known SOHs for each molecule. This final data set included 811 molecules, each with bonds adjacent to hydroxylations labeled as SOHs.
Descriptors
Our approach used information encoded in descriptors for each bond to assess its susceptibility to epoxidation. Each bond was associated with a total of 214 numerical descriptors, including atom-level, bond-level, and molecule-level descriptors. Descriptors were calculated by in-house software that took as input SDF files with explicit hydrogens and 3D coordinates created by Open Babel.42 The majority of our descriptors were atom-level descriptors previously developed for the XenoSite metabolism model28 and the XenoSite reactivity model.43 Each bond contained 89 descriptors from its “left” atom and its “right” atom. To prevent representation bias due to atom ordering, left and right atom assignment was randomized on a bond-by-bond basis. Twenty-three molecule-level descriptors, reported in our prior work, were also computed and used by the network to make predictions.
We supplemented these atom and molecule descriptors with bond descriptors developed specifically to capture the chemical properties of bonds. These 13 new bond descriptors are summarized in Table 1; a comprehensive table of the descriptors used in this study is available in the Supporting Information. There were two types of bond descriptors. First, topological bond descriptors summarized information from the molecular 2D structure. Second, quantum chemical descriptors were calculated from self-consistent field computations by MOPAC, a semiempirical quantum chemistry modeler, utilizing an implicit solvent model and the PM7 force field.44,45
Table 1. Condensed List of Bond Descriptors Developed for This Studya.
| Topological Bond Descriptors | |
|---|---|
| single bond | binary value indicating whether bond is a single bond |
| aromatic bond | binary value indicating whether bond is an aromatic bond |
| double bond | binary value indicating whether bond is a double bond |
| conjugated bond | binary value indicating whether bond is conjugated |
| triple bond | binary value indicating whether bond is a triple bond |
| topologically equivalent | number of topologically equivalent bonds in the same molecule |
| Quantum Chemical Bond Descriptors | |
|---|---|
| bond length | distance between the two atoms of a bond |
| σs occupancy | interaction of s-orbital σ bond electrons |
| σp occupancy | interaction of p-orbital σ bond electrons |
| πp occupancy | interaction of p-orbital π bond electrons |
| σs–σp occupancy | interaction of s-orbital σ bond and p-orbital σ bond electrons |
| σs–πp occupancy | interaction of s-orbital σ bond and p-orbital π bond electrons |
| σp–πp occupancy | interaction of p-orbital σ bond and p-orbital π bond electrons |
Descriptors were generated using both topological and quantum chemical information. A full list of descriptors used in this study is available in the Supporting Information.
In total, 214 numbers were used to describe each bond: 89 atom descriptors for the “left” atom, 89 for the “right” atom, 23 molecule descriptors, and 13 bond specific descriptors.
Combined Atom- and Molecule-Level Epoxidation Model
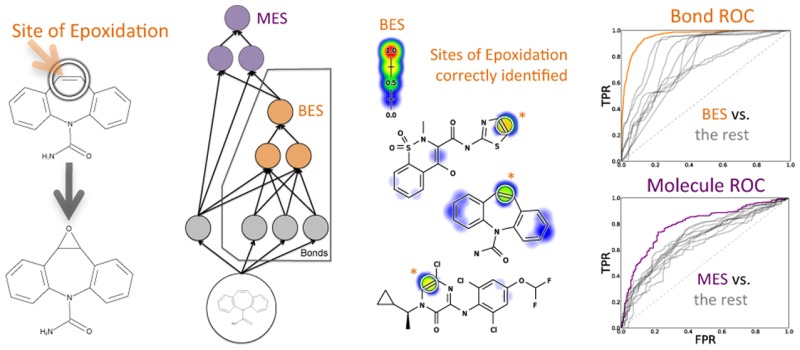
We built a model for bond and molecule epoxidation using a deep neural network with one input layer, two hidden layers, and two output layers (Figure 3). The top-level output layer computed molecule-level predictions called the molecule epoxidation scores (MES); the next output layer computed bond-level predictions called the bond epoxidation scores (BES). Here, the term “deep network” does not mean a deep autoencoder network as is being increasingly used.46 Instead, we mean a deep convolution network, with many more layers than a standard network and extensive weight sharing between replicates of the BES network.47 This network was trained in two stages.
Figure 3.

The structure of the epoxidation model. This diagram shows how information flowed through the model, which was composed of one input layer, two hidden layers, and two output layers. This model computed a molecule-level prediction for each test molecule as well as predictions for each bond within that test molecule. From the 3D structure of an input molecule, 23 molecule-level and 191 bond-associated descriptors were calculated. These inputs nodes are inputted into the first hidden layer (with 10 nodes), which outputs a bond epoxidation score (BES) for each bond in the molecule. The BES quantifies the probability that the bond is a site of epoxidation. The top five BES, and all molecule-level descriptors, flow into the second hidden layer (with 10 nodes), which outputs a single molecule epoxidation score (MES) for the input molecule, reflecting the probability that the molecule will be epoxidized. For conciseness, the diagram is abbreviated and only shows two nodes for each hidden layer, one molecule input node, two atom input nodes (for each atom associated with the bond), and one bond input node. The actual model had several additional nodes in the input and hidden layers.
First, we trained the bond-level network to compute accurate BES values. In this training, each bond within a molecule was considered a possible SOE. Each bond had a vector of numbers (descriptors), with each entry of the vector describing a chemical property of that bond. The data set was a matrix, structured as one column per descriptor, and one row per bond. A final binary target vector labeled experimentally observed SOEs with a 1. The weights of the network were trained using gradient descent on the cross-entropy error, so that SOEs scored higher BES than other bonds. These BES ranged from zero to one, representing the probability that a bond was an SOE.
Second, the molecule-level output layer was trained to compute MES values. Several versions of this output layer were considered, including another multilayer neural network, a logistic regressor, and a max layer that computed the MES as the maximum BES observed in the molecule. The logistic regressor and neural network took as input the top five BES, as well as all molecule-level descriptors. As we will see, both the neural network and the logistic regressor offer better scaled predictions with higher classification performances than the simpler max layer.
Results and Discussion
The following sections study the classification performance and inner workings of the epoxidation model. First, we evaluated the ability of BES to predict the SOE of epoxidized molecules. Second, we considered the credibility of the model by analyzing which descriptors are most important to the model’s performance. Third, we increased resolution on the quality of the model predictions by calculating classification performance on aromatic and double bonds individually. Fourth, we asked whether BES distinguish SOEs from sites of sp2 hydroxylation, because both epoxidation and sp2 hydroxylation are catalyzed by P450s but have significantly different implications for toxicity. Fifth, we tested how well MES separated epoxidized and non-epoxidized molecules. Finally, we studied how the model could direct drug modifications to reduce toxicity of known drugs.
Accuracy in Identifying Sites of Epoxidation
An important goal for designing drugs less prone to metabolic activation is to accurately identify the site (bond) within a molecule that undergoes epoxidation. In our study, SOE predictions gave a specific hypothesis about the mechanism of a molecule’s toxicity. Furthermore, knowledge of the SOE lays a strong foundation for guiding the modification of a molecule to make it less susceptible to epoxidation and thus less likely to cause protein and DNA adducts that lead to toxic effects. There are currently no other published computational methods that specifically predict SOEs among a diverse set of molecules.
The trained model predicts SOEs by computing a BES for each bond in a test molecule. These scores ranged between zero and one and reflected the probability that an epoxide will form on the two atoms within the corresponding bond. If accurate, BES should discriminate between SOEs and all other bonds within epoxidized molecules.
We assessed the generalization performance of our model using a cross-validation protocol. In this procedure, we separated molecules into metabolically related groups that represented metabolic networks in the database. Each group was comprised of epoxidized molecules and all parent and sibling molecules of those epoxidized molecules. One by one, each group of molecules derived from these networks was withheld from the training set. The rest of the molecules was used to train a model and make predictions on all the molecules present in the group left out of the training process. In each cross-validation fold, the model predictions for test molecules then did not depend on training data from identical or closely related molecules and thus provided a rigorous evaluation of the model. In this way, BES predictions were made on all molecules in the training data.
We used two metrics to quantitatively measure the classification performance of the cross-validated BES. First, we computed the “average site AUC” by calculating the area under the ROC curve (AUC) for each molecule and quantified the whole data set performance by averaging the AUCs for each molecule in the data set. Second, we used the “top-two” metric, which is often used in site of metabolism prediction.28,48,49 By this metric, a molecule was considered correctly predicted if any of its observed SOEs were predicted in the first- or second-rank position by a given model. Both metrics measure the separation of known SOEs from all other bonds within each molecule known to undergo epoxidation.
The BES reported by the neural network model accurately identified SOEs with an average site AUC performance of 94.9% and a top-two performance of 83.0% (Figure 4). The neural network outperformed a simpler logistic regressor model (BES[LR] in the figure), which had an average site AUC performance of 93.7% and a top-two performance of 80.5%. The neural network was significantly more accurate than the logistic regressor, reducing the error by 19.0% (average site AUC) and 12.8% (top-two). This improvement is significant according to a paired t-test, with p-values of 0.000454 (average site AUC) and 0.0328 (top-two).50 This improvement indicated nonlinearity in the epoxidation data that cannot be taken into account by a logistic regressor. This finding justified the use of the more complex neural network and was consistent with a previous study on site of metabolism prediction,51 as well as our previous work on sites of glutathione reactivity.43
Figure 4.

Bond epoxidation scores accurately (BES) identify sites of epoxidation (SOEs). Top left, for each prediction method, average site AUC was computed for 389 molecules extracted from the Accelrys Metabolite Database with their SOEs labeled. This metric reflected how often SOEs were ranked above other sites within these molecules. Bottom left, top-two classification performance was computed, by which a molecule was considered correctly predicted if any of its observed SOEs were predicted in the first- or second-rank position. By both metrics, the cross-validated predictions generated by a neural network (BES) outperformed the predictions of a logistic regressor (BES[LR]). The classification performance of BES also exceeded that of all raw descriptors, the five best of which are included in each panel. Right, examples from the data set are visualized with their predictions.52−54 In the bar graph axis, the two-center electron–nuclear attraction energy is abbreviated as electron–nuclear attraction. For each molecule, the colored shading represents BES, which range from 0 to 0.73. Each experimentally observed SOE is circled.
This model for epoxidation is the first of its kind, and thus there are no other published models to which performance can be compared. Instead, we tested the performance of each raw descriptor to provide a baseline for comparison. Each descriptor was treated as a very simple model limited to a single chemical attribute to predict SOE. The best performing descriptor was πp occupancy; however, this descriptor significantly underperformed our model, with accuracies of 90.8% (average site AUC) and 72.8% (top-two). Using machine learning to collectively consider many chemical attributes classified SOEs more accurately than any attribute considered in isolation.
Descriptors Driving Bond Epoxidation Score Performance
We identified which descriptors the model relied upon by using sensitivity analysis by using sensitivity analysis to further assess the sensibleness of the model. The contribution of individual descriptors for identifying SOEs was measurable with a permutation sensitivity analysis.43,55 First, a baseline model was built using the entire training data set, and its performance was calculated on this training data. The average site AUC performance was used for the sensitivity analysis, because it most closely measures performance in the intended use case. It quantifies how accurately the model identifies the correct SOEs within epoxidized test molecules, relative to all other potential sites, on a molecule-by-molecule basis. Reassuringly, very similar results from the sensitivity analysis are obtained using other metrics (data not shown). Next, the influence of individual descriptors, as well as groups of descriptors, was measured by recording the drop in the model’s performance on the training data when the descriptor values were shuffled randomly. For each descriptor set, the shuffling procedure was performed 10 times, and the mean performance drop reported. Descriptors more heavily relied upon by the model were associated with higher performance drops.
As seen in Figure 5, the model primarily relied on quantum chemical bond descriptors. Shuffling all quantum chemical bond descriptors (listed in Table 1) as a group resulted in a performance drop of 10.3%. The most important individual descriptor was πp occupancy; shuffling of this descriptor was associated with a performance drop of 4.8%. This observation was consistent with πp occupancy predicting SOEs reasonably well by itself, with the best performance among all lone descriptors (Figure 4). The model's heavy reliance on πp occupancy is logical given its role in epoxidation. In fact, a π-complex is the initial intermediate formed during epoxidation by cytochromes P450.37,56,57 While reasonable, πp occupancy has never been proposed as a way to identify SOE.
Figure 5.

The importance of specific descriptors to the bond epoxidation model. A permutation sensitivity analysis quantified the importance of descriptors for the final trained site of epoxidation model. Left, the 10 most important individual descriptors in decreasing order of importance from top to bottom. Right, the importance of four broad descriptor categories. The graph shows the model performance drop associated after permuting the associated descriptor values, averaging over 10 iterations.
The second most important descriptor was SMARTCyp reactivity, with a performance drop of 2.5%. The relevance of SMARTCyp reactivity is readily understandable, because it predicts the sites of cytochromes P450 metabolism of drug-like molecules.30 The remaining most important individual descriptors were topological. Previous studies by our group found topological descriptors to be important for many different types of chemical modeling.43 Topology encompasses fundamental information, such as atom element identity or bond type, which has been useful for finding many different types of patterns. Overall, the results of sensitivity analysis indicated that the model logically relied upon descriptors relevant to epoxidation.
Accuracy in Identifying Aromatic and Double Bond Sites of Epoxidation
Ideally, the model should be able to distinguish SOEs from all other bonds across the entire data set. This is not assessed by the average site AUC and top-two metrics used in prior sections, which only compare BES predictions on a molecule-by-molecule basis. In contrast, global AUC, computed across all atoms in the data set does measure this behavior. The model’s BES is very accurate across the whole data set, with a global AUC of 95.6%. The logistic regressor is slightly less accurate with a global AUC of 94.5%, but this performance drop is significant with a p-value of 10–8 computed with a paired t-test.50 Similarly, the best performing descriptor is the πp occupancy with a global AUC of 88.4%, which is also a significant performance drop from the BES with a p-value approaching zero.
We further assessed the model’s performance by ensuring it was able to distinguish SOEs from either aromatic and double bonds (Figure 6). These tests excluded (for example) single bonds, which are very rarely epoxidized and might artificially inflate performance if included in performance calculations. An aromatic bond AUC was computed by first extracting all aromatic bonds within epoxidized molecules and then calculating AUC. A double bond AUC was calculated similarly. Encouragingly, BES were very accurate in identifying both epoxidized aromatic bonds (92.5%) and epoxidized double bonds (95.1%) and also substantially outperformed all individual descriptors.
Figure 6.

Bond epoxidation scores (BES) accurately identified both aromatic and double bond sites of epoxidation. Across the 389 molecules that underwent epoxidation, the model accurately separated epoxidized and non-epoxidized aromatic bonds (left) and double bonds (right). Using cross-validated scores, classification performance was quantified by computing the AUC of the model on either the aromatic or the double bonds in the full data set. The AUC of the model was compared with similarly computed AUCs for individual descriptors. In both cases, the model BES outperformed all individual descriptors.
Distinguishing Epoxidation from Hydroxylation
Another key task was to accurately distinguish SOEs from SOHs, because epoxidation and hydroxylation may have significantly different implications for toxicity and downstream metabolism. Generally, SOEs are not obviously distinguishable from sites of sp2 hydroxylation, because either epoxidation or hydroxylation may occur at sp2 atoms. While several studies have already demonstrated that computational models can predict the sites where molecules are oxidized,28−33 they do not predict if the oxidation is an epoxidation or a hydroxylation.
For our study, we tested whether BES distinguished SOEs from SOHs. We initially built a hydroxylation data set of 3000 hydroxylation reactions that were randomly sampled from the AMD resource, as described in the Methods. This final data set included 811 molecules, in which atoms were marked if they are sites of sp2 hydroxylation.
In this study, an SOE was defined as a bond between the two carbons of the final epoxide, whereas an SOH is usually defined as the single atom targeted for hydroxylation. However, our model only makes predictions on bonds. So, for validation purposes, we labeled the bonds connecting to hydroxylated atoms as SOHs and asked whether these sites receive lower scores than SOEs. Only bonds between two sp2 carbon atoms were included. Each of the 811 molecules in the hydroxylation data set was tested by our model, and the predictions for each bond of hydroxylation were extracted. As previously explained, the hydroxylation reactions were sampled randomly from our database. Therefore, molecules subject to both hydroxylation and epoxidation data sets were included. Cross-validated predictions were used for molecules that were also part of the training set. Within these molecules, it was possible for the same site to be subject to both epoxidation and hydroxylation. These sites were labeled as SOEs.
We investigated whether these SOEs were distinguishable from SOHs. Encouragingly, BES separated SOEs from SOHs with an AUC of 83.3% (Figure 7). In contrast, the best performing raw descriptor among all tested was πp occupancy, with an AUC of only 77.0%. This is a critical result because it demonstrates that the model can distinguish SOEs from sites that are also acted on by P450s, but not epoxidized.
Figure 7.

Bond epoxidation scores (BES) distinguish sites of epoxidation (SOEs) from sites of hydroxylation (SOHs). Top, each prediction method was assessed by its ability to separate SOEs from SOHs. The cross-validated scores on the SOEs of 389 epoxidized training molecules were compared with the SOH scores on 811 test molecules with their sites of sp2 hydroxylation labeled. The scores for each SOE and SOH were extracted and performance was quantified by computing the AUC. The classification performance of the model was then compared with similarly computed AUCs for individual descriptors. The model’s BES outperformed all individual descriptors. Right, from top to bottom are 1-nitropyrene58 and ketoconazole,59 example molecules subject to both epoxidation and hydroxylation. Each SOE is indicated by solid circles, and SOHs are indicated by dashed circles. The colored shading indicates BES (which range from 0 to 0.45).
πp Occupancy and Epoxidation
One striking result from these experiments is the consistently high importance of πp occupancy in identifying SOEs. Although it has been known for a long time that a π-complex is the initial intermediate formed during epoxidation by cytochromes P450,37,56,57 no published literature has suggested πp occupancy is a marker for SOEs or quantitatively assessed its ability to identify SOEs.
To further investigate this observation, which may provide mechanistic clues, we studied the distribution of πp occupancy and BES as a function of epoxidation and bond type (Figure 8). From these distributions, it seems immediately clear that SOEs have higher πp occupancy than non-epoxidized sites. However, πp occupancy is also strongly correlated with the type of bond, and the optimal cutoff between SOEs and non-epoxidized sites is different for double and aromatic bonds. This result suggests that πp occupancy may not be the direct driver of the π-intermediate’s formation. Instead, πp occupancy may be a proxy for another factor that we do not directly capture in other descriptors. One possible factor may be the ability of neighboring groups to donate πp electrons, but directly testing this hypothesis is beyond the immediate scope of this study and will be left for future work.
Figure 8.

Bond epoxidation scores (BES) represent a well-scaled probability that a site will be epoxidized. Across the 389 molecules that underwent epoxidation, the normalized distribution of BES (bottom) and πp occupancy (top) across both aromatic bonds (left) and double bonds (right) are displayed for all epoxidized and non-epoxidized sites, indicated by the shaded bars. The solid lines represent the percentage of bonds that are epoxidized (using non-normalized frequencies). The diagonal dashed lines on the bottom plots indicate a hypothetical perfectly scaled prediction. This demonstrates that BES is much better scaled than πp occupancy.
These distributions also highlight another key feature of our approach; the model’s output is well-scaled and can be interpreted as a probability. In other words, bonds with a BES score of 0.8 have approximately a 80% chance of being epoxidized. In contrast, πp occupancy, though predictive, is not scaled to be an SOE probability.
Accuracy at Identifying Molecules that Undergo Epoxidation
We also assessed the ability of our model to separate epoxidized from non-expoxidized molecules. With high enough classification performance, our model might be a useful tool to rapidly screen drug candidates for potentially problematic molecules.7−11
In this assessment, we trained the model for epoxidation to distinguish between those molecules that underwent epoxidation and those that did not. We included in our training data set molecules that are structurally closely related to epoxidized molecules, but are not themselves epoxidized in our database. After training the model on the SOE level, we tested several methods of separating epoxidized and non-epoxidized molecules (Figure 9). In this case, classification performance was quantified by measuring the AUC across the entire data set.
Figure 9.

Molecule epoxidation scores accurately identify molecules subject to epoxidation. Left, several prediction methods were compared by their ability to identify molecules that underwent epoxidation. The data set included 524 molecules, 389 of which were epoxidized and 135 structurally similar but not epoxidized molecules. Model performance was measured by computing the AUC across epoxidized and non-epoxidized molecules (Molecule AUC), using cross-validated scores. By this metric, the best approach inputted the top five bond epoxidation scores (BES) and all molecule-level descriptors into a neural network (MES[NN]). This slightly outperformed the simpler methods of using a logistic regressor (MES[LR]) or merely taking the maximum bond epoxidation score (max[BES]). While this improvement is not statistically significant, on the basis of the reliability plots in Figure 10, the neural network (MES[NN]) was chosen to calculate molecule epoxidation scores (MES) for this study. Right, example pairs of epoxidized and closely related non-epoxidized molecules are visualized. From left to right, top to bottom: resveratrol (MES: 0.79),60 quinalbarbitone (MES: 0.88),61 glucuronidated resveratrol (MES: 0.37),62 and thiopental (MES: 0.60).63 Each experimentally observed site of epoxidation is circled. For each molecule, the colored shading represents BES, which range from 0 to 0.76.
The simplest method for predicting molecule epoxidation was to take the cross-validated maximum BES score within each molecule. Across the entire data set, this approach yielded MES that separate epoxidized and non-epoxidized molecules with an AUC of 78.6%. The addition of a training step to input the top five BES and molecule-level descriptors into a logistic regressor or neural network slightly improved classification performance. The cross-validated scores outputted by a logistic regressor (MES[LR]in the figure) had a higher AUC of 78.9%, and those of the neural network (MES[NN]) had an AUC of 79.3%. A false positive rate paired t-test50 indicated that MES[NN] was not significantly better than max[BES] (p-value 0.14) or MES[LR] (p-value 0.19).
However, MES[NN] provided a better scaled prediction than either max[BES] or MES[LR], as demonstrated by the reliability plots in Figure 10. The neural network closely approximated a perfectly well-scaled prediction, with an R2 value of 0.971, compared to 0.956 for the logistic regressor or 0.889 for max[BES]. The neural network’s reliability plot is superior to that of the logistic regressor, not only due to the higher R2 value, but also because it assigns significantly more non-epoxidized molecules low scores, and epoxidized molecules high scores, evidenced by the relative densities in Figure 10.
Figure 10.

MES[NN] offers a well-scaled probabilistic prediction of molecule epoxidation. The bar graphs plot the normalized distributions of max[BES], MES[LR], and MES[NN] across 525 epoxidized and non-epoxidized molecules. The solid lines plot the percentage of molecules that are epoxidized (using non-normalized frequencies) in each bin. The diagonal dashed lines indicate a hypothetical perfectly scaled prediction. MES[NN] offers the best scaled prediction of the three methods, with a strong correlation to a perfectly scaled prediction. This means that the MES[NN] is interpretable as the probability that a molecule is epoxidized.
Nevertheless, choosing between the logistic regressor and neural network is debatable. The logistic regressor offers a simpler model structure, whereas the neural network provides a slightly higher classification performance and better scaled prediction. Going forward, we decided to use the neural network, but we believe that the logistic regressor could also be used with similar results. For the rest of the study, we define MES to mean MES[NN].
The significantly lower AUC of the molecule-level MES compared to the site-level BES was a consequence of the lower quality of the molecule-level data, which included “non-epoxidized” molecules. This was based on our assumption that molecules were non-epoxidized if they were not subject to any epoxidation reaction in our literature-derived database. While necessary, this assumption was not strong evidence that molecules were not subject to epoxidation, because not all studies look for epoxidation products. As a consequence, some epoxidizable molecules were incorrectly labeled as non-epoxidized in our data. In contrast, our site-level epoxidation data is much less noisy, because it is drawn from experiments detecting epoxidation, and this is reflected in the higher site-level performance.
Nevertheless, MES separated epoxidized and non-epoxidized molecules with 79.3% AUC. This result is consistent with our presumption that most of the molecules labeled as non-epoxidized, are truly not epoxidized. If epoxidized and non-epoxidized molecules were drawn from the same chemical distribution, it would not be possible to separate them with any accuracy. Furthermore, MES outperformed all molecule-level descriptors in terms of classification performance. This result demonstrated that our model offers an informative prediction on the molecule level. The best performing descriptor was the negative of the total number of single bonds in a molecule, yet its AUC was only 72.3%, considerably worse than MES. In contrast to site-level epoxidation, for which πp occupancy was quite predictive (Figure 4), maximum πp occupancy predicts molecule epoxidation with only 57.7% AUC. The model MES much more accurately predicted which molecules will be epoxidized than any single chemical descriptor.
Case Studies
Knowledge of the SOE of a drug or drug candidate can direct rational drug design to avoid the formation of reactive metabolites and reduce the risk of adverse drug reactions. Case studies provide excellent examples of how our model could enable the development of safer drugs (Figure 11).
Figure 11.
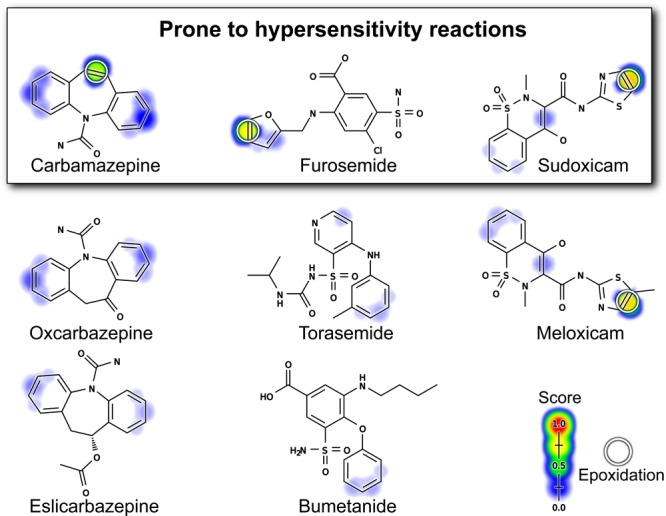
The epoxidation model recognizes sites of epoxidation within drugs that can be modified to reduce toxicity. The figure includes three groups of closely related drugs shaded by their BES scores; the top three are prone to hypersensitivity reactions while their analogues are not. The top three molecules and meloxicam are epoxidized and their sites of epoxidation are circled.21,23−25,66,67 The model’s BES correctly identifies the SOEs in these molecules. The model’s MES correctly identifies these molecules as epoxidized, with higher scores than the non-epoxidized molecules. For the top three molecules, epoxidation is the primary mechanism of their hypersensitivty.65 Encouragingly, the two analogues of carbamazepine are correctly identified as non-epoxidized and therefore non-hepatotoxic. This demonstrates how the model could be used to identify less toxic analogues. Furosemide does not have a close analogue on the market, but the model correctly identifies the furan ring as problematic. The other diuretics with the same active scaffold, but without this furan, are less toxic.65 Identifying meloxicam as less toxic is a more difficult task and would require more comprehensive metabolism modeling. Meloxicam is a safer analogue of sudoxicam because an alternate hydroxylation pathway is introduced by the modification that outcompetes the epoxidation pathway.21
Carbamazepine is an effective drug to treat epilepsy; however, it can cause severe adverse reactions mediated by reactive metabolites. Carbamazepine metabolism can form several reactive metabolites, including an iminoquinone,22 but the epoxide’s formation is the focus of this study and more correlated with adverse reactions.23−25 Analogues of carbamazepine that block the epoxidation have a lower incidence of adverse effects. Replacement of the problematic double bond with a ketone yielded oxcarbazepine, which lacks the metabolic activation to an epoxide and adverse events, yet retains similar efficacy.27 Similarly, eslicarbazepine does not contain the problematic double bond, is no longer epoxidized at this position, and also has a lower incidence of adverse reactions.25,64 The model correctly identifies carbamazepine’s SOE. Furthermore, the model correctly identified two carbamazepine analogous as less likely to be epoxidized: oxcarbazepine (MES: 0.38) and eslicarbazepine (MES: 0.20) compared with carbamazepine (MES: 0.88).
Furosemide is a commonly prescribed diuretic but is prone to hypersensitivity reactions and hepatotoxicity due to the epoxidation of its furan ring.65 The model correctly identifies this as an SOE. There are no close analogues of furosemide on the market. However, there are three other drugs in the same class that contain the same sulfamyl-based active scaffold: piretanide, bumetanide, and torasemide. None of these drugs contain the problematic furan, are all predicted not to form epoxides (MES: 0.21, 0.19, and 0.21, respectively, compared with 0.94 for furosemide), and all are less prone to hypersensitivity driven reactions than furosemide.65
The case of hepatotoxic sudoxicam and its non-hepatotoxic analogue, meloxicam, is more complicated.65 Sudoxicam is a NSAID that was withdrawn from testing due to hepatotoxicity caused by epoxidation of its thiazole ring; the unstable epoxide causes ring scission and formation of a reactive acylthiourea metabolite.21,65 This reaction pathway is suppressed by the addition of a single methyl group to sudoxicam’s SOE. The resulting drug meloxicam is less prone to epoxidation, although the epoxide still forms.21 Instead, meloxicam is primarily hydroxylated at the added carbon.21 As a result, the reactive acylthiourea urea metabolite forms less often, and consequently meloxicam is not hepatotoxic, despite being prescribed at a similar dose to the hepatotoxic sudoxicam.21,65
The model correctly predicts the SOEs of both sudoxicam and meloxicam, and assigns them high MES of 0.95 and 0.96, respectively. However, the model does not identify meloxicam as the less toxic molecule. This is exactly what we should expect, because both molecules are epoxidized by P450s.21 Meloxicam’s modification introduces an alternative hydroxylation pathway that reduces the amount of epoxide formed, and this change is responsible for its reduced toxicity. This highlights the limitations of considering the epoxidation pathway in isolation. A better risk assessment might combine epoxidation predictions with more comprehensive models of metabolism to predict if epoxides are a major metabolite. Building this system is exactly our long-range goal, but beyond the scope of the current study.
Nevertheless, our findings provide a critical step in the right direction: the first reported model that predicts the formation of reactive epoxides from drug candidates and the accurate identification of the specific epoxidized bonds. As is clear in all three of these cases, the model can be used to identify SOEs that can then be modified to make drugs safer.
Conclusion
This study establishes a new system to predict the formation of reactive epoxide metabolites. The epoxidation model—trained on SOE data—identifies with 94.9% AUC performance the SOEs within epoxidized molecules. The model also classifies epoxidized and non-epoxidized molecules with 79.3% AUC. This method needs to be combined with additional tools to be useful for predicting the toxicity of drugs. For example, while this model predicts the formation of epoxides, it does not score the reactivity of these epoxides. Epoxide reactivity can vary widely, with half-lives ranging from one second to several hours,37 and this variation may have significant implications for toxicity. To address this, we plan to combine this epoxidation model with a model of reactivity already developed.43 Furthermore, we will expand to model quinone formation, another motif of potentially high reactivity that frequently causes adverse drug reactions.15,68,69 Ultimately, we envision a powerful model for predicting adverse drug reactions that integrates metabolism models, reactivity models, and dosage information. By accurately modeling epoxidation, this study provides a key piece of this ultimate goal.
Acknowledgments
The authors thank Cynthia Lo, Jed Zaretzki, and Na Le Dang for helpful discussions, and Matthew Matlock for previous development of the XenoSite algorithm, which is expanded in this study. Figures 1–3 used OEDepict, version 1.7.4.5, OpenEye Scientific Software, Inc., Santa Fe, NM, USA, www.eyesopen.com, 2015. We also thank the developers of the open-source cheminformatics tools Open Babel and RDKit, of which we made broad use. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by NIH Grants Nos. 1S10RR022984-01A1 and 1S10OD018091-01.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00131.
Figure S1 depicts the global AUC of BES compared to a logistic regressor and raw descriptors, and Tables S1–S3 define the molecule, bond, and atom level descriptors, respectively, used by the XenoSite epoxidation model. The AMD Registry Numbers file lists the reaction and molecule registry numbers of the epoxidation and hydroxylation data sets, extracted from the December 2014 AMD release. (ZIP)
We thank the Department of Immunology and Pathology at the Washington University School of Medicine for its generous support of this work.
The authors declare no competing financial interest.
Supplementary Material
References
- DiMasi J. A. Success rates for new drugs entering clinical testing in the United States. Clin. Pharmacol. Ther. 1995, 58, 1–14. [DOI] [PubMed] [Google Scholar]
- Srivastava A.; Maggs J.; Antoine D.; Williams D., Smith D., Park B.. Role of Reactive Metabolites in Drug-Induced Hepatotoxicity. In Adverse Drug Reactions; Uetrecht J., Ed.; Springer: Berlin, 2009; pp 165–194. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Didiuk M. T. Structural alerts, reactive metabolites, and protein covalent binding: how reliable are these attributes as predictors of drug toxicity?. Chem. Biodiversity 2009, 6, 2115–2137. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Han M.; Chen T.; Yan W.; Ning Q. Acute liver failure: mechanisms of immune-mediated liver injury. Liver Int. 2010, 30, 782–794. [DOI] [PubMed] [Google Scholar]
- Knowles S. R.; Uetrecht J.; Shear N. H. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet 2000, 356, 1587–1591. [DOI] [PubMed] [Google Scholar]
- Numata K.; Kubo M.; Watanabe H.; Takagi K.; Mizuta H.; Okada S.; Kunkel S. L.; Ito T.; Matsukawa A. Overexpression of suppressor of cytokine signaling-3 in T cells exacerbates acetaminophen-induced hepatotoxicity. J. Immunol. 2007, 178, 3777–3785. [DOI] [PubMed] [Google Scholar]
- Morisseau C. Role of epoxide hydrolases in lipid metabolism. Biochimie 2013, 95, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R.-E.; Isaacs N. Mechanisms of epoxide reactions. Chem. Rev. 1959, 59, 737–799. [Google Scholar]
- Tang W. Drug metabolite profiling and elucidation of drug-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2007, 3, 407–420. [DOI] [PubMed] [Google Scholar]
- Amacher D. E. The primary role of hepatic metabolism in idiosyncratic drug-induced liver injury. Expert Opin. Drug Metab. Toxicol. 2012, 8, 335–347. [DOI] [PubMed] [Google Scholar]
- Fretland A. J.; Omiecinski C. J. Epoxide hydrolases: biochemistry and molecular biology. Chem. Biol. Interact. 2000, 129, 41–59. [DOI] [PubMed] [Google Scholar]
- Lamb D. C.; Waterman M. R.; Zhao B. Streptomyces cytochromes P450: applications in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1279–1294. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Lonsdale R.; Harvey J. N.; Mulholland A. J.; Olsen L. Trends in predicted chemoselectivity of cytochrome P450 oxidation: B3LYP barrier heights for epoxidation and hydroxylation reactions. J. Mol. Graphics Model. 2014, 52, 30–35. [DOI] [PubMed] [Google Scholar]
- Rendic S.; Guengerich F. P. Contributions of human enzymes in carcinogen metabolism. Chemical Res. Toxicol. 2012, 25, 1316–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa B.; Pedretti A.; Vistoli G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discovery Today 2012, 17, 549–560. [DOI] [PubMed] [Google Scholar]
- Seidegård J.; Ekström G. The role of human glutathione transferases and epoxide hydrolases in the metabolism of xenobiotics. Environ. Health Perspect. 1997, 105, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S. K.; Watkins S. C.; Schuetz E.; Singh S. V. Role of glutathione conjugate efflux in cellular protection against benzo [a] pyrene-7, 8-diol-9, 10-epoxide-induced DNA damage. Mol. Carcinog. 2002, 33, 156–162. [PubMed] [Google Scholar]
- Begriche K.; Massart J.; Robin M.-A.; Borgne-Sanchez A.; Fromenty B. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa J. C.; Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [DOI] [PubMed] [Google Scholar]
- Moghaddam M. F.; Grant D. F.; Cheek J. M.; Greene J. F.; Williamson K. C.; Hammock B. D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach R. S.; Kalgutkar A. S.; Ryder T. F.; Walker G. S. In vitro metabolism and covalent binding of enol-carboxamide derivatives and anti-inflammatory agents sudoxicam and meloxicam: insights into the hepatotoxicity of sudoxicam. Chem. Res. Toxicol. 2008, 21, 1890–1899. [DOI] [PubMed] [Google Scholar]
- Pearce R. E.; Uetrecht J. P.; Leeder J. S. Pathways of carbamazepine bioactivation in vitro: II. The role of human cytochrome P450 enzymes in the formation of 2-hydroxyiminostilbene. Drug Metab. Dispos. 2005, 33, 1819–1826. [DOI] [PubMed] [Google Scholar]
- Kalis M. M.; Huff N. A. Oxcarbazepine, an antiepileptic agent. Clin. Ther. 2001, 23, 680–700. [DOI] [PubMed] [Google Scholar]
- Harder J. L.; Heung M.; Vilay A. M.; Mueller B. A.; Segal J. H. Carbamazepine and the active epoxide metabolite are effectively cleared by hemodialysis followed by continuous venovenous hemodialysis in an acute overdose. Hemodial. Int. 2011, 15, 412–415. [DOI] [PubMed] [Google Scholar]
- Jayasutha S.; Bhargavdilip S.; Kishore K.; Ramasamy C. Comparision of efficacy and safety of carbamazepine and eslicarbazepine in adult partial and generalized seizures. Asian J. Pharm. Clin. Res 2014, 7, 144–147. [Google Scholar]
- Yip V.; Maggs J.; Meng X.; Marson A.; Park K.; Pirmohamed M. Covalent adduction of carbamazepine 10, 11-epoxide with human serum albumin and glutathione S-transferase pi: implications for carbamazepine hypersensitivity. Lancet 2014, 383, S114. [Google Scholar]
- Dam M.; Ekberg R.; Løyning Y.; Waltimo O.; Jakobsen K. A double-blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Res. 1989, 3, 70–76. [DOI] [PubMed] [Google Scholar]
- Zaretzki J.; Matlock M.; Swamidass S. J. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [DOI] [PubMed] [Google Scholar]
- Campagna-Slater V.; Pottel J.; Therrien E.; Cantin L.-D.; Moitessier N. Development of a Computational Tool to Rival Experts in the Prediction of Sites of Metabolism of Xenobiotics by P450s. J. Chem. Inf. Model. 2012, 52, 2471–2481. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Gloriam D. E.; Zaretzki J.; Breneman C.; Olsen L. SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretzki J.; Rydberg P.; Bergeron C.; Bennett K. P.; Olsen L.; Breneman C. M. RS-Predictor Models Augmented with SMARTCyp Reactivities: Robust Metabolic Regioselectivity Predictions for Nine CYP Isozymes. J. Chem. Inf. Model. 2012, 52, 1637–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. P.; Mysinger M.; Korzekwa K. R. Computational models for cytochrome P450: a predictive electronic model for aromatic oxidation and hydrogen atom abstraction. Drug Metab. Dispos. 2002, 30, 7–12. [DOI] [PubMed] [Google Scholar]
- Kim D. N.; Cho K.-H.; Oh W. S.; Lee C. J.; Lee S. K.; Jung J.; No K. T. EaMEAD: Activation energy prediction of cytochrome P450 mediated metabolism with effective atomic descriptors. J. Chem. Inf. Model. 2009, 49, 1643–1654. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [DOI] [PubMed] [Google Scholar]
- Galeone D.; Lamontanara G.; Torelli D. Acute hepatitis in a patient treated with carbamazepine. J. Neurol. 1985, 232, 301–303. [DOI] [PubMed] [Google Scholar]
- Green V. J.; Pirmohamed M.; Kitteringham N. R.; Gaedigk A.; Grant D. M.; Boxer M.; Burchell B.; Park B. K. Genetic analysis of microsomal epoxide hydrolase in patients with carbamazepine hypersensitivity. Biochem. Pharmacol. 1995, 50, 1353–1359. [DOI] [PubMed] [Google Scholar]
- Peter Guengerich F. Cytochrome P450 oxidations in the generation of reactive electrophiles: epoxidation and related reactions. Arch. Biochem. Biophys. 2003, 409, 59–71. [DOI] [PubMed] [Google Scholar]
- Rojas M.; Alexandrov K.; Auburtin G.; Wastiaux-Denamur A.; Mayer L.; Mahieu B.; Sebastien P.; Bartsch H. Anti-benzo [a] pyrene diolepoxide–DNA adduct levels in peripheral mononuclear cells from coke oven workers and the enhancing effect of smoking. Carcinogenesis 1995, 16, 1373–1376. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M., Morley C., Hutchison G. R.. Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit Chem. Cent J.. 20082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A.; Lian L.-Y.; Maggs J. L.; Chaponda M.; Pirmohamed M.; Williams D. P.; Park B. K. Quantifying the metabolic activation of nevirapine in patients by integrated applications of NMR and mass spectrometries. Drug Metab. Dispos. 2010, 38, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alton K.; Grimes R.; Shaw C.; Patrick J.; McGuire J. Biotransformation of a 1, 5-benzodiazepine, triflubazam, by man. Drug Metab. Dispos. 1975, 3, 352–360. [PubMed] [Google Scholar]
- OLBoyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. B.; Miller G. P.; Swamidass S. J. Site of Reactivity Models Predict Molecular Reactivity of Diverse Chemicals with Glutathione. Chem. Res. Toxicol. 2015, 28, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. MOPAC: a semiempirical molecular orbital program. J. Comput.-Aided Mol. Des. 1990, 4, 1–103. [DOI] [PubMed] [Google Scholar]
- Hostaš J.; Řezáč J.; Hobza P. On the performance of the semiempirical quantum mechanical PM6 and PM7 methods for noncovalent interactions. Chem. Phys. Lett. 2013, 568, 161–166. [Google Scholar]
- Chicco D.; Sadowski P.; Baldi P.. Deep autoencoder neural networks for gene ontology annotation predictions. In Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics; Newport Beach, CA, USA, September 20–23, 2014; Association for Computing Machinery: New York, 2014; pp 533–540.
- Sainath T. N.; Mohamed A.-r.; Kingsbury B.; Ramabhadran B.. Deep convolutional neural networks for LVCSR. IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP); Vancouver, British Columbia, May 26–31, 2013; IEEE: Piscataway, NJ, 2013; pp 8614–8618.
- Cruciani G.; Baroni M.; Benedetti P.; Goracci L.; Fortuna C. G. Exposition and reactivity optimization to predict sites of metabolism in chemicals. Drug Discovery Today: Technol. 2013, 10, e155–e165. [DOI] [PubMed] [Google Scholar]
- Zamora I.; Afzelius L.; Cruciani G. Predicting drug metabolism: a site of metabolism prediction tool applied to the cytochrome P450 2C9. J. Med. Chem. 2003, 46, 2313–2324. [DOI] [PubMed] [Google Scholar]
- Swamidass S. J.; Azencott C.-A.; Daily K.; Baldi P. A CROC stronger than ROC: measuring, visualizing and optimizing early retrieval. Bioinformatics 2010, 26, 1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorich M. J.; Miners J. O.; McKinnon R. A.; Winkler D. A.; Burden F. R.; Smith P. A. Comparison of linear and nonlinear classification algorithms for the prediction of drug and chemical metabolism by human UDP-glucuronosyltransferase isoforms. J. Chem. Inf. Comput. Sci. 2003, 43, 2019–2024. [DOI] [PubMed] [Google Scholar]
- Mahadevan B.; Luch A.; Atkin J.; Nguyen T.; Sharma A. K.; Amin S.; Baird W. M. Investigation of the genotoxicity of dibenzo [c, p] chrysene in human carcinoma MCF-7 cells in culture. Chem. Biol. Interact. 2006, 164, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo X.; Hartz R. A.; Bronson J. J.; Wong H.; Ahuja V. T.; Vrudhula V. M.; Leet J. E.; Huang S.; Macor J. E.; Shu Y.-Z. Comparative biotransformation of pyrazinone-containing corticotropin-releasing factor receptor-1 antagonists: minimizing the reactive metabolite formation. Drug Metab. Dispos. 2010, 38, 5–15. [DOI] [PubMed] [Google Scholar]
- Subramanian R.; et al. Cytochrome P450-mediated epoxidation of 2-aminothiazole-based AKT inhibitors: identification of novel GSH adducts and reduction of metabolic activation through structural changes guided by in silico and in vitro screening. Chem. Res. Toxicol. 2010, 23, 653–663. [DOI] [PubMed] [Google Scholar]
- Hunter A.; Kennedy L.; Henry J.; Ferguson I. Application of neural networks and sensitivity analysis to improved prediction of trauma survival. Comput. Methods Programs Biomed. 2000, 62, 11–19. [DOI] [PubMed] [Google Scholar]
- Ostovic D.; Bruice T. C. Intermediates in the epoxidation of alkenes by cytochrome P-450 models. 5. Epoxidation of alkenes catalyzed by a sterically hindered (meso-tetrakis (2, 6-dibromophenyl) porphinato) iron (III) chloride. J. Am. Chem. Soc. 1989, 111, 6511–6517. [Google Scholar]
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [DOI] [PubMed] [Google Scholar]
- Sun Y.-W.; Guengerich F. P.; Sharma A. K.; Boyiri T.; Amin S.; El-Bayoumy K. Human cytochromes P450 1A1 and 1B1 catalyze ring oxidation but not nitroreduction of environmental pollutant mononitropyrene isomers in primary cultures of human breast cells and cultured MCF-10A and MCF-7 cell lines. Chem. Res. Toxicol. 2004, 17, 1077–1085. [DOI] [PubMed] [Google Scholar]
- Fitch W. L.; Tran T.; Young M.; Liu L.; Chen Y. Revisiting the metabolism of ketoconazole using accurate mass. Drug Metab. Lett. 2009, 3, 191–198. [DOI] [PubMed] [Google Scholar]
- Steenwyk R.; Tan B. In vitro evidence for the formation of reactive intermediates of resveratrol in human liver microsomes. Xenobiotica 2010, 40, 62–71. [DOI] [PubMed] [Google Scholar]
- Gilbert J.; Hetherington W.; Powell J.; Whalley W. Metabolism of quinalbarbitone. J. Pharm. Pharmacol. 1975, 27, 343–347. [DOI] [PubMed] [Google Scholar]
- Wang D.; Zhang Z.; Ju J.; Wang X.; Qiu W. Investigation of piceid metabolites in rat by liquid chromatography tandem mass spectrometry. J. Chromatogr., B 2011, 879, 69–74. [DOI] [PubMed] [Google Scholar]
- Namera A.; Yashiki M.; Okada K.; Iwasaki Y.; Ohtani M.; Kojima T. Automated preparation and analysis of barbiturates in human urine using the combined system of PrepStation and gas chromatography-mass spectrometry. J. Chromatogr., B: Biomed. Sci. Appl. 1998, 706, 253–259. [DOI] [PubMed] [Google Scholar]
- Brown M. E.; El-Mallakh R. S. Role of eslicarbazepine in the treatment of epilepsy in adult patients with partial-onset seizures. Ther. Clin. Risk Manage. 2010, 6, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepan A. F.; Walker D. P.; Bauman J.; Price D. A.; Baillie T. A.; Kalgutkar A. S.; Aleo M. D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. [DOI] [PubMed] [Google Scholar]
- Mitchell J. R.; Snodgrass W. R.; Gillette J. R. The role of biotransformation in chemical-induced liver injury. Environ. Health Perspect. 1976, 15, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J., Potter W., Jollow D.. Furosemide-induced hepatic and renal tubular necrosis. I. Effects of treatments which alter drug-metabolizing enzymes Fed. Proc. 1973, 32, 305. [Google Scholar]
- Attia S. M. Deleterious effects of reactive metabolites. Oxid. Med. Cell. Longevity 2010, 3, 238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. K.; et al. Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discovery 2011, 10, 292–306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.