

Abstract
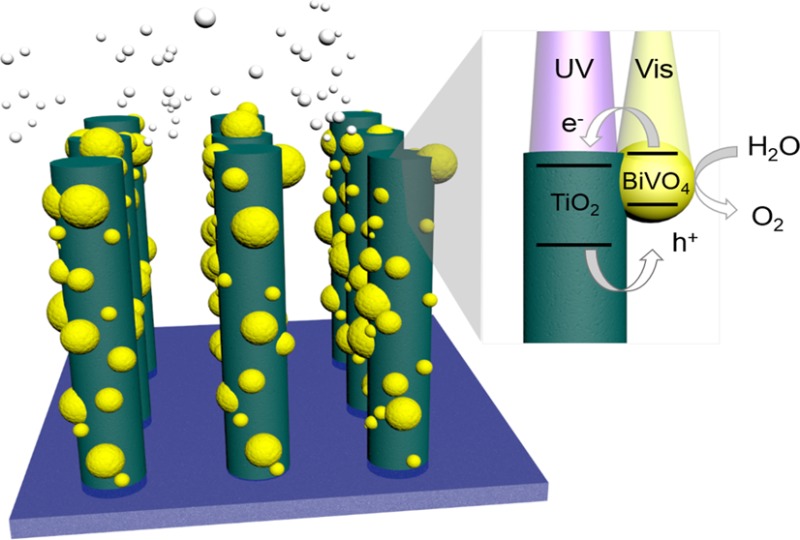
Metal oxides that absorb visible light are attractive for use as photoanodes in photoelectrosynthetic cells. However, their performance is often limited by poor charge carrier transport. We show that this problem can be addressed by using separate materials for light absorption and carrier transport. Here, we report a Ta:TiO2|BiVO4 nanowire photoanode, in which BiVO4 acts as a visible light-absorber and Ta:TiO2 acts as a high surface area electron conductor. Electrochemical and spectroscopic measurements provide experimental evidence for the type II band alignment necessary for favorable electron transfer from BiVO4 to TiO2. The host–guest nanowire architecture presented here allows for simultaneously high light absorption and carrier collection efficiency, with an onset of anodic photocurrent near 0.2 V vs RHE, and a photocurrent density of 2.1 mA/cm2 at 1.23 V vs RHE.
Short abstract
We report the use of Ta:TiO2|BiVO4 as a photoanode for use in solar water splitting cells. This host−guest system makes use of the favorable band alignment between the two semiconductors. The nanowire architecture allows for simultaneously high light absorption and carrier collection for efficient solar water oxidation.
Introduction
Harnessing energy from sunlight is a means of meeting the large global energy demand in a cost-effective and environmentally benign manner. However, to provide constant and stable power on demand, it is necessary to convert sunlight into an energy storage medium.1 An example of such a method is the production of hydrogen by photoelectrochemical (PEC) water splitting. The direct splitting of water can be achieved using a single semiconductor; however, due to the voltage requirement of the water splitting reaction and the associated kinetic overpotentials, only wide-band-gap materials can perform overall water splitting, limiting the efficiency due to insufficient light absorption.2 To address this issue, a dual-band-gap z-scheme system can be utilized, with a semiconductor photoanode and photocathode to perform the respective oxidation and reduction reactions.3 This approach allows for the use of lower-band-gap materials that can absorb complementary portions of the solar spectrum and yield higher solar-to-fuel efficiencies.4,5 In this integrated system, the charge flux is matched in both light absorbers of the photoelectrochemical cell. Therefore, the overall performance is determined by the limiting component. In most photoelectrosynthetic cells, this limiting component is the semiconductor photoanode.6
Metal oxides have been heavily researched as photoanode materials since few conventional light absorber materials are stable at the highly oxidizing conditions required for water oxidation.7 However, the most commonly studied binary oxide, TiO2, has a band gap that is too large to absorb sunlight efficiently (∼3.0 eV), consequently limiting its achievable photocurrent.8 While promising work has recently been done on stabilizing conventional light absorbers such as Si,9 GaAs,10 and InP,11 the photovoltage obtained by these materials thus far has been insufficient to match with smaller-band-gap photocathode materials such as Si and InP in a dual absorber photoelectrosynthetic cell.12,13 Additionally, these materials have high production and processing costs. Small-band-gap metal oxides that absorb visible light and can be inexpensively synthesized, such as WO3, Fe2O3, and BiVO4, are alternative materials that hold promise to overcome these limitations.14−16 Among these metal oxides, BiVO4 has emerged as one of the most promising materials due to its relatively small optical band gap of ∼2.5 eV and its negative conduction band edge (∼0 V versus RHE).17,18 Under air mass 1.5 global (AM1.5G) solar illumination, the maximum achievable photocurrent for water oxidation using BiVO4 is ∼7 mA/cm2.16 However, the water oxidation photocurrent obtained in practice for BiVO4 is substantially lower than this value, mainly due to poor carrier transport properties, with electron diffusion lengths shorter than the film thickness necessary to absorb a substantial fraction of light.17
One approach for addressing this problem is to use two separate materials for the tasks of light absorption and carrier transport. To maximize performance, a conductive and high surface area support material (“host”) is used, which is coated with a highly dispersed visible light absorber (“guest”). This architecture allows for efficient use of absorbed photons due to the proximity of the semiconductor liquid junction (SCLJ). This strategy has been employed in dye sensitized (DSSC) and quantum dot sensitized solar cells (QDSSC).19,20 Using a host–guest scheme can improve the performance of photoabsorbing materials with poor carrier transport but relies upon appropriate band alignment between the host and guest. Namely, the electron affinity of the host should be larger, to favor electron transfer from guest to host without causing a significant loss in open-circuit voltage.21 Nanowire arrays provide several advantages for use as the host material as they allow high surface area loading of the guest material, enhanced light scattering for improved absorption, and one-dimensional electron transport to the back electrode.22 Therefore, nanowire arrays have been used as host materials in DSSCs, QDSSCs, and hybrid perovskite solar cells.23−25 In photoelectrosynthetic cells, host–guest architectures have been utilized for oxide photoanodes such as Fe2O3|TiSi2,26 Fe2O3|WO3,27 Fe2O3|SnO2,28 and Fe2TiO5|TiO2.29 For BiVO4, it has been studied primarily with WO3|BiVO4,30−32 ZnO|BiVO4,33 and anatase TiO2|BiVO4.34 While attractive for its electronic transport properties, ZnO is unstable in aqueous environments, and WO3 has the disadvantage of having a relatively positive flatband potential (∼0.4 V vs RHE)14 resulting in potential energy losses for electrons as they are transferred from BiVO4 to WO3, thereby limiting the photovoltage of the combined system. Performance in the low potential region is critical for obtaining high efficiency in photoelectrosynthetic cells when coupled to typical p-type photocathode materials such as Si or InP.12,13 TiO2 is stable in a wide range of pH and has a relatively negative flat band potential (∼0.2 V vs RHE)7 which does not significantly limit the photovoltage obtainable from BiVO4, while still providing a driving force for electron transfer. While TiO2 has intrinsically low mobility, doping TiO2 with donor type defects could increase the carrier concentration and thus the conductivity. Indeed, niobium and tantalum doped TiO2 have recently been investigated as potential transparent conductive oxide (TCO) materials.35,36 A host material with high carrier concentration could also ensure low contact resistance with the guest material.37
Using a solid state diffusion approach based on atomic layer deposition (ALD), we have previously demonstrated the ability to controllably and uniformly dope TiO2.38 In this study we demonstrate a host–guest approach using Ta-doped TiO2 (Ta:TiO2) nanowires as a host and BiVO4 as a guest material. This host–guest nanowire architecture allows for simultaneously high light absorption and carrier collection efficiency, with an onset of anodic photocurrent near 0.2 V vs RHE, and a photocurrent of 2.1 mA/cm2 at 1.23 V vs RHE. We show that the synergistic effect of the host–guest structure results in higher performance than either pure TiO2 or BiVO4. We also experimentally demonstrate thermodynamically favorable band alignment between TiO2 and BiVO4 using spectroscopic and electrochemical methods, and study the band edge electronic structure of the TiO2 and BiVO4 using X-ray absorption and emission spectroscopies.
Development of a Nanowire Heterostructure Photoanode
A schematic of the desired host–guest structure is shown in Figure 1, in which a high surface area TiO2 nanowire array is used as an electron conductor, and small, highly dispersed BiVO4 nanoparticles coat the surface as visible light sensitizers. The small size of these nanoparticles allows high collection efficiency of electrons by the nanowires, and the proximity of the semiconductor liquid junction allows holes to reach the surface to perform the water oxidation reaction. The type II band alignment required for electron transfer between TiO2 and BiVO4 is illustrated, in which electrons produced by photoexcitation in BiVO4 are transferred to the conduction band of TiO2. Additionally, holes produced in TiO2 can move through the valence band of BiVO4 and to the semiconductor surface to oxidize water. Scanning electron microscopy (SEM) images of the as-grown nanowire arrays and the optimized type II photoanode heterostructures are shown in Figure 1b,c. The following sections discuss the development of these optimized structures.
Figure 1.

Nanowire heterostructure scheme. (a) Schematic of the photoanode architecture. The nanowire morphology provides an increased path length for absorption of visible photons by BiVO4 , as well as a pathway for efficient electron transfer. The small size of the BiVO4 particles maintains close proximity of the semiconductor liquid junction for holes to carry out the oxygen evolution reaction. Type II band alignment allows electron transfer from BiVO4 to TiO2. (b, c) SEM images of designed nanowire heterostructure before and after BiVO4 loading. Scale bar 500 nm.
Synthesis of Ta-Doped TiO2 Nanowire Arrays
To provide a high surface area scaffold for the BiVO4, rutile TiO2 nanowire arrays were grown on glass substrates coated with fluorine-doped tin oxide (FTO) by using a hydrothermal method.39 High resolution transmission electron microscopy (HRTEM) analysis demonstrated that the nanowires are single crystalline and grow along the [001] direction (Figure S1). The nanowires are ∼2.0 μm in length and 150 nm in diameter. Again, for use as an electron transporting scaffold, the carrier concentration of the TiO2 nanowires must be increased. This can be accomplished by doping with Ta5+, which acts as a donor defect when occupying substitutional Ti4+ sites.35 Previously, synthesis of transparent conductive oxides with resistivities of ∼2.5 × 10–4 Ω cm have been demonstrated using Ta- and Nb-doped anatase TiO2 compared to resistivities of ∼1 × 10–1 Ω cm for undoped TiO2.35,36 Following a previously developed solid state diffusion method,38 the doping was accomplished by forming a core–shell TiO2|Ta2O5 structure using ALD, followed by high temperature annealing. ALD of Ta2O5 was performed using pentakis(dimethylamino)tantalum and water.40
Two important characteristics of an ALD reaction are the saturation of the surface reaction after each precursor pulse and a linear relationship between film thickness and number of cycles after a sufficient number of cycles.41 To measure film thicknesses, ALD Ta2O5 was deposited on planar Si substrates and measured by ellipsometry. Saturating, linear growth was demonstrated indicating an ALD growth mode with a growth rate of ∼0.6 Å/cycle. Using X-ray photoelectron spectroscopy (XPS), no measurable carbon or nitrogen was observed in the spectrum following 1 min of Ar sputtering. Because the ligands of the Ta precursor contain carbon and nitrogen, the lack of any organic contamination in the film indicates that the ALD surface reaction was complete, and the ligands were completely substituted by oxygen from the H2O precursor pulse. The TiO2 nanowire arrays were coated with tantalum oxide using ALD producing conformal core–shell nanowires with uniform shell thicknesses that could be tuned with subnanometer precision. Characterization of the ALD process can be found in Figures S2–S4.
The coated nanowire arrays were then annealed at 600 °C in an argon environment to facilitate diffusion of tantalum atoms into the rutile lattice, yielding Ta-doped TiO2 nanowires. The solid state diffusion process maintains the nanowire morphology and crystallinity as evidenced by X-ray diffraction (XRD) and electron microscopy (Figure S5). To confirm the presence of Ta atoms and obtain a visualization of their distribution within the TiO2 lattice, energy dispersive X-ray spectroscopy (EDS) mapping was conducted using scanning transmission electron microscopy (STEM). The elemental maps show a homogeneous incorporation of the Ta atoms, in contrast with the core–shell nanowires observed prior to annealing (Figure 2a). To determine the effect of the Ta doping on the carrier concentration of the nanowires, electrochemical impedance spectroscopy was performed. From the slopes of the Mott–Schottky plots (Figures 2b), carrier densities of undoped and 1% Ta-doped TiO2 nanowires were estimated to be ∼1018 and ∼1020 cm–3, respectively, using the Mott–Schottky relation,
where Csc is the capacitance of the space charge region, e is the elementary charge of an electron, ε0 is the permittivity of vacuum, ε is the dielectric constant of rutile TiO2 (ε = 86),42A is the electrochemically active surface area, V is the applied voltage, Vfb is the flatband potential, and Nd is the donor density.43 The positive slope indicates n-type behavior of the rutile nanowires. Mott–Schottky plots for varying Ta concentrations showed a monotonic increase in carrier concentration with the amount of Ta (Figure S6).
Figure 2.
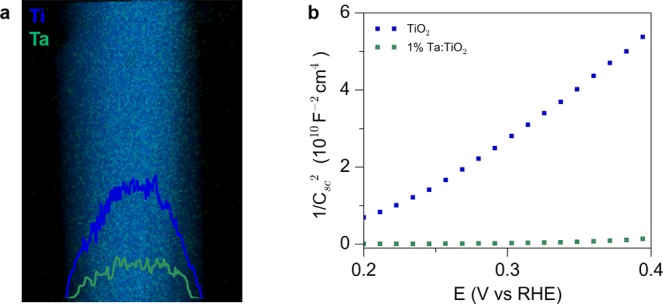
Modification of the electronic properties of TiO2 nanowires. (a) Elemental maps and line scans for Ta:TiO2 nanowires. Diffusion of Ta yields a uniformly doped nanowire. (b) Mott–Schottky plots of undoped and Ta-doped TiO2 showing an increase in carrier concentration with doping.
Deposition of BiVO4
A conductive high surface area support having been obtained, undoped BiVO4 was deposited on the nanowire array by thermal evaporation of Bi metal, followed by heating in the presence of vanadyl acetylacetonate (VO(C5H7O2)2).44 During heating Bi and VO2+ are oxidized and react to form BiVO4. Excess V2O5 was dissolved after heating by soaking the substrates in 1 M KOH. Based solely on the density of the respective materials, 1 nm of Bi metal is converted to ∼2.5 nm of BiVO4. The thermal conversion process yields isolated BiVO4 particles in the size range of 20–100 nm (Figure S7), which is favorable for photoelectrochemical performance due to the short electron diffusion length of BiVO4 (L < 100 nm).17 The density and size of the BiVO4 particles could be controlled by changing the thickness of the initial Bi seed layer (Figures S7, S8). At Bi seed layer thickness greater than a planar equivalent of 100 nm, the nanowire array becomes filled, and BiVO4 that does not infiltrate the array forms a thin film on top of the nanowire array. XRD confirms that the monoclinic scheelite BiVO4 phase is formed (Figure S9), and no changes in the original rutile TiO2 or F:SnO2 peaks are observed (Figure 3a). Elemental mapping also demonstrates that Bi and V signals are observed on the particles and Ti signal is observed on the core of the nanowires (Figure 3b).
Figure 3.
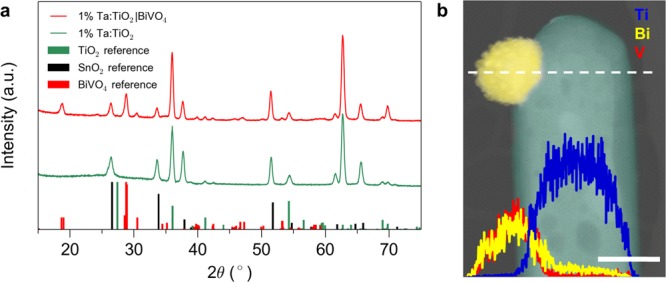
Loading of BiVO4 onto TiO2. (a) XRD patterns for doped TiO2 nanowire arrays before and after BiVO4 deposition. (b) Elemental mapping of a TiO2 nanowire decorated with a BiVO4 particle. Scale bar 100 nm.
Photoelectrochemistry
The effect of Ta doping on the photoelectrochemical performance was first investigated without BiVO4 coating by controlling the Ta dopant source via the thickness of the Ta2O5 shell. Although series resistance losses are likely decreased due to the higher carrier concentration, the introduction of the Ta dopant is detrimental to the overall performance of the TiO2 nanowire arrays (Figures S10, S11). As a relatively small fraction of Ta atoms in the rutile structure act as active dopants, it is possible that the high concentration of Ta dopants could result in increased defects in the nanowires and higher rates of carrier recombination. However, when a fixed amount (40 nm planar equivalent Bi seed layer) of BiVO4 is deposited on the wire arrays, the doped heterostructures clearly outperform their undoped analogues. Although electron transfer occurs in the desired direction in both cases, undoped TiO2 coated with BiVO4 actually shows a decrease in performance, possibly indicating that a substantial fraction of charge carriers are lost at the TiO2|BiVO4 interface (Figure S12). The higher carrier concentration in Ta:TiO2 could result in a decrease in the depletion width in TiO2, resulting in improved electron transfer from the BiVO4 to the TiO2. It is evident that although Ta doping is detrimental to the carrier transport properties of the TiO2 nanowires, it is necessary to allow favorable electron transfer at the TiO2|BiVO4 interface (Figures S13, S14). The highest performance was obtained with ∼1% Ta doping, which was used for all subsequent measurements for Ta:TiO2|BiVO4 heterostructures (Figure S11).
The effect of the BiVO4 loading amount was investigated by controlling the Bi metal seed layer thickness. The addition of the BiVO4 particles extends the absorption of the nanowire structures into the visible light region (Figure 4a). An onset of absorption is observed at ∼520 nm, consistent with the 2.5 eV band gap of monoclinic BiVO4 (Figure S15). As the Bi metal thickness is increased from a planar equivalent of 10–50 nm, the absorption in the 400–500 nm region increases, and samples with thickness greater than 40 nm show near unity absorption in this region. This highlights the advantage of the high surface area scaffold, as an identically prepared BiVO4 thin film on FTO shows ∼50% absorption at 450 nm (Figure S15). The scaffold also prevents the formation of much larger BiVO4 particles which are detrimental to performance. From voltammetric and incident photon to current efficiency (IPCE) measurements, it can be seen that an optimum thickness exists, with 40 nm Bi thickness having optimal performance. (Figures 4b, 4c, S16) The absorbed photon to current efficiency (APCE) shows similar internal quantum efficiency for low loading amounts, after which the APCE decreases (Figure S17). At higher loadings, the BiVO4 particle sizes become too large and begin to form films that do not penetrate the nanowire array and correspondingly the electron collection efficiency is decreased. The collection efficiency for large particles is particularly low for these undoped BiVO4 nanoparticles. We see that comparatively high APCE values can be obtained for loading amounts which give near unity light absorption. This shows the ability of the host–guest design to decouple light absorption and carrier collection efficiencies, which is not possible with thin film or nanostructured BiVO4 electrodes. However, it is also observed that the APCE values for the BiVO4 deposited on the nanowire arrays are lower than the corresponding thickness of BiVO4 deposited on a FTO substrate. This can be explained by the substantial loss of charge carriers in the TiO2 nanowire array, due to the defects caused by Ta doping. The higher absorption in the nanowire case compensates this loss in APCE yielding higher activity; however, improving the carrier collection in the nanowire array could result in simultaneously high absorption and collection efficiencies. One potential strategy would be the use of more effective dopants or codopants which have previously been shown to increase the carrier concentration of TiO2 nanowires without sacrificing performance.45,46 Additionally, the photoelectrochemical performance of the Ta:TiO2|BiVO4 nanowire photoanodes was investigated in electrolyte containing sodium sulfite (Na2SO3), which acts as an efficient hole scavenger. As the efficiency of hole capture by sulfite ions can be assumed to be near unity, measuring photocurrent for sulfite oxidation allows for determination of the photoelectrochemical activity of a photoanode irrespective of its water oxidation kinetics. It is clear that there is a significant difference between the water oxidation and sulfite oxidation photocurrent (Figure 4d), demonstrating that slow kinetics for water oxidation substantially limit the performance of the photoanodes. To address this issue, a cobalt based catalyst was deposited on the Ta:TiO2|BiVO4 samples by photoassisted electrodepostion.47 Cobalt oxides have also been shown to have high activity for water oxidation in near neutral pH with near unity charge transfer efficiencies.48−50 The performance of the Ta:TiO2|BiVO4 electrodes before and after the deposition of the Co-Pi catalyst is shown in Figure 4d. The performance is greatly enhanced, especially in the low potential region, and the similarity with the performance for sulfite oxidation shows the efficiency of the cobalt catalyst for decreasing surface recombination and improving water oxidation kinetics.
Figure 4.

Photoelectrochemical performance of the Ta:TiO2/BiVO4 nanowire heterostructures. (a, b) Absorption and IPCE for Ta:TiO2|BiVO4 samples with increasing loading. Near complete absorption and optimal activity are observed for planar equivalent thickness of 40 nm of Bi. (c) IPCE for Ta:TiO2 and Ta:TiO2|BiVO4 showing visible contribution from BiVO4. (d) Current–voltage curves in 0.5 M potassium phosphate electrolyte buffered to pH 7 with and without the presence of a hole scavenger. (e) Current–voltage curves in 0.5 M potassium phosphate electrolyte buffered to pH 7 for Ta:TiO2 and Ta:TiO2|BiVO4.
The optimized Ta:TiO2|BiVO4|Co-Pi photoanodes show an onset of photocurrent at a potential near 0.2 V vs RHE, and a photocurrent of 2.1 mA/cm2 at 1.23 V vs RHE. By comparison, an equal loading of BiVO4|Co-Pi deposited on FTO shows an onset potential of 0.25 V vs RHE and a photocurrent of 1.4 mA/cm2 at 1.23 V vs RHE (Figure S18). As previously discussed, the planar film is primarily limited by insufficient light absorption, which is substantially increased in the nanowire array. An identically prepared Ta:TiO2|Co-Pi nanowire array shows an onset potential of 0.25 V vs RHE, and a photocurrent of 0.5 mA/cm2 at 1.23 V vs RHE. From the IPCE spectra, we can therefore see that the type II structure allows for BiVO4 to act as a sensitizer for TiO2 to allow visible light utilization, and that the nanowire array provides a higher path length for light absorption, resulting in much higher light harvesting efficiency than planar BiVO4 (Figure 4c). Improvements to the host material could result in higher collection efficiency from the BiVO4 guest. Additionally, as these BiVO4 particles are undoped, further improvements in performance could be made by taking advantage of the extensive previous studies on improvements in BiVO4 performance by W or Mo doping.50−52
Band Alignment
The host–guest photoanode design relies on a type II band alignment existing between BiVO4 and TiO2, such that electron transfer from the BiVO4 to the TiO2 is favorable. To determine the relative band positions of TiO2 and BiVO4 and experimentally demonstrate this type II band alignment, electrochemical and spectroscopic measurements were performed.
The flat band potential (Vfb) can give an estimate of the band positions of a semiconductor in an aqueous environment. The primary method for determining Vfb is based on electrochemical impedance spectroscopy and appears as the x-intercept of the linear portion of a Mott–Schottky plot.43 In addition, the Vfb of a semiconductor will change as a function of pH due to surface charging by adsorption of H+ and OH– ions. The Vfb was measured as a function of pH and shown to change by approximately 59 mV/pH (Figure S19). Therefore, as shown in Figure 5a, the Vfbs of TiO2 and BiVO4 are determined to be 0.19 and 0.08 V vs RHE, respectively. These results are in agreement with previous measurements of the Vfb of the two semiconductors.17,53 Assuming that the gap between the Vfb and the bottom edge of the conduction band is small (∼0.2 eV) and similar for TiO2 and BiVO4, this result is consistent with a type II band alignment between the two semiconductors.54
Figure 5.

Band alignment between TiO2 and BiVO4. (a) Mott–Schottky plots showing that the flat band potential of BiVO4 is more negative than that of TiO2. (b) UPS spectra of TiO2 and BiVO4 with a fixed vacuum level. The valence band maximum and Fermi level of the two semiconductors are indicated. (c) Band diagram from electrochemical and spectroscopic data, confirming the type II alignment between BiVO4 and TiO2.
For further confirmation of the band positions, ultraviolet photoelectron spectroscopy (UPS) was used in addition to Mott–Schottky measurements. UPS provides high energy resolution (<0.1 eV) due to the small line width of the He discharge source and is therefore a powerful tool for investigating the band positions of semiconductors. UPS allows one to obtain both the work function of the material, which is the energetic difference between the photon energy of He I (21.22 eV) and the low kinetic energy cutoff of the spectrum, and the valence band maximum with respect to the Fermi level (EF).55 The position of EF is referenced to a metal standard. The relationship between these energy levels is depicted with the UPS spectrum of TiO2 and BiVO4 in Figure 5b. Here, we are interested in the position of the band edges of the two semiconductors with respect to the Evac. Thus, we plot the spectra for TiO2 and BiVO4 with Evac at the same position. The positions of the band edges are found using the Fermi level of a reference standard and linear extrapolation of the valence band and secondary electron cutoff regions. The work functions for TiO2 and BiVO4 were found to be 4.64 and 4.56 eV respectively, and the valence band positions were found to be 7.61 and 6.99 eV vs the Evac, or 3.17 and 2.55 V on the RHE scale.
To give a complete picture of the band alignment of the two semiconductors, the conduction band position was calculated by addition of the band gap to the valence band position. The optical band gap of the two semiconductors was determined by constructing Tauc plots from optical absorption data (Figures S18, S20, S21). Extrapolation of the Tauc plot onto the x-intercept gives an optical band gap of ∼3.0 eV and ∼2.5 eV for TiO2 and BiVO4, respectively (Figures S20, S21).56 Optical absorption for TiO2 and Ta:TiO2 nanowires was identical (Figure S22). Additionally, the band gap was confirmed by measuring the difference between the O K-edge X-ray emission and X-ray absorption edge positions. This analysis yielded values of 2.5 and 3.0 eV, consistent with the optical measurements (Figures S23, S24). Addition of the optical band gap to the valence band position gives the conduction band position and completes the full band diagram (Figure 5c). This analysis yielded conduction band values of 0.17 V vs RHE for TiO2 and 0.04 V vs RHE for BiVO4. These spectroscopic measurements are consistent with the type II band alignment between TiO2 and BiVO4, and the electrochemical flat band potential measurements.
In addition to the UPS measurements, the work function was also confirmed by ambient pressure photoelectron spectroscopy. Briefly, the binding energy of Ar 2p in the vicinity of the sample surface can be used to measure the local work function. This is an emergent technique for measuring work functions and has been demonstrated to measure work function differences in ligand capped PbS quantum dots.57 This analysis found the work function of TiO2 to be ∼0.2 eV higher than that of BiVO4, consistent with the previous spectroscopic and electrochemical measurements (Figure S25).
Using these results, we can show the band alignment relative to vacuum of the two semiconductors before contact. Consistent with our photoelectrochemical data, the results of both our spectroscopic and electrochemical measurements demonstrate that TiO2 and BiVO4 form a type II band alignment and that electron transfer from BiVO4 to TiO2 is indeed favorable. After equilibration, band bending at the interface of the two semiconductors will determine the ease with which charge carriers can cross the interface, and unfavorable band bending can create barriers which reduce electron transfer, even if it is energetically favorable overall. We observed this through the reduced photocurrents of TiO2|BiVO4 samples with low Ta concentrations, indicating significant loss of charge carriers at the TiO2|BiVO4 interface. Further investigations into the efficiency of electron transfer between the two materials could yield insights to improve the photoelectrochemical performance.
Electron Transfer
X-ray absorption and emission spectroscopy were used to gain insight into the orbital character of the energy levels involved in the electron transfer process. Previous studies indicate that the TiO2 conduction band is composed primarily of Ti 3d orbital contributions while the valence band has primarily O 2p character.58 For BiVO4, the most important contributions to the conduction band come from V 3d orbitals and O 2p orbitals for the valence band.56 To further investigate the band edge electronic structure of the two semiconductors, valence band photoemission spectroscopy and resonant inelastic X-ray scattering (RIXS) was used. Our results were in agreement with previous studies indicating that the electron transfer occurs via a d–d transition between V 3d states in the conduction band of BiVO4 and Ti 3d states in TiO2 (Figures S26–S32). Further studies to investigate the kinetics of this electron transfer process and how they affect the system performance are underway.
Conclusions
Host–guest systems are useful for achieving high performance in dual absorber photoelectrosynthetic cells when using photoanode materials with poor intrinsic carrier transport properties such as Fe2O3 and BiVO4. In this study we have demonstrated the use of TiO2 as a host material for BiVO4, to provide simultaneous high light absorption and carrier collection efficiency. We have shown how modifications of the host and guest materials can lead to higher performance, namely, by doping the TiO2 guest material, and improving the charge transfer efficiencies of the BiVO4 by addition of a Co-based catalyst. Electrochemical and spectroscopic measurements give evidence for the type II band alignment necessary for favorable electron transfer from BiVO4 to TiO2. We believe this architecture can be extended to improve the performance of related photoanode materials. Achieving higher photocurrents at low potentials will allow for efficient coupling with well-developed photocathode materials for higher bias-free solar to hydrogen efficiencies.
Experimental Methods
Synthesis of TiO2 Nanowire Arrays
Titanium dioxide (TiO2) nanowires were synthesized by a hydrothermal method.39 In a typical synthesis, 83 μL of titanium isopropoxide was mixed with 5 mL of 6 M hydrochloric acid in a 40 mL Teflon vessel. A fluorine-doped tin oxide (FTO) coated glass substrate was angled against the wall of the Teflon vessel such that the FTO surface was facing down. The Teflon vessel was loaded in a stainless steel autoclave and heated to 180 °C for 180 min. After cooling, the substrates were thoroughly rinsed in deionized water. Following growth, the substrates were annealed in a tube furnace in air at 450 °C for 30 min following a 30 min ramp from room temperature. The substrates were allowed to cool, after which a 20 nm TiO2 layer was deposited on the nanowire arrays by ALD.
Atomic Layer Deposition (ALD)
Deposition of Ta2O5 thin films was performed in a customized thermal ALD reactor. The precursors used for Ta2O5 deposition were pentakis(dimethylamido)tantalum (Strem purity 99%) and water.40 The tantalum precursor was held in a stainless steel cylinder, which was maintained at 105 °C. The water source was maintained at room temperature. The substrate temperature for Ta2O5 deposition was 150 °C. Typical pulse times for the tantalum precursor and water were 2.0 and 1.0 s, respectively. The precursors used for TiO2 deposition were titanium(IV) chloride and water. The titanium precursor and the water source were both maintained at room temperature. The substrate temperature for TiO2 deposition was 300 °C. Typical pulse times for the titanium precursor and water were 0.2 and 0.1 s, respectively. Nitrogen was used as a carrier and purge gas in both processes at a flow rate of 10 sccm.
Conversion Process
After the ALD process, the nanowire substrates were loaded into the center of a 1 in. diameter quartz tube and heated in a tube furnace under an Ar atmosphere at 823 K for 12 h.38
Synthesis of BiVO4
A home-built thermal evaporation system was used to deposit varying amounts of Bi metal as measured by the film thickness of evaporated metal incident on a quartz crystal monitor. To convert the Bi metal to BiVO4, ∼50 μL/cm2 of dimethyl sulfoxide (DMSO) containing 150 mM vanadyl acetylacetonate (VO(C5H7O2)2) was dropcast onto the Bi coated TiO2 samples to fully cover their surface. The samples were then heated to 450 °C for 2 h in air following a ramp from room temperature at a rate of ∼1.8 °C/min. During heating, Bi metal and VO2+ ions oxidize and react to form BiVO4. An excess of the vanadium source is used to ensure complete conversion of Bi to BiVO4. Excess V2O5 formed during heating can be removed by soaking samples in 1 M KOH solution for 30 min.44
Physical and Chemical Characterization
The morphology of the nanowire substrates was studied using scanning electron microscopy. SEM images were collected using a Zeiss Gemini Ultra-55 analytical field emission scanning electron microscope operating at 10 kV. Phase information was obtained using XRD. XRD patterns were collected using a Bruker D8 Advance diffractometer with Cu Kα radiation. Individual nanowires were imaged by transmission electron microscopy (TEM) using a JEOL 2100-F field emission analytical TEM operated at 200 kV equipped with an analytical pole piece, a high solid-angle EDS system, and a HAADF (high-angle annular dark field) scanning TEM (STEM) detector. Chemical information was investigated using X-ray photoelectron spectroscopy (XPS). XPS spectra were collected using a PHI 5400 X-ray photoelectron spectrometer equipped with a 4 kV argon ion gun, with Al Kα radiation. The angle between the source and detector was 35°. The measurement chamber was maintained at ∼10–9 Torr during measurement, and measurements were taken at a pass energy of 17.9 eV. All energies were calibrated to spurious carbon at 284.6 eV. Ultraviolet photoelectron spectroscopy measurements were carried out on an Omicron SPHERA spectrometer with a He discharge lamp (He I line, 21.22 eV) as the excitation source. To ensure that the secondary electron cutoff was captured, a sample bias of −3 V was applied to compensate for the instrument work function difference repelling low kinetic energy electrons. The base pressure of the analyzer chamber was below 6 × 10–9 Torr. APXPS and XPS experiments are performed at beamline 9.3.2 of the Advanced Light Source, which employs a differentially pumped electrostatic lens system that allows for measurements at pressures on the order of 100 mTorr.59 Unless otherwise noted, all photoelectron spectra were recorded at photon energies of 490 eV. XAS, XES, and RIXS measurements were performed at beamline 8.0.1 of the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory (LBNL). Absorption data were collected in total electron yield (TEY), total fluorescence yield (TFY), and partial fluorescence yield (PFY). The resolution for XAS spectra was better than 0.15 eV at the O K-edge. The spectra were normalized to the incident photon flux monitored by measuring the photocurrent from a clean gold mesh. The instrumental resolution for X-ray emission was 0.4 eV, and the emission energy was calibrated using the positions of the elastic features in the emission spectra.
Electrochemical Characterization
Electrochemical properties of the nanowire samples were studied in a three-electrode electrochemical cell using a Bio-Logic potentiostat/galvanostat with a built-in electrochemical impedance spectroscopy (EIS) analyzer. Unless otherwise noted, all measurements were performed at room temperature in 0.5 M potassium phosphate electrolyte buffered to pH 7, using a platinum wire counter electrode and a Ag/AgCl reference electrode. The potential scale was calibrated to a reversible hydrogen electrode. A 300 W Xe lamp equipped with an air mass 1.5G filter (Newport) was used as the light source. Prior to measurement, light intensity was standardized using a calibrated silicon photodiode. For IPCE measurements, light from the xenon lamp was dispersed by a monochromator (Newport Corp.), and the photocurrent was recorded at a constant bias with a spectral step of 10 nm. The measured photocurrent was converted to IPCE by normalizing to a standard photodiode with a known quantum efficiency.
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231(P-Chem). We would like to thank Dr. Ethan Crumlin at the Advanced Light Source for assistance with APXPS measurements and helpful discussions. J.R. gratefully acknowledges the support of the National Science Foundation Graduate Research Fellowship Program (NSF GRFP) under Grant No. DGE-0802270 and the UC Berkeley Chancellor’s fellowship. H.L. and A.L.B. thank the Center for Hierarchical Manufacturing (CMMI-0531171). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00402.
Additional characterization of structural, electrochemical, and electronic properties (PDF)
Author Contributions
J.R. and P.Y. devised the project and wrote the paper. J.R. carried out the experiments. J.R. and H.Z. performed synthesis of photoanode samples. J.R., N.K., and N.B. carried out XAS, XES, RIXS, and APXPS measurements. J.R. and H.L. conducted UPS measurements. J.G., A.B., and P.Y. guided the work. All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Walter M. G.; Warren E. L.; McKone J. R.; Boettcher S. W.; Mi Q.; Santori E. A.; Lewis N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Bolton J. R.; Strickler S. J.; Connolly J. S. Limiting and realizable efficiencies of solar photolysis of water. Nature 1985, 316, 495–500. 10.1038/316495a0. [DOI] [Google Scholar]
- Nozik A. J. Photochemical diodes. Appl. Phys. Lett. 1977, 30, 567. 10.1063/1.89262. [DOI] [Google Scholar]
- Seitz L. C.; Chen Z.; Forman A. J.; Pinaud B. A.; Benck J. D.; Jaramillo T. F. Modeling practical performance limits of photoelectrochemical water splitting based on the current state of materials research. ChemSusChem 2014, 7, 1372–1385. 10.1002/cssc.201301030. [DOI] [PubMed] [Google Scholar]
- Hu S.; Xiang C.; Haussener S.; Berger A. D.; Lewis N. S. An analysis of the optimal band gaps of light absorbers in integrated tandem photoelectrochemical water-splitting systems. Energy Environ. Sci. 2013, 6, 2984. 10.1039/c3ee40453f. [DOI] [Google Scholar]
- Liu C.; Dasgupta N. P.; Yang P. Semiconductor Nanowires for Artificial Photosynthesis. Chem. Mater. 2014, 26, 415–422. 10.1021/cm4023198. [DOI] [Google Scholar]
- Ni M.; Leung M. K. H.; Leung D. Y. C.; Sumathy K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renewable Sustainable Energy Rev. 2007, 11, 401–425. 10.1016/j.rser.2005.01.009. [DOI] [Google Scholar]
- Chen Z.; Jaramillo T. F.; Deutsch T. G.; Kleiman-Shwarsctein A.; Forman A. J.; Gaillard N.; Garland R.; Takanabe K.; Heske C.; Sunkara M.; McFarland E. W.; Domen K.; Miller E. L.; Turner J. A.; Dinh H. N. Accelerating materials development for photoelectrochemical hydrogen production: Standards for methods, definitions, and reporting protocols. J. Mater. Res. 2010, 25, 3–16. 10.1557/JMR.2010.0020. [DOI] [Google Scholar]
- Chen Y. W.; Prange J. D.; Duhnen S.; Park Y.; Gunji M.; Chidsey C. E.; McIntyre P. C. Atomic layer-deposited tunnel oxide stabilizes silicon photoanodes for water oxidation. Nat. Mater. 2011, 10, 539–544. 10.1038/nmat3047. [DOI] [PubMed] [Google Scholar]
- Hu S.; Shaner M. R.; Beardslee J. A.; Lichterman M.; Brunschwig B. S.; Lewis N. S. Amorphous TiO2 coatings stabilize Si, GaAs, and GaP photoanodes for efficient water oxidation. Science 2014, 344, 1005–1009. 10.1126/science.1251428. [DOI] [PubMed] [Google Scholar]
- Sun K.; Kuang Y.; Verlage E.; Brunschwig B. S.; Tu C. W.; Lewis N. S. Sputtered NiOx Films for Stabilization of p+n-InP Photoanodes for Solar-Driven Water Oxidation. Adv. Energ Mater. 2015, 5, 1402276. 10.1002/aenm.201402276. [DOI] [Google Scholar]
- Boettcher S. W.; Warren E. L.; Putnam M. C.; Santori E. A.; Turner-Evans D.; Kelzenberg M. D.; Walter M. G.; McKone J. R.; Brunschwig B. S.; Atwater H. A.; Lewis N. S. Photoelectrochemical hydrogen evolution using Si microwire arrays. J. Am. Chem. Soc. 2011, 133, 1216–1219. 10.1021/ja108801m. [DOI] [PubMed] [Google Scholar]
- Lee M. H.; Takei K.; Zhang J.; Kapadia R.; Zheng M.; Chen Y. Z.; Nah J.; Matthews T. S.; Chueh Y. L.; Ager J. W.; Javey A. p-Type InP nanopillar photocathodes for efficient solar-driven hydrogen production. Angew. Chem., Int. Ed. 2012, 51, 10760–10764. 10.1002/anie.201203174. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wang F.; Wang Q. Nanostructure-based WO3 photoanodes for photoelectrochemical water splitting. Phys. Chem. Chem. Phys. 2012, 14, 7894–7911. 10.1039/c2cp40976c. [DOI] [PubMed] [Google Scholar]
- Sivula K.; Le Formal F.; Gratzel M. Solar water splitting: progress using hematite (alpha-Fe(2) O(3)) photoelectrodes. ChemSusChem 2011, 4, 432–449. 10.1002/cssc.201000416. [DOI] [PubMed] [Google Scholar]
- Park Y.; McDonald K. J.; Choi K. S. Progress in bismuth vanadate photoanodes for use in solar water oxidation. Chem. Soc. Rev. 2013, 42, 2321–2337. 10.1039/C2CS35260E. [DOI] [PubMed] [Google Scholar]
- Rettie A. J. E.; Lee H. C.; Marshall L. G.; Lin J.-F.; Capan C.; Lindemuth J.; McCloy J. S.; Zhou J.; Bard A. J.; Mullins C. B. Combined Charge Carrier Transport and Photoelectrochemical Characterization of BiVO4 Single Crystals: Intrinsic Behavior of a Complex Metal Oxide. J. Am. Chem. Soc. 2013, 135, 11389–11396. 10.1021/ja405550k. [DOI] [PubMed] [Google Scholar]
- Kim T. W.; Choi K.-S. Nanoporous BiVO4 Photoanodes with Dual-Layer Oxygen Evolution Catalysts for Solar Water Splitting. Science 2014, 343, 990–994. 10.1126/science.1246913. [DOI] [PubMed] [Google Scholar]
- Robel I.; Subramanian V.; Kuno M.; Kamat P. V. Quantum Dot Solar Cells. Harvesting Light Energy with CdSe Nanocrystals Molecularly Linked to Mesoscopic TiO2 Films. J. Am. Chem. Soc. 2006, 128, 2385–2393. 10.1021/ja056494n. [DOI] [PubMed] [Google Scholar]
- Grätzel M. Dye-sensitized solar cells. J. Photochem. Photobiol., C 2003, 4, 145–153. 10.1016/S1389-5567(03)00026-1. [DOI] [Google Scholar]
- Liu H.; Tang J.; Kramer I. J.; Debnath R.; Koleilat G. I.; Wang X.; Fisher A.; Li R.; Brzozowski L.; Levina L.; Sargent E. H. Electron Acceptor Materials Engineering in Colloidal Quantum Dot Solar Cells. Adv. Mater. 2011, 23, 3832–3837. 10.1002/adma.201101783. [DOI] [PubMed] [Google Scholar]
- Dasgupta N. P.; Sun J.; Liu C.; Brittman S.; Andrews S. C.; Lim J.; Gao H.; Yan R.; Yang P. 25th Anniversary Article: Semiconductor Nanowires – Synthesis, Characterization, and Applications. Adv. Mater. 2014, 26, 2137–2184. 10.1002/adma.201305929. [DOI] [PubMed] [Google Scholar]
- Law M.; Greene L. E.; Johnson J. C.; Saykally R.; Yang P. Nanowire dye-sensitized solar cells. Nat. Mater. 2005, 4, 455–459. 10.1038/nmat1387. [DOI] [PubMed] [Google Scholar]
- Kim H. S.; Lee J. W.; Yantara N.; Boix P. P.; Kulkarni S. A.; Mhaisalkar S.; Gratzel M.; Park N. G. High efficiency solid-state sensitized solar cell-based on submicrometer rutile TiO2 nanorod and CH3NH3PbI3 perovskite sensitizer. Nano Lett. 2013, 13, 2412–2417. 10.1021/nl400286w. [DOI] [PubMed] [Google Scholar]
- Leschkies K. S.; Divakar R.; Basu J.; Enache-Pommer E.; Boercker J. E.; Carter C. B.; Kortshagen U. R.; Norris D. J.; Aydil E. S. Photosensitization of ZnO Nanowires with CdSe Quantum Dots for Photovoltaic Devices. Nano Lett. 2007, 7, 1793–1798. 10.1021/nl070430o. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Zhou S.; Sheehan S. W.; Wang D. Nanonet-Based Hematite Heteronanostructures for Efficient Solar Water Splitting. J. Am. Chem. Soc. 2011, 133, 2398–2401. 10.1021/ja110741z. [DOI] [PubMed] [Google Scholar]
- Sivula K.; Formal F. L.; Grätzel M. WO3–Fe2O3 Photoanodes for Water Splitting: A Host Scaffold, Guest Absorber Approach. Chem. Mater. 2009, 21, 2862–2867. 10.1021/cm900565a. [DOI] [Google Scholar]
- Stefik M.; Cornuz M.; Mathews N.; Hisatomi T.; Mhaisalkar S.; Grätzel M. Transparent, Conducting Nb:SnO2 for Host–Guest Photoelectrochemistry. Nano Lett. 2012, 12, 5431–5435. 10.1021/nl303101n. [DOI] [PubMed] [Google Scholar]
- Liu Q.; He J.; Yao T.; Sun Z.; Cheng W.; He S.; Xie Y.; Peng Y.; Cheng H.; Sun Y.; Jiang Y.; Hu F.; Xie Z.; Yan W.; Pan Z.; Wu Z.; Wei S. Aligned Fe2TiO5-containing nanotube arrays with low onset potential for visible-light water oxidation. Nat. Commun. 2014, 5, 5122. 10.1038/ncomms6122. [DOI] [PubMed] [Google Scholar]
- Pilli S. K.; Janarthanan R.; Deutsch T. G.; Furtak T. E.; Brown L. D.; Turner J. A.; Herring A. M. Efficient photoelectrochemical water oxidation over cobalt-phosphate (Co-Pi) catalyst modified BiVO4/1D-WO3 heterojunction electrodes. Phys. Chem. Chem. Phys. 2013, 15, 14723–14728. 10.1039/c3cp52401a. [DOI] [PubMed] [Google Scholar]
- Rao P. M.; Cai L.; Liu C.; Cho I. S.; Lee C. H.; Weisse J. M.; Yang P.; Zheng X. Simultaneously Efficient Light Absorption and Charge Separation in WO3/BiVO4 Core/Shell Nanowire Photoanode for Photoelectrochemical Water Oxidation. Nano Lett. 2014, 14, 1099–1105. 10.1021/nl500022z. [DOI] [PubMed] [Google Scholar]
- Shi X.; Choi I. Y.; Zhang K.; Kwon J.; Kim D. Y.; Lee J. K.; Oh S. H.; Kim J. K.; Park J. H. Efficient photoelectrochemical hydrogen production from bismuth vanadate-decorated tungsten trioxide helix nanostructures. Nat. Commun. 2014, 5, 4775. 10.1038/ncomms5775. [DOI] [PubMed] [Google Scholar]
- Moniz S. J. A.; Zhu J.; Tang J. 1D Co-Pi Modified BiVO4/ZnO Junction Cascade for Efficient Photoelectrochemical Water Cleavage. Adv. Energ Mater. 2014, 4, 1301590. 10.1002/aenm.201301590. [DOI] [Google Scholar]
- Ho-Kimura S.; Moniz S. J. A.; Handoko A. D.; Tang J. Enhanced photoelectrochemical water splitting by nanostructured BiVO4-TiO2 composite electrodes. J. Mater. Chem. A 2014, 2, 3948–3953. 10.1039/c3ta15268e. [DOI] [Google Scholar]
- Furubayashi Y.; Hitosugi T.; Yamamoto Y.; Inaba K.; Kinoda G.; Hirose Y.; Shimada T.; Hasegawa T. A transparent metal: Nb-doped anatase TiO2. Appl. Phys. Lett. 2005, 86, 252101. 10.1063/1.1949728. [DOI] [Google Scholar]
- Hitosugi T.; Furubayashi Y.; Ueda A.; Itabashi K.; Inaba K.; Hirose Y.; Kinoda G.; Yamamoto Y.; Shimada T.; Hasegawa T. Ta-doped Anatase TiO 2 Epitaxial Film as Transparent Conducting Oxide. Jpn. J. Appl. Phys. 2005, 44, L1063. 10.1143/JJAP.44.L1063. [DOI] [Google Scholar]
- Shockley W. The Theory of p-n Junctions in Semiconductors and p-n Junction Transistors. Bell Syst. Tech. J. 1949, 28, 435–489. 10.1002/j.1538-7305.1949.tb03645.x. [DOI] [Google Scholar]
- Resasco J.; Dasgupta N. P.; Rosell J. R.; Guo J.; Yang P. Uniform Doping of Metal Oxide Nanowires Using Solid State Diffusion. J. Am. Chem. Soc. 2014, 136, 10521–10526. 10.1021/ja505734s. [DOI] [PubMed] [Google Scholar]
- Liu B.; Aydil E. S. Growth of Oriented Single-Crystalline Rutile TiO2 Nanorods on Transparent Conducting Substrates for Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2009, 131, 3985–3990. 10.1021/ja8078972. [DOI] [PubMed] [Google Scholar]
- Hausmann D. M.; de Rouffignac P.; Smith A.; Gordon R.; Monsma D. Highly conformal atomic layer deposition of tantalum oxide using alkylamide precursors. Thin Solid Films 2003, 443, 1–4. 10.1016/S0040-6090(03)00502-9. [DOI] [Google Scholar]
- George S. M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. 10.1021/cr900056b. [DOI] [PubMed] [Google Scholar]
- Parker R. A. Static Dielectric Constant of Rutile TiO2. Phys. Rev. 1961, 124, 1719–1722. 10.1103/PhysRev.124.1719. [DOI] [Google Scholar]
- Gelderman K.; Lee L.; Donne S. W. Flat-Band Potential of a Semiconductor: Using the Mott–Schottky Equation. J. Chem. Educ. 2007, 84, 685. 10.1021/ed084p685. [DOI] [Google Scholar]
- Kang D.; Park Y.; Hill J. C.; Choi K.-S. Preparation of Bi-Based Ternary Oxide Photoanodes BiVO4, Bi2WO6, and Bi2Mo3O12 Using Dendritic Bi Metal Electrodes. J. Phys. Chem. Lett. 2014, 5, 2994–2999. 10.1021/jz501544k. [DOI] [PubMed] [Google Scholar]
- Cho I. S.; Lee C. H.; Feng Y.; Logar M.; Rao P. M.; Cai L.; Kim D. R.; Sinclair R.; Zheng X. Codoping titanium dioxide nanowires with tungsten and carbon for enhanced photoelectrochemical performance. Nat. Commun. 2013, 4, 1723. 10.1038/ncomms2729. [DOI] [PubMed] [Google Scholar]
- Xu M.; Da P.; Wu H.; Zhao D.; Zheng G. Controlled Sn-Doping in TiO2 Nanowire Photoanodes with Enhanced Photoelectrochemical Conversion. Nano Lett. 2012, 12, 1503–1508. 10.1021/nl2042968. [DOI] [PubMed] [Google Scholar]
- McDonald K. J.; Choi K.-S. Photodeposition of Co-Based Oxygen Evolution Catalysts on α-Fe2O3 Photoanodes. Chem. Mater. 2011, 23, 1686–1693. 10.1021/cm1020614. [DOI] [Google Scholar]
- Zhong D. K.; Choi S.; Gamelin D. R. Near-Complete Suppression of Surface Recombination in Solar Photoelectrolysis by “Co-Pi” Catalyst-Modified W:BiVO4. J. Am. Chem. Soc. 2011, 133, 18370–18377. 10.1021/ja207348x. [DOI] [PubMed] [Google Scholar]
- Kanan M. W.; Nocera D. G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- Abdi F. F.; Firet N.; van de Krol R. Efficient BiVO4 Thin Film Photoanodes Modified with Cobalt Phosphate Catalyst and W-doping. ChemCatChem 2013, 5, 490–496. 10.1002/cctc.201200472. [DOI] [Google Scholar]
- Park H. S.; Kweon K. E.; Ye H.; Paek E.; Hwang G. S.; Bard A. J. Factors in the Metal Doping of BiVO4 for Improved Photoelectrocatalytic Activity as Studied by Scanning Electrochemical Microscopy and First-Principles Density-Functional Calculation. J. Phys. Chem. C 2011, 115, 17870–17879. 10.1021/jp204492r. [DOI] [Google Scholar]
- Abdi F. F.; Han L.; Smets A. H.; Zeman M.; Dam B.; van de Krol R. Efficient solar water splitting by enhanced charge separation in a bismuth vanadate-silicon tandem photoelectrode. Nat. Commun. 2013, 4, 2195. 10.1038/ncomms3195. [DOI] [PubMed] [Google Scholar]
- Cooper G.; Turner J. A.; Nozik A. J. Mott-Schottky Plots and Flatband Potentials for Single Crystal Rutile Electrodes. J. Electrochem. Soc. 1982, 129, 1973–1977. 10.1149/1.2124334. [DOI] [Google Scholar]
- Xu Y.; Schoonen M. A. A. The absolute energy positions of conduction and valence bands of selected semiconducting minerals. Am. Mineral. 2000, 85, 543–556. 10.2138/am-2000-0416. [DOI] [Google Scholar]
- Goscinski O. Principles of Ultraviolet Photoelectron Spectroscopy. J. W. Rabalais. Wiley-Interscience Monographs in Chemical Physics, New York, 1977. Int. J. Quantum Chem. 1978, 13, 263–263. 10.1002/qua.560130213. [DOI] [Google Scholar]
- Cooper J. K.; Gul S.; Toma F. M.; Chen L.; Glans P.-A.; Guo J.; Ager J. W.; Yano J.; Sharp I. D. Electronic Structure of Monoclinic BiVO4. Chem. Mater. 2014, 26, 5365–5373. 10.1021/cm5025074. [DOI] [Google Scholar]
- Axnanda S.; Scheele M.; Crumlin E.; Mao B.; Chang R.; Rani S.; Faiz M.; Wang S.; Alivisatos A. P.; Liu Z. Direct Work Function Measurement by Gas Phase Photoelectron Spectroscopy and Its Application on PbS Nanoparticles. Nano Lett. 2013, 13, 6176–6182. 10.1021/nl403524a. [DOI] [PubMed] [Google Scholar]
- Landmann M.; Rauls E.; Schmidt W. The electronic structure and optical response of rutile, anatase and brookite TiO2. J. Phys.: Condens. Matter 2012, 24, 195503. 10.1088/0953-8984/24/19/195503. [DOI] [PubMed] [Google Scholar]
- Grass M. E.; Karlsson P. G.; Aksoy F.; Lundqvist M.; Wannberg B.; Mun B. S.; Hussain Z.; Liu Z. New ambient pressure photoemission endstation at Advanced Light Source beamline 9.3.2. Rev. Sci. Instrum. 2010, 81, 053106. 10.1063/1.3427218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.