

Abstract
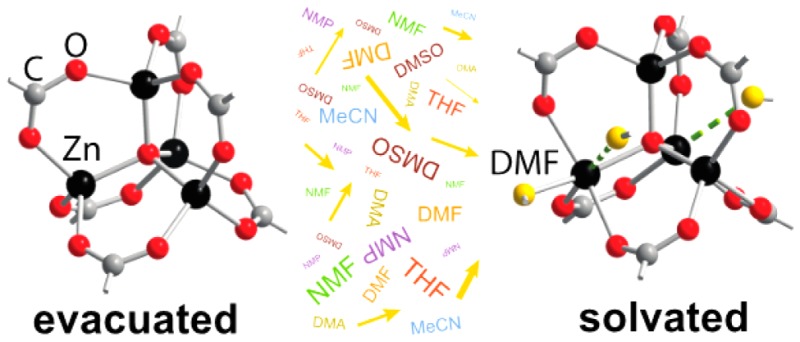
Multinuclear solid-state nuclear magnetic resonance, mass spectrometry, first-principles molecular dynamics simulations, and other complementary evidence reveal that the coordination environment around the Zn2+ ions in MOF-5, one of the most iconic materials among metal–organic frameworks (MOFs), is not rigid. The Zn2+ ions bind solvent molecules, thereby increasing their coordination number, and dynamically dissociate from the framework itself. On average, one ion in each cluster has at least one coordinated N,N-dimethylformamide (DMF) molecule, such that the formula of as-synthesized MOF-5 is defined as Zn4O(BDC)3(DMF)x (x = 1–2). Understanding the dynamic behavior of MOF-5 leads to a rational low-temperature cation exchange approach for the synthesis of metastable Zn4–xCoxO(terephthalate)3 (x > 1) materials, which have not been accessible through typical high-temperature solvothermal routes thus far.
Short abstract
Typically viewed as rigid and static, the metal centers in MOF-5 are shown to interact dynamically with coordinating solvent. With this insight, we accessed unprecedented metastable Co-substituted analogues.
Introduction
Dynamic motion is pervasive and functionally critical in natural and synthetic chemical systems. Enzymes, such as methane monooxygenase, bend and contort interior channels to ensure that substrates, like methane, arrive at the active site just on time.1 Heterogeneous catalysts are no less dynamic: the oxidation of H2 on Pt(111) requires that the surface Pt atoms spontaneously shuffle into a new morphology.2 Even in energy storage systems such as Al-ion batteries, recently reported ultrafast charging likely implies unusual transport of [AlCl4]− anions through dynamic, flexible three-dimensional pores.3 In these and many other examples, crystal structures are not adequate descriptors of the dynamic motions occurring in a given molecule or material. However, whereas probing the dynamic motion in soft materials such as proteins and enzymes is well established,4 an appreciation for the dynamism of harder materials, and its critical role in the function of these materials, has been gaining momentum only more recently.
One class of materials that has seen tremendous growth in this space is MOFs. Although snapshots of dynamic, mechanical motion in these materials can sometimes be gleaned from crystallography, as is the case with “breathing” frameworks,5−7 many materials in this class exhibit properties that are inconsistent with the static view typically conveyed by their crystal structures. For instance, guest molecules that are significantly larger than the pore openings, such as enzymes, can sometimes be adsorbed into the pores.8,9 Furthermore, the organic ligands and secondary building units (SBUs) can be exchanged in numerous MOFs by simply soaking them in solutions of the inserting components.10,11 In another body of literature, catalysis occurs at SBUs where metal centers have no available binding sites, yet the catalytic transformation involves inner-sphere reactivity and must proceed through bond formation between a substrate and a metal center.12−16 Defects aside,17 for these and other phenomena to occur, the metal–ligand bonds in MOFs likely dissociate and, in the case of ligand or metal exchange or SBU-based catalysis, new metal–ligand bonds are formed.
Herein, we report experimental evidence that the Zn4O SBUs in MOF-5, an iconic example in this class of materials, interact dynamically with solvent molecules even in native MOF-5, in liquid DMF. In the presence of coordinating solvents, this material contains not just tetrahedral Zn ions but also octahedral metals. We also report that the surprisingly dynamic coordination environment of Zn ions in MOF-5 allows the synthesis of metastable MOF-5 analogues that are not accessible by typical solvothermal routes. Due to its ubiquity in the field, demonstrating that MOF-5 is a dynamic structure, with Zn ions that can shuffle fast between four- and six-coordinate geometries, suggests that the SBUs in other MOFs may also be less rigid than previously believed.
Results and Discussion
We first suspected that Zn2+ ions of MOF-5 interact with solvent molecules during a routine characterization of an as-synthesized sample. We followed a previously reported procedure that was optimized to remove excess solvent molecules from the pores and to maximize surface area.18 Zn(NO3)2·6H2O and 1,4-benzenedicarboxylate were dissolved in DMF containing 2% deionized water and heated for 7 h at 100 °C. The crystals were collected and washed with fresh DMF every 8 h for 2 days and then soaked in CH2Cl2 with similar repetitions. Surprisingly, although this treatment was reported to remove excess DMF, a Fourier transform infrared spectrum (FT-IR) of a sample that had been fully washed with CH2Cl2, but not evacuated, showed a resonance at 1665 cm–1 corresponding to the C=O stretch of DMF (inset of Figure 1). Furthermore, a TGA profile of the same sample exhibited a well-defined mass loss around 50 °C (shown in Figure 1).19,20 Because of its unambiguous and reproducible “step-like” change, we were able to quantify this mass loss and discovered that it corresponds to exactly two molecules of DMF per formula unit, Zn4O(BDC)3. The mass loss was further identified as DMF by measuring a TGA of CH2Cl2-soaked MOF-5 in-line with a mass spectrometer (MS). This confirmed that the weight loss step between 50 and 150 °C corresponds to the release of DMF (Figure 1). Together, the FT-IR and TGA-MS data showed that unactivated or as-synthesized MOF-5 contained up to two bound DMF molecules per formula unit.
Figure 1.
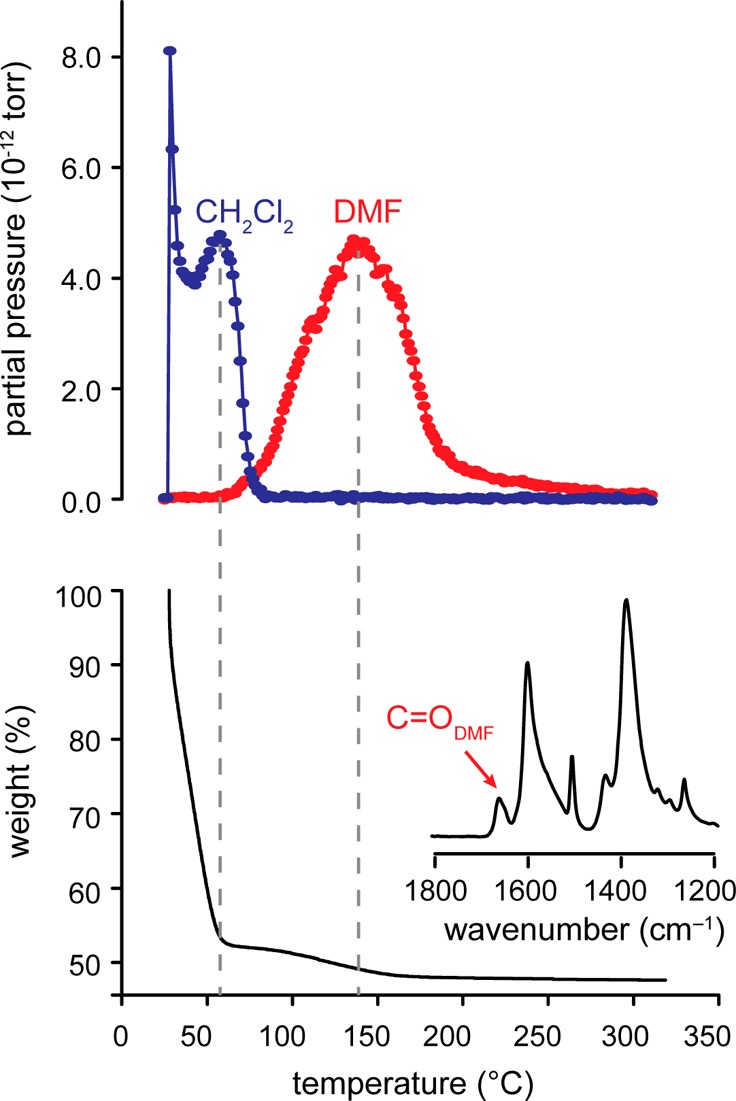
A TGA analysis (bottom) shown alongside mass spectrometry (top) of CH2Cl2- and DMF-soaked MOF-5. A selected portion of the FT-IR spectrum of this sample is shown as an inset.
Surmising that the most likely binding sites for DMF are Zn2+ ions in the Zn4O SBUs, we sought structural evidence for this surprising solvent–SBU interaction. Although X-ray diffraction would be an obvious choice for such studies, solvated crystals of MOF-5 diffracted very poorly. The diffraction quality of MOF-5 crystals improved only upon heating, suggesting again that as-synthesized MOF-5 suffers from long-range structural disorder or otherwise fast dynamic processes that can only be eliminated by evacuating the solvent molecules.21 In the absence of X-ray diffraction data, we sought to obtain structural information on DMF interacting with the Zn4O SBUs from solid-state 67Zn nuclear magnetic resonance spectroscopy (67Zn NMR).22−24 A previous 67Zn NMR study of MOF-5 yielded high quality spectra, but focused only on the fully evacuated samples or samples that were first fully evacuated and then subsequently soaked in noncoordinating solvents such as chloroform.25 Our 67Zn NMR examination of a sample of DMF-soaked MOF-5 with natural abundance of 67Zn (4.1%) for 20 h at 21.1 T and under magic-angle spinning (MAS) conditions revealed no discernible NMR signal (Figure 2A (bottom)). However, a fully evacuated sample produced a well-resolved signal under otherwise identical conditions (Figure 2A (top) and Figure S3). This comparison clearly indicated that the presence of DMF affected the 67Zn NMR parameters in MOF-5, but further experiments and a significantly improved signal-to-noise ratio were needed to identify the exact nature of the interaction of DMF with solvated MOF-5.
Figure 2.

(A) 67Zn NMR spectra of MOF-5 taken under magic-angle spinning conditions at 21.1 T: fully evacuated (top) and when solvated with DMF using enriched (middle) and natural abundant (bottom) zinc. Asterisks denote spinning side bands. The spectral inset (blue) illustrates the secondary site present within the solvated system. (B) 67Zn nonspinning NMR spectra of experimental 97% enriched sample soaked in DMF alongside spectra for two simulated sites collected at 11.7 T. (C) 13C MAS NMR spectra taken of CH2Cl2- and DMF-soaked MOF-5 with only the methyl region displayed using direct-polarization (DP) and 13C[1H] cross-polarization (CP).
To increase the sensitivity of the 67Zn NMR signal, we synthesized MOF-5 from 97%-enriched 67Zn metal. A new spectrum of 67Zn-enriched, DMF-solvated MOF-5, acquired at 21.1 T revealed a strong resonance with some finer structural details at the edges. Careful examination between the nonspinning and MAS spectra (Figure S1) suggested a second zinc site. As a first approximation, this spectrum (Figure S1A and Figure 2A, middle) can be described using a two site model: a resonance centered at approximately 100 ppm with a full-width at half-maximum (fwhm) of 14 kHz, and a second site with a larger quadrupolar interaction (CQ < 6.5 MHz, η < 0.2; to assist the reader a simulation (blue inset) of this larger site is shown in Figure 2A). Because the first site has the smaller quadrupolar coupling constant and is similar to what is observed with evacuated MOF-5, we refer to it as the “unsolvated site”, i.e., four-coordinated, pseudotetrahedral site. Accordingly, the second site, clearly different from that observed in evacuated MOF-5, is named the “solvated site”.
To confirm our two-site model and deconvolute the NMR parameters for each Zn2+ site, we repeated the 67Zn NMR measurements at a lower magnetic field because the line width due to second order quadrupolar interactions scales inversely with magnetic field (i.e., the solvated site would become more apparent as the broadening would become more obvious at 500 MHz vs 900 MHz). Nonspinning experiments for the 67Zn-enriched solvated sample performed at 11.7 T revealed two distinguishable Zn2+ sites with lineshapes dominated by second order quadrupolar interactions, shown in Figure 2B. Using collectively the 11.7 and 21.1 T data, the unsolvated site was simulated with CQ ∼ 4 MHz, η ≤ 0.65, and δiso = 100 (50) ppm and the solvated site with CQ = 5.90 (0.25) MHz, η ≤ 0.1, and δiso = 165 (10) ppm. Due to the lack of resolution between the sites and unsuccessful attempts with multiple quantum experimental methods due to nonfavorable nuclear spin–spin relaxation, further constraints regarding the NMR parameters could not be obtained. Importantly, the parameters fitting these data show that the unsolvated and solvated sites exist in a ratio of 70(8):30(8). In other words, DMF molecules bind to one of the four Zn2+ sites in each Zn4O cluster of MOF-5, as corroborated by the TGA-MS and FT-IR data.
As a complement to our structural information obtained from the 67Zn NMR data, we also probed the DMF–MOF-5 interaction by performing 13C MAS NMR. Thus, MOF-5 solvated with DMF was examined by a Bloch decay experiment to observe all 13C resonances within the sample. The resulting spectrum, shown in Figure S2, displays resonances between 130 and 140 ppm and at 175 ppm, corresponding to aromatic and carboxylate carbons, respectively. Additional resonances between 30 and 40 ppm suggested that at least two DMF species exist in the sample. To test whether these signals result from bound and unbound solvent, we performed a 13C{1H} cross-polarization experiment, intended to reveal only rigid species, i.e., terephthalate and bound DMF. Indeed, Figures S2 and 2C indicate that the signal at 31.4 ppm, likely stemming from unbound DMF, drops out under cross-polarization conditions, leaving the two inequivalent methyl groups at 32.5 and 37.6 ppm locked in position, as would be expected for bound DMF. We also attempted a solution-based refocused-INEPT experiment (Figure S2) and temperature-dependent 2H NMR (Figure S5) using d7-DMF in the hope of probing the dynamics of the Zn–DMF interaction. Due to the time scale of these measurements, however, they were sensitive only to the internal dynamics of methyl rotation of the bound DMF. Nevertheless, the spectra acquired from the 2H NMR confirm that the DMF molecules were bound since they depict a Pake doublet below 255 K, which is only observed for rigid powder-type species.
With experimental evidence that the Zn4O clusters interact with coordinating solvent, DMF in particular, we employed density functional theory to explore how this interaction might occur. Several previous reports used molecular dynamics simulations to understand the water-induced degradation of MOF-5.26−28 Inspired by these studies, we used Born–Oppenheimer molecular dynamics (BOMD) to take into consideration bond formation and configurational contributions from the interaction between MOF-5 and DMF. We employed a cell containing two rotated Zn4(μ4-O)(1,4-benzenedicarboxylate)3 units in the presence of 15 DMF molecules, equivalent to liquid DMF (see Figure 3A). The trajectories of all 15 DMF molecules were calculated at 300 K over 25 ps using 1 fs steps. Starting from an optimized structure, we explored two scenarios: one Zn4O node (Core 1 containing Zn atoms Zn1 through Zn4) was constructed without bound DMF molecules, whereas a second Zn4O node (Core 2, containing Zn atoms Zn5 through Zn8) was specified with one pseudo-octahedral site (Zn8) with two bound DMF molecules at the onset.
Figure 3.
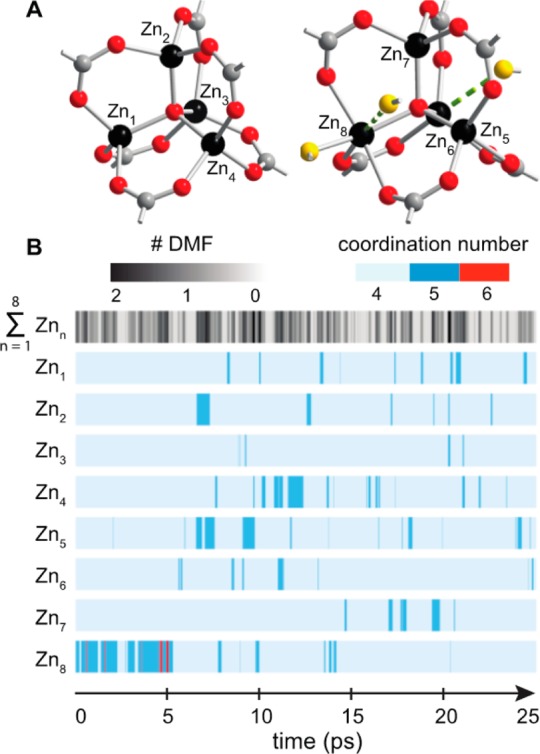
(A) Cluster 1 (left) and cluster 2 (right) of the two Zn4(μ4-O)(1,4-benzenedicarboxylate)3 units employed for the simulation as they appear at time = 0 ps. Carbonyl oxygen atoms from DMF solvent are depicted in yellow. (B) Coordination number for individual Zn sites (Zn1 through Zn8) along the BOMD 25 ps simulation. Light blue denotes a coordination number of 4, dark blue 5, and red 6. The total number of DMF molecules bound to any of the eight Zn sites, Zn1 through Zn8, is shown at the top on a spectrum from zero to two, denoted by white to black, respectively.
In line with our experimental observations, our simulations predict that the MOF-5 SBUs dynamically bind and release DMF molecules without compromising the structural integrity of the framework within the 25 ps time frame of our simulation. Although Core 1 is initially unsolvated, its Zn2+ ions repeatedly exchange DMF molecules after the first 5 ps (Zn1 through Zn4 in Figure 3). Remarkably, our simulations suggest that all Zn atoms participate in DMF binding events over the simulated time frame, but just one site at a time interacts with solvent. Overall, the node containing atoms Zn1–Zn4 spends 28% of the simulation time with coordinated DMF. Even more strikingly, these calculations illustrate that the Zn2+ ions in Core 2 (Zn5 through Zn8) not only dynamically coordinate DMF but also release the organic linker (shown in red in Figure 4). The Zn2+ ion that begins with two solvent ligands, Zn8, quickly releases one DMF to solution, and after only 2.5 ps a framework carboxylate detaches. Yet, by 5 ps the Zn8 site regains both DMF molecules and the organic linker, thereby assuming its original pseudo-octahedral geometry, only to finally release the DMF back to solution (right panel of Figure 4). The carboxylate group initially bound to the pseudo-octahedral Zn8 ion starts coordinated in η2 fashion to Zn8 and Zn6 atoms, then detaches and coordinates η1 only to Zn6, and ends as η2-bound to the same. Overall, Core 2 spends 53% of the simulation time with a Zn site bound to DMF. Most significantly, these simulations show that DMF binding to the Zn4O clusters is favorable and occurs dynamically in liquid DMF at a rate faster than 109 Hz (1 ns).
Figure 4.

Summary of the BOMD simulated trajectories corresponding to the pseudo-octahedral Zn site, Zn8, for the first 6 ps. Zn–O distances are plotted at the top for the framework carboxylates and at the bottom for the two nearest DMF molecules. Snapshots of the simulation are shown at time points (ps) on the right. The carboxylate oxygen that dissociates completely is shown as the red trace in the Ocarboxylate panel. The two colored traces in the ODMF panel represent the positions of the oxygen atoms of two DMF molecules in the vicinity of Zn8. The Zn–O bonding regions are defined by the colored rectangular regions starting at 2.2 Å for both carboxylate and DMF oxygen atoms.
The fact that the Zn4O clusters bind DMF is surprising. Yet, the fact that only one Zn2+ ion in each SBU may exhibit a coordination number above four at any given time is in line with our previous observation of cation exchange in MOF-5: when Ni2+ ions replace Zn2+ in this material, they assume octahedral geometry, and accordingly no more than one Ni2+ ion can be inserted in each SBU.29,30 Importantly, however, the experiments above demonstrate that DMF binding to MOF-5 clusters is dynamic. In other words, as long as the metal ions within the SBUs can support fast exchange between 4-, 5-, and 6-coordinate species, DMF could bind indiscriminately to any one of the four metal ions and exchange between these ions rapidly regardless of their identity. Because four-coordinate pseudotetrahedral Ni2+ is unfavorable in this ligand field environment, insertion of a single Ni2+ ion locks the two DMF molecules onto that ion, preventing exchange with the remaining three Zn2+ ions and limiting exchange to a single Ni2+ per cluster. This hypothesis suggests that a metal ion that could accommodate both tetrahedral and octahedral geometries, such as Co2+, should exchange DMF molecules with the remaining Zn2+ ions in a given SBU, leading to substitution of more than one Zn2+ per cluster, according to the mechanism proposed in Figure 5A. If true, such a mechanism could afford unprecedented “oversubstituted” MOF-5 analogues of formula Zn4–xMxO(BDC)3 (x > 1; M = substituting metal ion).
Figure 5.

(A) The sequence of Co2+ exchanges within the Zn4O cluster of MOF-5, as suggested by the Co2+ symmetry inferred from the UV–vis traces. (B) The results of ICP-AES analysis of MOF-5 soaked in CoCl2 for periods between 1 min and 1 month. (C) Optical photographs of the Co-MOF-5 materials after cation exchange for periods denoted by the labels in white lettering. (D) Normalized diffused reflectance UV–visible traces of the various Co-MOF-5 materials with the length of cation exchange labeled by each trace.
To test this hypothesis, crystals of MOF-5 were soaked in DMF solutions of anhydrous CoCl2 for 1 min, 30 min, 1 h, 1 day, 1 week, 2 weeks, or 1 month. After isolating and washing the resulting materials, they displayed a striking color progression from light pink to deep blue (Figure 5C), indicating the presence of Oh Co2+ and increasing quantities of Td Co2+ with increasing time. A more precise kinetic analysis is complicated by Co–Co and Zn–Zn self-exchange processes, as well as Zn–Co back-exchange processes. ICP-AES analysis of materials isolated at each data point in Figure 5A confirmed that one Co2+ exchanged into each cluster of MOF-5 after just 1 day, and that three Zn2+ ions were replaced after one month (Figure 5B). Diffuse reflectance UV–vis spectra of all materials showed traces that were initially consistent with Co2+ in Oh geometry, but which became increasingly characteristic of Td geometry, validating the scheme shown in Figure 5A. PXRD analysis confirmed that the material soaked for one month retains the MOF-5 structure (Figure S7), while BET analysis of its N2 isotherm at 77 K indicates a surface area of 3170 m2 g–1 (Figure S8).
Whereas isolating these “oversubstituted” analogues by cation exchange was facile, attempts to synthesize them by direct solvothermal synthesis were unsuccessful. Although Co4O(BDC)3 is known,31 and substoichiometrically substituted analogues Zn4–xCoxO(BDC)3 (x ≤ 1) have also been reported,32 the intermediate compounds have been unknown. Our attempts to access these by solvothermal routes included keeping the overall metal content in a typical MOF-5 synthesis mixture constant, while using both 1:1 and 3:1 ratios of Co:Zn under conditions that mimicked the original preparation of MOF-5. A summary of the attempted reaction conditions is given in Table S1. Only by using Co(NO3)2·6H2O in a 1:1 ratio with Zn(NO3)2·6H2O did the resulting material demonstrate the PXRD pattern expected for MOF-5 analogues (Figure S9), but the cobalt content was still less than 25% of the total metal content, indicating the incorporation of less than one Co2+ per SBU. In an alternative strategy, we employed the typical conditions for synthesizing MOF-5, but added additional equivalents of metal in the form of Co(NO3)2·6H2O with Co:Zn ratios of 1:1 or 10:1 in the reaction mixture. In both cases, the resulting materials showed weak diffraction by PXRD analysis that did not resemble the pattern expected of MOF-5 (Figure S10). Although investigations of solvothermal routes can never be exhaustive, these results highlight cation exchange, enabled by dynamic binding of DMF to the MOF-5 SBUs, as a rational and tunable route to potentially metastable MOF-5 analogues that have thus far been inaccessible by solvothermal routes.
Although MOF-5 has previously been depicted as a coordinatively saturated and rigid MOF even in its solvated form, our observation that DMF binds to MOF-5 is consistent with crystal structures of molecules and other MOFs with Zn4O clusters where at least one Zn2+ site features pendant solvent molecules. For instance, Bury et al. reported crystal structures of molecular Zn4O clusters with benzoate or 9-anthracenecarboxylate ligands and a Zn site featuring additional bonds to tetrahydrofuran or water.33 A number of MOFs with Zn4O clusters also exhibit bound solvent molecules, but no reports exist describing MOF-5 itself with bound solvent molecules. The most relevant is a structure that is analogous to MOF-5 and includes a Zn2+ site with two bound water molecules, but is made from naphthalene linkers.34 Consistent with our TGA-MS, NMR, and simulations of MOF-5, the Zn4O clusters in these crystal structures accommodate only one 6-coordinate Zn site.35−47 This evidence suggests that while the MOF-5 node undergoes dynamic interactions, the number of Zn2+ sites that can bind DMF in each Zn4O cluster is limited by how much the framework lattice can distort.
Having this insight into the structural limitations of the Zn4O cluster in MOF-5 allowed us to synthesize materials that would otherwise be difficult to obtain. Knowing that just a single metal site in the Zn4O binds additional ligands provided insight into how to exchange out more Zn sites than had been possible previously.29,30 In deliberately side-stepping this structural limitation by taking advantage of the ability of Co2+ to assume Td symmetry, cation exchange becomes a rational synthetic tool. These results also set the general precedent that we can view the dynamic interactions at the SBUs of MOFs from the perspective of molecular coordination chemistry.
The ability of Zn2+ sites in MOF-5 to alter their coordination environment and detach from ligands may explain other interesting phenomena observed in MOFs. Of the materials reported to undergo cation exchange, many contain metal ions that are coordinatively saturated and are integral to the framework integrity. It is likely that cation exchange preserves the crystallinity in these systems because their SBUs reversibly bind solvent molecules, as in MOF-5. The dynamic behavior of the MOF-5 SBUs revealed in this study could also explain the growing body of literature describing redox catalysis at coordinatively saturated metal sites. Although defect sites can never be discounted in heterogeneous catalysis, it is possible that MOFs able to catalyze reactions at metal sites that are seemingly coordinatively saturated do so by distorting their SBUs when binding substrates.12−14,16 Future studies will explore these concepts in more detail.
Conclusion
Several seminal findings for MOFs were established by first demonstrating they were true for MOF-5. When reported in 1999,48 MOF-5 proved that materials in this class could be permanently porous and provided design principles for the emerging field. Early studies on modifying the composition of MOF-5 inspired the now common notion that the pore size and physicochemical properties of a material might be tailored systematically while preserving the overall topology, through reticular synthesis.49 Gas storage in these materials has become a large area of sustainable energy research and is rooted in the early reports on hydrogen and methane sorption in MOF-5.50 When the binding sites of N2 and Ar within a MOF were first established by single crystal X-ray diffraction51 and when Monte Carlo simulations were employed to evaluate the accuracy of the Brunauer–Emmett–Teller (BET) theory in calculating apparent internal surface areas,52 MOF-5 was again the material of choice.
The results described here add to this list of seminal findings by showing, surprisingly, that the geometry of the Zn4O SBUs in as-synthesized MOF-5 is not rigid. An immediate application of these fundamental studies is the synthesis of new metastable MOF-5 analogues, enabled by the detailed understanding of the dynamic coordination environment of the Zn ions in this material. MOF-5 thus continues to offer critical insight into porous frameworks in general: viewing SBUs in other MOFs as dynamic entities will inform the design principles used for future reactivity studies, thus possibly enabling new applications for these materials. Using this perspective to review even mature fields, such as gas storage and separation, may uncover new insights into how guest molecules adsorb onto pore surfaces. How MOFs are formed, how they interact with their guest molecules, and other fundamental questions might also be answered from viewing even the more classically rigid SBUs as dynamic and potentially coordinatively unsaturated molecular entities.
Experimental Section
Experimental Conditions for Solid-State Nuclear Magnetic Resonance
High field 67Zn nuclear magnetic resonance (NMR) spectra were acquired using a Bruker Avance III 500 (11.7 T) equipped with a 4 mm magic-angle spinning (MAS) single resonance low-gamma Bruker probe using a quadrupolar echo experiment.53 Pulses were calibrated using a 2 M aqueous solution of Zn(NO3)2 (2.3 μs, π/2 pulses, γB1/2π = 108 kHz), an echo delay of 60 μs, an optimized recycle delay of 1 s, and 512,000 coadded transients. The frequency axis is referenced to 0 ppm using a 1 M solution of Zn(NO3)2.54
Ultrahigh field 67Zn NMR spectra were acquired using a Bruker Avance II 900 (21.1 T) spectrometer. Spectra were acquired using various probes including 4 mm MAS double resonance (H/X), home-built single resonance solenoid 5 mm low-gamma nonspinning, 7 mm MAS double resonance (H/X), and 7 mm solenoid-coil single resonance nonspinning probes. Spectra were acquired using either the Bloch decay,55 the quadrupole echo,53 or the CPMG22 (quadrupolar CPMG) pulse sequence. Pulses were calibrated using a 1 M aqueous solution of Zn(NO3)2 for the 4 mm (3 μs, π/2 pulses, γB1/2π = 83 kHz), the 5 mm (2 μs, π/2 pulses, γB1/2π = 125 kHz), and the 7 mm probes (5 μs, π/2 pulses, γB1/2π = 50 kHz (H/X), and 3 μs, π/2 pulses, γB1/2π = 83 kHz (X)). Experiments were acquired using either a 0.5 or 1 s recycle delay, between 8,192 and 128,000 coadded transients, and an echo delay between 60 and 100 μs. MAS NMR spectra were acquired using a spinning frequency between 5 and 10 kHz. The frequency axis is referenced to 0 ppm using 1 M solution of Zn(NO3)2.54 Spectral parameters were simulated using the WSOLIDS software package.56
13C MAS NMR data were acquired using a home-built 500 MHz (11.7 T) spectrometer (courtesy of Dr. D. Ruben, FBML-MIT) equipped with a Magnex superconducting magnet. Spectra were acquired using a 4 mm MAS Chemagnetics triple resonance (H/C/N) probe. Variable temperature 13C MAS NMR data were acquired using Bloch,55 cross-polarization (CP),57 and refocused-INEPT58 experiments. A 1.5 ms contact time was used for the CP experiment, and all experiments were collected with high-power (γB1/2π = 100 kHz) TPPM591H decoupling during acquisition. Spectra were acquired using 3 to 20 s recycle delays, between 8,192 and 32,768 coadded transients, temperatures between 273 and 308 K, and a spinning frequency, ωr/2π = 8,000 (2) Hz. The 13C chemical shift axis was referenced to 40.49 ppm using solid adamantane with respect to DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid, 0 ppm).
Nonspinning 2H NMR experiments were acquired on a custom-built 400 MHz spectrometer (courtesy of Dr. D. Ruben, FBML-MIT) using a custom-built single channel transmission line probe equipped with cryogenic temperature capabilities. All 2H NMR spectra were obtained using the quadrupolar echo sequence60 with 8-step phase cycling61 using a π/2 pulse of 2.0 μs and a delay of 30 μs between the two pulses. The recycle delay was 30 s for all experiments, and the number of coadded transients was between 4,000 and 20,000 depending on the signal-to-noise.
Born–Oppenheimer Molecular Dynamics
A rhombohedral cell was used to model the MOF-5 lattice, consisting of two rotated Zn4(μ4-O)(μ-BDC)3 units. This corresponds to 25% of the cubic crystallographic cell, [Zn4(μ4-O)(μ-BDC)3]8, and has been successfully employed in previous studies.62
Static calculations (optimization) were performed using density functional theory (DFT) as implemented in the Vienna Ab Initio Simulation Package (VASP) plane wave code, version 5.2.63,64 We employed the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional65 that was shown to reproduce binding energies well,66,67 with dispersion corrections added via the Grimme DFT-D2 semiempirical approach.68 Inner electrons were replaced by all-electron frozen cores (projector augmented wave) PAW method,69,70 whereas monoelectronic valence electrons were expanded in plane waves with a kinetic cutoff energy of 350 eV. The threshold for the geometry optimizations was set to 0.015 eV/Å. All the calculations were performed at the Γ point.
This cell was then employed in a first-principles Molecular Dynamics calculation, since it is the only method capable of describing the electronic structure and the dynamics simultaneously. In particular, we used Born–Oppenheimer molecular dynamics (BOMD) to investigate the dynamics of the cage in the presence of DMF, with a liquid phase density. For the BOMD, the convergence criterion in the electronic density of each minimization was set to 10–7 eV. This setup minimizes the energy drift of the extended system including ions, electrons, and the thermostat, for an average value smaller than 0.003 eV/ps per unit cell. The runs were performed in the NVT canonical ensemble; the temperature was controlled using a Nosé thermostat, mass parameter 0.01 amu, and set to 300 K. The equilibration step was performed over 1 ps, whereas each production run was 24 ps long with a time step of 1 fs.
To address the issue of DMF coordination to the metal centers in the MOF-5 structure we employed a continuous coordination number, c. We define two atoms being bonded, c = 1, when their distance is smaller than or equal to the sum of the ionic radii of the cation and anion. The coordination is then calculated through a Gaussian function that follows the harmonic potential between the two atoms and set to zero for distances longer than the sum of the radii plus 0.9 Å (i.e., longer than the van der Waals corresponding distances). The values rounded to the closest integer were then plotted in Figure 3.
Acknowledgments
Synthetic and experimental work except NMR spectroscopy was supported by a CAREER award to M.D. from the National Science Foundation (DMR-1452612). Computational studies (ICIQ) were supported through MINECO (CTQ2012-33826/BQU) and the generous computing resources of BSC-RES. C.K.B. acknowledges support from the NSF through a Graduate Research Fellowship through Grant 1122374. NMR was supported through the National Institutes of Health (EB002026). V.K.M. is grateful to the Natural Sciences and Engineering Research Council of Canada and the Government of Canada for a Banting postdoctoral fellowship. The authors thank Drs. S. Kroeker and K. Marat for access to the Prairie Regional NMR Centre (University of Manitoba, Winnipeg, Canada), and Dr. V. Terskikh (National Research Council, Ottawa, Canada) for his assistance at the Ultrahigh Field NMR Facility for Solids. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh-Field NMR Facility for Solids (Ottawa, Canada), a national research facility funded by CFI, the Ontario Innovation Trust, Recherche Québec, NSERC, Bruker BioSpin, and managed by the University of Ottawa (www.nmr900.ca). NSERC is acknowledged for a Major Resources Support grant. We are grateful to Karthik Narsimhan and the Román group (MIT) for use of their mass spectrometer, and to Dr. C. J. Turner (MIT) for insightful conversations about R-INEPT.
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00247.
Materials, synthetic details, additional analysis of NMR experiments, NMR spectra, PXRD patterns, and N2 isotherms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lee S. J.; McCormick M. S.; Lippard S. J.; Cho U.-S. Control of Substrate Access to the Active Site in Methane Monooxygenase. Nature 2013, 494, 380. 10.1038/nature11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs C.; Hildebrand M.; Volkening S.; Wintterlin J.; Ertl G. Spatiotemporal Self-Organization in a Surface Reaction: From the Atomic to the Mesoscopic Scale. Science 2001, 293, 1635. 10.1126/science.1062883. [DOI] [PubMed] [Google Scholar]
- Lin M.-C.; Gong M.; Lu B.; Wu Y.; Wang D.-Y.; Guan M.; Angell M.; Chen C.; Yang J.; Hwang B.-J.; et al. An Ultrafast Rechargeable Aluminium-Ion Battery. Nature 2015, 520, 324. 10.1038/nature14340. [DOI] [PubMed] [Google Scholar]
- Lewandowski J. R.; Halse M. E.; Blackledge M.; Emsley L. Direct Observation of Hierarchical Protein Dynamics. Science 2015, 348, 578. 10.1126/science.aaa6111. [DOI] [PubMed] [Google Scholar]
- Kitaura R.; Fujimoto K.; Noro S. I.; Kondo M.; Kitagawa S. A Pillared-Layer Coordination Polymer Network Displaying Hysteretic Sorption: [Cu2(pzdc)2(dpyg)]n (pzdc= Pyrazine-2,3-Dicarboxylate; Dpyg = 1,2-Di(4-Pyridyl)glycol. Angew. Chem., Int. Ed. 2002, 41, 133. 10.1002/1521-3773(20020104)41:1<133::AID-ANIE133>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Bourrelly S.; Llewellyn P. L.; Serre C.; Millange F.; Loiseau T.; Férey G. Different Adsorption Behaviors of Methane and Carbon Dioxide in the Isotypic Nanoporous Metal Terephthalates MIL-53 and MIL-47. J. Am. Chem. Soc. 2005, 127, 13519. 10.1021/ja054668v. [DOI] [PubMed] [Google Scholar]
- Sun L.; Hendon C. H.; Minier M. A.; Walsh A.; Dincă M. Million-Fold Electrical Conductivity Enhancement in Fe2(DEBDC) versus Mn2(DEBDC) (E = S, O). J. Am. Chem. Soc. 2015, 137, 6164. 10.1021/jacs.5b02897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D.; Liu T.-F.; Su J.; Bosch M.; Wei Z.; Wan W.; Yuan D.; Chen Y.-P.; Wang X.; Wang K.; et al. Stable Metal-Organic Frameworks Containing Single-Molecule Traps for Enzyme Encapsulation. Nat. Commun. 2015, 6, 5979. 10.1038/ncomms6979. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Lykourinou V.; Vetromile C.; Hoang T.; Ming L. J.; Larsen R. W.; Ma S. How Can Proteins Enter the Interior of a MOF: Investigation of Cytochrome c Translocation into a MOF Consisting of Mesoporous Cages with Microporous Windows. J. Am. Chem. Soc. 2012, 134, 13188. 10.1021/ja305144x. [DOI] [PubMed] [Google Scholar]
- Brozek C. K.; Dincă M. Cation Exchange at the Secondary Building Units of Metal-Organic Frameworks. Chem. Soc. Rev. 2014, 43, 5456. 10.1039/C4CS00002A. [DOI] [PubMed] [Google Scholar]
- Deria P.; Mondloch J. E.; Karagiaridi O.; Bury W.; Hupp J. T.; Farha O. K. Beyond Post-Synthesis Modification: Evolution of Metal-Organic Frameworks via Building Block Replacement. Chem. Soc. Rev. 2014, 43, 5896. 10.1039/C4CS00067F. [DOI] [PubMed] [Google Scholar]
- Beier M. J.; Kleist W.; Wharmby M. T.; Kissner R.; Kimmerle B.; Wright P. A.; Grunwaldt J. D.; Baiker A. Aerobic Epoxidation of Olefins Catalyzed by the Cobalt-Based Metal-Organic Framework STA-12(Co). Chem. - Eur. J. 2012, 18, 887. 10.1002/chem.201101223. [DOI] [PubMed] [Google Scholar]
- Leus K.; Muylaert I.; Vandichel M.; Marin G. B.; Waroquier M.; Van Speybroeck V.; Van der Voort P. The Remarkable Catalytic Activity of the Saturated Metal Organic Framework V-MIL-47 in the Cyclohexene Oxidation. Chem. Commun. 2010, 46, 5085. 10.1039/c0cc01506g. [DOI] [PubMed] [Google Scholar]
- Liu Y.-Y.; Leus K.; Grzywa M.; Weinberger D.; Strubbe K.; Vrielinck H.; Van Deun R.; Volkmer D.; Van Speybroeck V.; Van Der Voort P. Synthesis, Structural Characterization, and Catalytic Performance of a Vanadium-Based Metal-Organic Framework (COMOC-3). Eur. J. Inorg. Chem. 2012, 2012, 2819. 10.1002/ejic.201101099. [DOI] [Google Scholar]
- Ravon U.; Domine M. E.; Gaudillère C.; Desmartin-Chomel A.; Farrusseng D. MOFs as Acid Catalysts with Shape Selectivity Properties. New J. Chem. 2008, 32, 937. 10.1039/b803953b. [DOI] [Google Scholar]
- Sen R.; Saha D.; Mal D.; Brandão P.; Lin Z. Cobalt-Based 3D Metal-Organic Frameworks: Useful Candidates for Olefin Epoxidation at Ambient Temperature by H2O2. Eur. J. Inorg. Chem. 2013, 2013, 5103. 10.1002/ejic.201300560. [DOI] [Google Scholar]
- Fang Z.; Bueken B.; De Vos D. E.; Fischer R. A. Defect-Engineered Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2015, 54, 7234. 10.1002/anie.201411540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye S. S.; Dailly A.; Yaghi O. M.; Long J. R. Impact of Preparation and Handling on the Hydrogen Storage Properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5). J. Am. Chem. Soc. 2007, 129, 14176. 10.1021/ja076877g. [DOI] [PubMed] [Google Scholar]
- Hafizovic J.; Bjørgen M.; Olsbye U.; Dietzel P. D. C.; Bordiga S.; Prestipino C.; Lamberti C.; Lillerud K. P. The Inconsistency in Adsorption Properties and Powder XRD Data of MOF-5 Is Rationalized by Framework Interpenetration and the Presence of Organic and Inorganic Species in the Nanocavities. J. Am. Chem. Soc. 2007, 129, 3612. 10.1021/ja0675447. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ng Z.; Khan E. A.; Jeong H.-K.; Ching C.-b.; Lai Z. Synthesis of Continuous MOF-5 Membranes on Porous alpha-Alumina Substrates. Microporous Mesoporous Mater. 2009, 118, 296–301 10.1016/j.micromeso.2008.08.054. [DOI] [Google Scholar]
- We note that, to our knowledge, there are no reported X-ray structures of as-synthesized or DMF-solvated MOF-5.
- Larsen F. H.; Jakobsen H. J.; Ellis P. D.; Nielsen N. C. Sensitivity-Enhanced Quadrupolar-Echo NMR of Half-Integer Quadrupolar Nuclei. Magnitudes and Relative Orientation of Chemical Shielding and Quadrupolar Coupling Tensors. J. Phys. Chem. A 1997, 101, 8597. 10.1021/jp971547b. [DOI] [Google Scholar]
- Lipton A. S.; Heck R. W.; Ellis P. D. Zinc Solid-State NMR Spectroscopy of Human Carbonic Anhydrase: Implications for the Enzymatic Mechanism. J. Am. Chem. Soc. 2004, 126, 4735. 10.1021/ja0305609. [DOI] [PubMed] [Google Scholar]
- Hibble S. J.; Chippindale A. M.; Marelli E.; Kroeker S.; Michaelis V. K.; Greer B. J.; Aguiar P. M.; Bilbé E. J.; Barney E. R.; Hannon A. C. Local and Average Structure in Zinc Cyanide: Toward an Understanding of the Atomistic Origin of Negative Thermal Expansion. J. Am. Chem. Soc. 2013, 135, 16478. 10.1021/ja406848s. [DOI] [PubMed] [Google Scholar]
- Sutrisno A.; Terskikh V. V.; Shi Q.; Song Z.; Dong J.; Ding S. Y.; Wang W.; Provost B. R.; Daff T. D.; Woo T. K.; et al. Characterization of Zn-Containing Metal-Organic Frameworks by Solid-State 67Zn NMR Spectroscopy and Computational Modeling. Chem. - Eur. J. 2012, 18, 12251. 10.1002/chem.201201563. [DOI] [PubMed] [Google Scholar]
- Bellarosa L.; Castillo J. M.; Vlugt T.; Calero S.; López N. On the Mechanism behind the Instability of Isoreticular Metal-Organic Frameworks (IRMOFs) in Humid Environments. Chem. - Eur. J. 2012, 18, 12260. 10.1002/chem.201201212. [DOI] [PubMed] [Google Scholar]
- Han S. S.; Choi S.-H.; van Duin A. C. T. Molecular Dynamics Simulations of Stability of Metal-Organic Frameworks against H2O Using the ReaxFF Reactive Force Field. Chem. Commun. 2010, 46, 5713. 10.1039/c0cc01132k. [DOI] [PubMed] [Google Scholar]
- Greathouse J. A.; Allendorf M. D. The Interaction of Water with MOF-5 Simulated by Molecular Dynamics. J. Am. Chem. Soc. 2006, 128, 10678. 10.1021/ja063506b. [DOI] [PubMed] [Google Scholar]
- Brozek C. K.; Dincă M. Ti3+-, V2+/3+-, Cr2+/3+-, Mn2+-, and Fe2+-Substituted MOF-5 and Redox Reactivity in Cr- and Fe-MOF-5. J. Am. Chem. Soc. 2013, 135, 12886. 10.1021/ja4064475. [DOI] [PubMed] [Google Scholar]
- a Brozek C. K.; Dincă M. Lattice-Imposed Geometry in Metal–organic Frameworks: Lacunary Zn4O Clusters in MOF-5 Serve as Tripodal Chelating Ligands for Ni2+. Chem. Sci. 2012, 3, 2110. 10.1039/c2sc20306e. [DOI] [Google Scholar]; b Bellarosa L.; Brozek C. K.; García-Melchor M.; Dincă M.; López N. When the Solvent Locks the Cage: Theoretical Insight into the Transmetalation of MOF-5 Lattices and Its Kinetic Limitations. Chem. Mater. 2015, 27, 3422. 10.1021/acs.chemmater.5b00723. [DOI] [Google Scholar]
- Hausdorf S.; Baitalow F.; Böhle T.; Rafaja D.; Mertens F. O. R. L. Main-Group and Transition-Element IRMOF Homologues. J. Am. Chem. Soc. 2010, 132, 10978. 10.1021/ja1028777. [DOI] [PubMed] [Google Scholar]
- Botas J. A.; Calleja G.; Sánchez-Sánchez M.; Orcajo M. G. Cobalt Doping of the MOF-5 Framework and Its Effect on Gas-Adsorption Properties. Langmuir 2010, 26, 5300. 10.1021/la100423a. [DOI] [PubMed] [Google Scholar]
- Bury W.; Justyniak I.; Prochowicz D.; Wróbel Z.; Lewiński J. Oxozinc Carboxylates: A Predesigned Platform for Modelling Prototypical Zn-MOFs’ Reactivity toward Water and Donor Solvents. Chem. Commun. 2012, 48, 7362. 10.1039/c2cc33094f. [DOI] [PubMed] [Google Scholar]
- Yao Q.; Su J.; Cheung O.; Liu Q.; Hedin N.; Zou X. Interpenetrated Metal–Organic Frameworks and Their Uptake of CO2 at Relatively Low Pressures. J. Mater. Chem. 2012, 22, 10345. 10.1039/c2jm15933c. [DOI] [Google Scholar]
- Perry J. J. IV; Feng P. L.; Meek S. T.; Leong K.; Doty F. P.; Allendorf M. D. Connecting Structure with Function in Metal–Organic Frameworks to Design Novel Photo- and Radioluminescent Materials. J. Mater. Chem. 2012, 22, 10235. 10.1039/c2jm16627e. [DOI] [Google Scholar]
- Zhou X. P.; Xu Z.; Zeller M.; Hunter A. D.; Chui S. S. Y.; Che C. M. Coordination Networks from a Bifunctional Molecule Containing Carboxyl and Thioether Groups. Inorg. Chem. 2008, 47, 7459. 10.1021/ic800952z. [DOI] [PubMed] [Google Scholar]
- Gedrich K.; Senkovska I.; Baburin I. A.; Mueller U.; Trapp O.; Kaskel S. New Chiral and Flexible Metal-Organic Framework with a Bifunctional Spiro Linker and Zn4O-Nodes. Inorg. Chem. 2010, 49, 4440. 10.1021/ic9022085. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Xuan W.; Cui Y. Luminescent Microporous Metal-Metallosalen Frameworks with the Primitive Cubic Net. Dalton Trans. 2012, 41, 3928. 10.1039/C1DT11584G. [DOI] [PubMed] [Google Scholar]
- Han S.; Ma Z.; Wei Y.; Kravtsov V. C.; Luisi B. S.; Kulaots I.; Moulton B. A Single-Crystalline Microporous Coordination Polymer with Mixed Parallel and Diagonal Interpenetrating α-Po Networks. CrystEngComm 2011, 13, 4838. 10.1039/c1ce05354j. [DOI] [Google Scholar]
- Burrows A. D.; Fisher L. C.; Hodgson D.; Mahon M. F.; Cessford N. F.; Duren T.; Richardson C.; Rigby S. P. The Synthesis, Structures and Reactions of Zinc and Cobalt Metal-Organic Frameworks Incorporating an Alkyne-Based Dicarboxylate Linker. CrystEngComm 2012, 14, 188. 10.1039/C1CE06044A. [DOI] [Google Scholar]
- Gole B.; Bar A. K.; Mukherjee P. S. Fluorescent Metal–organic Framework for Selective Sensing of Nitroaromatic Explosives. Chem. Commun. 2011, 47, 12137. 10.1039/c1cc15594f. [DOI] [PubMed] [Google Scholar]
- Yang G.-S.; Lang Z.-L.; Zang H.-Y.; Lan Y.-Q.; He W.-W.; Zhao X.-L.; Yan L.-K.; Wang X.-L.; Su Z.-M. Control of Interpenetration in S-Containing Metal-Organic Frameworks for Selective Separation of Transition Metal Ions. Chem. Commun. 2013, 49, 1088. 10.1039/c2cc36894c. [DOI] [PubMed] [Google Scholar]
- Burrows A. D.; Hunter S. O.; Mahon M. F.; Richardson C. A Reagentless Thermal Post-Synthetic Rearrangement of an Allyloxy-Tagged Metal-Organic Framework. Chem. Commun. 2013, 49, 990. 10.1039/C2CC38176A. [DOI] [PubMed] [Google Scholar]
- Barman S.; Furukawa H.; Blacque O.; Venkatesan K.; Yaghi O. M.; Berke H. Azulene Based Metal-Organic Frameworks for Strong Adsorption of H2. Chem. Commun. 2010, 46, 7981. 10.1039/c0cc02589e. [DOI] [PubMed] [Google Scholar]
- Burrows A. D.; Frost C. G.; Mahon M. F.; Richardson C. Sulfur-Tagged Metal-Organic Frameworks and Their Post-Synthetic Oxidation. Chem. Commun. 2009, 4218. 10.1039/b906170c. [DOI] [PubMed] [Google Scholar]
- Burrows A. D.; Frost C. G.; Mahon M. F.; Richardson C. Post-Synthetic Modification of Tagged Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2008, 47, 8482. 10.1002/anie.200802908. [DOI] [PubMed] [Google Scholar]
- Kesanli B.; Cui Y.; Smith M. R.; Bittner E. W.; Bockrath B. C.; Lin W. Highly Interpenetrated Metal-Organic Frameworks for Hydrogen Storage. Angew. Chem., Int. Ed. 2004, 44, 72. 10.1002/anie.200461214. [DOI] [PubMed] [Google Scholar]
- Li H.; Eddaoudi M.; O’Keeffe M.; Yaghi O. M. Design and Synthesis of an Exceptionally Stable and Highly Porous Metal-Organic Framework. Nature 1999, 402, 276. 10.1038/46248. [DOI] [Google Scholar]
- Rosi N. L.; Eckert J.; Eddaoudi M.; Vodak D. T.; Kim J.; O’Keeffe M.; Yaghi O. M. Hydrogen Storage in Microporous Metal-Organic Frameworks. Science 2003, 300, 1127. 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- Eddaoudi M.; Kim J.; Rosi N.; Vodak D.; Wachter J.; O’Keeffe M.; Yaghi O. M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469. 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- Rowsell J. L. C.; Spencer E. C.; Eckert J.; Howard J. A. K.; Yaghi O. M. Gas Adsorption Sites in a Large-Pore Metal-Organic Framework. Science 2005, 309, 1350. 10.1126/science.1113247. [DOI] [PubMed] [Google Scholar]
- Walton K. S.; Snurr R. Q. Applicability of the BET Method for Determining Surface Areas of Microporous Metal-Organic Frameworks. J. Am. Chem. Soc. 2007, 129, 8552. 10.1021/ja071174k. [DOI] [PubMed] [Google Scholar]
- Davis J.; Jeffrey K.; Bloom M.; Valic M. I.; Higgs T. P. Quadrupolar Echo Deuteron Magnetic Resonance Spectroscopy in Ordered Hydrocarbon Chains. Chem. Phys. Lett. 1976, 42, 390. 10.1016/0009-2614(76)80392-2. [DOI] [Google Scholar]
- Harris R. K.; Becker E. D. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts—IUPAC Recommendations. J. Magn. Reson. 2002, 156, 323. 10.1006/jmre.2002.2554. [DOI] [PubMed] [Google Scholar]
- Bloch F.; Hansen W. W.; Packard M. The Nuclear Induction Experiment. Phys. Rev. 1946, 70, 474. 10.1103/PhysRev.70.474. [DOI] [Google Scholar]
- Eichele K.WSOLIDS NMR Simulation Package; 2013. [Google Scholar]
- Pines A.; Gibby M. G.; Waugh J. S. Proton-Enhanced Nuclear Induction Spectroscopy. A Method for High Resolution NMR of Dilute Spins in Solids. J. Chem. Phys. 1972, 56, 1776. 10.1063/1.1677439. [DOI] [Google Scholar]
- Burum D. .; Ernst R. Net Polarization Transfer via a J-Ordered State for Signal Enhancement of Low-Sensitivity Nuclei. J. Magn. Reson. 1980, 39, 163. 10.1016/0022-2364(80)90168-7. [DOI] [Google Scholar]
- Bennett A. E.; Rienstra C. M.; Auger M.; Lakshmi K. V.; Griffin R. G. Heteronuclear Decoupling in Rotating Solids. J. Chem. Phys. 1995, 103, 6951. 10.1063/1.470372. [DOI] [Google Scholar]
- Solomon I. Multiple Echoes in Solids. Phys. Rev. 1958, 110, 61. 10.1103/PhysRev.110.61. [DOI] [Google Scholar]
- Mananga E. S.; Rumala Y. S.; Boutis G. S. Finite Pulse Width Artifact Suppression in Spin-1 Quadrupolar Echo Spectra by Phase Cycling. J. Magn. Reson. 2006, 181, 296. 10.1016/j.jmr.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Mulder F. M.; Dingemans T. J.; Wagemaker M.; Kearley G. J. Modelling of Hydrogen Adsorption in the Metal Organic Framework MOF-5. Chem. Phys. 2005, 317, 113. 10.1016/j.chemphys.2005.06.003. [DOI] [Google Scholar]
- Kresse G. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Perdew J.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Pan W.; Yang W. Describing van Der Waals Interaction in Diatomic Molecules with Generalized Gradient Approximations: The Role of the Exchange Functional. J. Chem. Phys. 1997, 107, 7921. 10.1063/1.475105. [DOI] [Google Scholar]
- Patton D.; Pederson M. Application of the Generalized-Gradient Approximation to Rare-Gas Dimers. Phys. Rev. A: At., Mol., Opt. Phys. 1997, 56, R2495. 10.1103/PhysRevA.56.R2495. [DOI] [Google Scholar]
- Grimme S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Kresse G. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Blöchl P. E. Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.