

Abstract
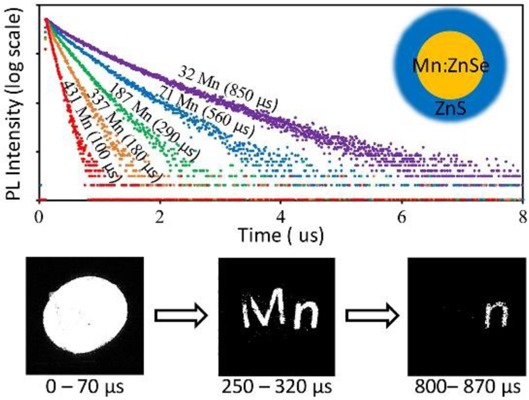
Transition metal doped semiconductor nanocrystals (d-dots) possess fundamentally different emission properties upon photo- or electroexcitation, which render them as unique emitters for special applications. However, in comparison with intrinsic semiconductor nanocrystals, the potential of d-dots has been barely realized, because many of their unique emission properties mostly rely on precise control of their photoluminescence (PL) decay dynamics. Results in this work revealed that it would be possible to obtain bright d-dots with nearly single-exponential PL decay dynamics. By tuning the number of Mn2+ ions per dot from ∼500 to 20 in Mn2+ doped ZnSe nanocrystals (Mn:ZnSe d-dots), the single-exponential PL decay lifetime was continuously tuned from ∼50 to 1000 μs. A synthetic scheme was further developed for uniform and epitaxial growth of thick ZnS shell, ∼7 monolayers. The resulting Mn:ZnSe/ZnS core/shell d-dots were found to be essential for necessary environmental durability of the PL properties, both steady-state and transient ones, for the d-dot emitters. These characteristics combined with intense absorption and high PL quantum yields (70 ± 5%) enabled greatly simplified schemes for various applications of PL lifetime multiplexing using Mn:ZnSe/ZnS core/shell d-dots.
Short abstract
Stable Mn:ZnSe d-dots with single-exponential PL decay and large tunable PL lifetime from 50 to 1000 μs were synthesized, which made them ideal emitters for PL lifetime multiplexing.
Colloidal semiconductor nanocrystals have been pursued as unique emitting materials in various applications because of their broad yet intensive absorption, high emitting brightness, and size-dependent emission colors. Emission from semiconductor nanocrystals can be classified into two main classes, namely, exciton emission and dopant emission. Exciton is an electron–hole pair generated by photo- or electro-excitation and bonded together through Columba interaction, which delocalizes in the entire nanocrystal in the quantum confinement size regime.1 Recombination of exciton in a semiconductor nanocrystal releases a photon with its energy near the absorption band edge, which is thus also known as either band-edge emission or intrinsic emission. Dopant emission is usually associated with the d-shell orbitals of intentionally doped ions in the lattice of a host semiconductor nanocrystal (d-dot),2−4 such as the 3d orbitals of Mn2+ ions in Mn2+ doped ZnSe nanocrystals (Mn:ZnSe d-dots). Different emission mechanisms render these two classes of emitters with complementary luminescence properties. Unlike significant overlapping between emission and absorption in exciton emitters, dopant emission of a d-dot is always noticeably lower in energy than the absorption edge of the host nanocrystal.4,5 Such a large Stokes shift prevents energy transfer and reabsorption for applications using high concentrations of nanocrystal emitters. Different from delocalization of exciton, dopant emission is associated with one atom, implying its low sensitivity to challenging environments.3,6 Opposite to optically allowed exciton emission, dopant emission usually involves optically forbidden transitions of d orbitals of transition metal.7,8 As a result, the typical photoluminescence (PL) decay lifetime for exciton emission is on the order of several tens of nanoseconds,9 which is beneficial for applications requiring “nanofluorophores”. Conversely, the PL decay lifetime of the dopant emission is in the range between microseconds and milliseconds,7,10−13 which fits those applications requiring “nanophosphors”. Finally, transition metal dopants are often magnetically active, which offers unique magneto-optical properties to d-dots.13−15
The potential of exciton emitters—mostly based on CdSe and their core/shell nanocrystals—have been developed to a level ready for certain real-life applications. Though research of d-dots remains in academic exploration at present, their unique emission properties have been actively studied for a variety of applications. D-dots with pure dopant emission were achieved with decent quantum yield about ten years ago, and color-tunable emission of d-dots was reported by several research groups as non-Cd alternative emitters of CdSe nanocrystals.3−5,16−18 Taking the advantages of long PL decay lifetime of dopant emission at low energy, various groups reported nanothermometers based on temperature-dependent dual emission of d-dots.19,20 The long PL lifetime of d-dot emitters also enabled time-resolved fluorometry to remove biological autoluminescence that possesses a typical PL decay lifetime in a few nanoseconds.21
If one could obtain a series of d-dots with single-exponential PL decay dynamics in their typical lifetime window, in principle, it should be possible to realize PL lifetime multiplexing. By labeling each target with an emitter with fixed and single-exponential PL lifetime, PL lifetime multiplexing could potentially read out multiple targets by single excitation. PL lifetime multiplexing possesses numerous advantages over PL color multiplexing and offers a new dimension—time—for PL multiplexing.9,22 While time-resolved fluorometry and time-gated imaging only require the labels emitting in a significantly longer time window than the background luminescence, PL lifetime multiplexing is substantially more stringent in terms of PL decay dynamics of the emitters. This is so because PL lifetime multiplexing requires full deconvolution of all emissive components in the system, each of which should possess single-exponential PL decay dynamics. Exciton emission of intrinsic semiconductor nanocrystals was demonstrated as a possible component for PL color multiplexing ∼15 years ago,23 but their nanosecond and environmentally sensitive PL lifetime would prevent them from being ideal for PL lifetime multiplexing.9,24 Conversely, the microsecond lifetime of d-dots would potentially overcome these problems.
However, to realize their unique applications, such as PL lifetime multiplexing and magneto-optical applications, there are several challenges facing d-dot emitters. For the most developed Mn:ZnSe d-dots, though several groups reported variation of PL lifetime of the dopant emission—the spin forbidden 4T1 to 6T1 transition within the d-orbitals of a Mn2+ ion, the tunable window of the lifetime was insufficient and the PL decay dynamics was usually multi-exponential.7,12,25 The PL decay lifetime of the 4T1 to 6T1 transition of Mn2+ ions in bulk II–VI semiconductor crystals has been known to depend on the dopant concentration, which was considered to be a result of Mn–Mn coupling within the host.26 Recently, Gamelin’s group found that it was possible to obtain single-exponential PL decay (up to ∼2 orders of magnitude of intensity) of the Mn2+ dopant emission with very low Mn2+ concentration (∼0.1%, about 1 Mn2+ ion per dot) though an additional fast component became evident upon increase of Mn concentration in the host ZnSe nanocrystal.13 The authors believed that such multiexponential PL decay dynamics at increased Mn concentration was due to inhomogeneous spatial distribution of Mn2+ ions in each ZnSe nanocrystal. Similarly, with a low Mn2+ concentration (∼6 Mn2+ ions per dot), Cao’s group obtained Mn:CdS/ZnS d-dots with single-exponential PL decay.27 The single-exponential PL decay dynamics should be associated with monodisperse chemical environment of the Mn2+ ions within a sample, including Mn–Mn distance, chemical coordination to Mn2+ ions, surface exposure of Mn2+ ions, uniform concentration among all d-dots in a sample, etc. Although thermal stability of Mn:ZnSe d-dots was well documented, their chemical and photochemical stability was questionable.28 Similar to the intrinsic semiconductor nanocrystals, an epitaxial shell of another wide-bandgap semiconductor would be necessary to meet the durability requirements.29 Furthermore, solving these challenges should simultaneously achieve efficient yet pure dopant emission.
To summarize the above analysis, synthesis of Mn:ZnSe d-dots with the ideal characteristics of dopant emission includes two consecutive steps. In the first step, we need to solve a series of issues for plain core Mn:ZnSe d-dots. In the second step, a wide-bandgap semiconductor should be epitaxially grown onto the Mn:ZnSe d-dots with the conduction (valence) band of the shell higher (lower) than that of ZnSe.
Nonbalanced reactivity of the host and dopant cationic precursors,30 self-purification,31 and rapid diffusion of the dopant ions32 were considered as technical hurdles to achieve controlled and monodisperse chemical environment of Mn2+ ions in d-dots. Especially, this work targeted an extremely large number of Mn2+ ions per nanocrystal—up to ∼500 Mn2+ ions per dot—to cover a sufficiently broad time window of their PL decay lifetime. The resulting d-dots, in terms of composition and structure, are closely related to alloy nanocrystals. In synthesis of alloy nanocrystals using hot injection methods, these problems were known to result in inhomogeneity in composition, surface exposure of all components, and/or isolated nucleation of each component,33 which would in turn cause multiexponential PL decay dynamics for d-dots.
To match the reactivity of zinc stearate coexisting in the reaction solution, we first varied the chain length of Mn2+ fatty acid salts, which did not offer much improvement (data not shown). We found that an active Se precursor—Se suspension in ODE (Se-SUS)34—coupled with a very high anionic to cationic precursor ratio would be necessary. In principle, highly reactive Se precursor with a high relative concentration should overcome the reactivity difference of Zn2+ stearate and Mn2+ stearate, which solved the problem of separate nucleation of pure nanocrystals (Figure S1). Previous work on Mn2+ doped CdS/ZnS core/shell d-dots also demonstrated that excess sulfur could enhance dopant growth ratio.35 To solve the problem of multiexponential decay of the d-dots with high concentration of Mn ions in a dot, a certain amount of fatty amine was found to be necessary in the Se-SUS prior to its injection into the hot mixture of Mn2+ stearate and Zn2+ stearate in octadecene (Figure S1). Under typical reaction conditions, formation of the initial Mn:ZnSe d-dots—about 5 nm in size with all Mn2+ incorporated—must be fast (within ∼5 min) to largely suppress self-purification31 and ion diffusion.32 To make sure that all Mn2+ ions are away from the surface of ZnSe host, additional growth of pure ZnSe onto those initial d-dots was carried out by two doses of the Zn precursor solution, which grew the plain core d-dots to ∼6.5 nm for all samples with different amounts of Mn2+ ions per dot (see transmission electron microscope (TEM) images in Figure S2). A typical procedure is provided in the Supporting Information, and detailed study of the reaction parameters of this new scheme shall be published elsewhere.
Figure 1a illustrates PL and UV spectra of a series of Mn:ZnSe d-dot samples with various doped amounts of Mn2+ ions per nanocrystal, namely, 32, 71, 187, 337, and 431 Mn2+ ions per nanocrystal (see method in Supporting Information). The number of Mn2+ ions per nanocrystal for each sample was determined by energy dispersive X-ray (EDX) spectroscopy and TEM as described in the Supporting Information. The absorption spectra of all five representative samples in Figure 1a were similar, with a peak around ∼425 nm. It should be pointed out that, unlike working in the strong quantum confinement regime of the host semiconductor,36 the sizes of the d-dots were in weak quantum confinement regime of ZnSe. Thus, the influence of dopant concentrations on absorption spectra should be small. PL spectra of these d-dots showed pure dopant emission at ∼583 nm, which was confirmed as the 4T1–6A1 transition of the Mn2+ ions in ZnSe nanocrystals.7 As long as Mn2+ ions per dot was higher than ∼20 in average, no sign of exciton emission from undoped ZnSe nanocrystals was observed in this new synthetic scheme. The number of Mn2+ ions per nanocrystal determined by EDX and TEM in Figure 1, i.e., ∼30 to 450 Mn2+ ions per nanocrystal of the Mn:ZnSe d-dots, corresponded ∼2.5% to 36% of the cations in each initial Mn:ZnSe d-dot (∼5 nm in size) for the five representative samples. Both the number of Mn2+ ions per nanocrystal and the percentage of cations were quite high in comparison to what has been reported in the literature.
Figure 1.
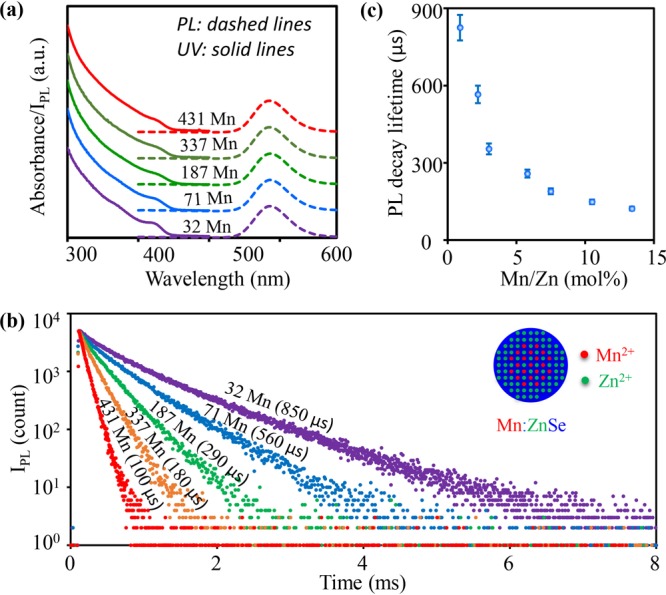
(a) Absorption, and PL spectra and (b) PL decay curves of Mn:ZnSe d-dots with different amounts of Mn2+ ions. (c) The relationship between single-channel PL decay lifetime and Mn2+ concentration. The error bars were determined by 6 sets of repeating experiments.
Different from absorption and steady-state PL spectra, the PL decay dynamics of five representative samples were different from each other (Figure 1b). The PL decay curves of all representative Mn:ZnSe d-dots were nearly single-exponential within 3–4 orders of magnitude of intensity. Moreover, by varying the Mn2+ ions per nanocrystal from about 500 to 20, the single-exponential PL decay lifetime could be varied from ∼50 to 1000 μs (Figure 1c). Reproducibility of the Mn2+ ions concentration dependence of the single-exponential PL decay lifetime was rather good (Figure 1c).
The highly tunable and nearly single-exponential PL decay dynamics of the dopant emission in Figure 1 implies several structure features, if we assumed the PL decay dynamics depending on the spin coupling of Mn–Mn ions in a nanocrystal.13 First, the Mn–Mn distance in d-dots decreased as the number of Mn2+ ions per nanocrystal increased. Second, the Mn2+ ions in each nanocrystal should be close to homogeneous distribution and should not form MnSe clusters within the d-dots. To further confirm the second point, control experiments revealed that, if MnSe cluster(s) existed within a d-dot, a very short component of PL decay lifetime coupled with a complicated PL decay would be the case (Figure S3). Third, the chemical environments of Mn2+ ions in all Mn:ZnSe d-dots in a given sample should be very similar to each other, and there should be no surface exposure of the Mn2+ ions. Detailed structure characterization is ongoing to confirm these conclusions.
Mn:ZnSe d-dots were found to be photochemically not sufficiently stable (Figure 2a). Importantly, along with decrease of PL quantum yield shown in Figure 2a (inset), continuous illumination also caused appearance of a noticeable short-lifetime component in the PL decay dynamics (Figure 2a). Presumably, the new short-lifetime component should be associated with the inefficient surface states created by photobleaching. A similar phenomenon was also reported in the literature, that surface states could influence the PL decay dynamics of the dopant emission of Mn:ZnSe d-dots.37
Figure 2.

(a) Change of PL decay curves of Mn:ZnSe d-dots before (black) and after (blue) UV irradiation. Inset: evolution of PL intensity of Mn:ZnSe and Mn:ZnSe/ZnS d-dots under UV irradiation. (b) Absorption (solid lines) and PL (dotted lines) of Mn:ZnSe (blue) and Mn:ZnSe/ZnS (red) d-dots. (c) TEM images of Mn:ZnSe d-dots before (top) and after (bottom) epitaxial growth of ZnS shell. (d) XRD patterns and (e) PL decay curves of Mn:ZnSe d-dots before (blue) and after (red) epitaxial growth of ZnS shell.
Epitaxial growth of relatively thick ZnS shell solved the photochemical stability problem. Figure 2a (inset) shows that, under the same conditions, the resulting Mn:ZnSe/ZnS core/shell d-dots were practically resistant to photochemical bleaching. Upon growth of the ZnS shell, the absorption spectrum revealed an apparent increase in the ZnS optical window without noticeable red shift of the absorption edge (Figure 2b). TEM measurements (Figure 2c) revealed that epitaxial growth of the ZnS shell increased the size of the nanocrystal significantly—from 6.5 to 11 nm—without worsening the size distribution. High resolution TEM images (Figure S4) revealed that the resulting core/shell d-dots were single crystalline in nature. X-ray powder diffraction measurements (Figure 2d) indicated that growth of the ZnS shell did not cause a new phase. Instead, the entire pattern shifted from that for ZnSe nanocrystals in standard zinc-blende structure to a pattern similar to that for pure ZnS nanocrystals with the same crystal structure. Given the large volume fraction of ZnS, this shift—corresponding to a lattice compression for ZnSe host—was found to be reasonable. Compression of ZnSe host lattice was consistent with the significant red shift of the dopant PL in Figure 2b.38 PL quantum yield of the Mn:ZnSe/ZnS core/shell d-dots was reproducibly ∼70 ± 5% for all dopant concentrations (Figure S5), which was similar to that of the plain core Mn:ZnSe d-dots.
Figure 2e shows that, upon growth of the ZnS shell, PL decay dynamics remained nearly single-exponential with high-count measurements (3–4 orders of magnitude of intensity). The PL decay lifetime decreased from 137 to 114 μs for the specific sample in Figure 2e. For Mn:ZnSe d-dots with different amounts of Mn2+ ions, their PL decay dynamics changed in a similar manner after epitaxial growth of the ZnS shell. This was found to be consistent with the compression of ZnSe lattice upon ZnS growth (Figure 2d), which shortened the Mn–Mn distance in a ZnSe host nanocrystal.38 According to the literature,7,39 the PL decay lifetime of Mn2+ ions in ZnS lattice should be about 1 order of magnitude longer than that in ZnSe lattice. Furthermore, literature results revealed that the PL decay lifetime of Mn2+ ions in the alloy of ZnSe and ZnS increased steadily as the increase of ZnS content.39 We thus considered that the systematic decrease of the single-exponential lifetime upon ZnS shell growth (see Figure 2e as an example) strongly supported no significant diffusion of Mn2+ ions into the ZnS shell and no noticeable alloy formation during the shell growth.
It should be pointed out that epitaxial growth of ZnS shell onto ZnSe (or Mn:ZnSe) nanocrystals with needed thickness for protection of ZnSe host nanocrystals is not well developed, especially for large size core nanocrystals used in this work. Insufficient growth of the shell, formation of pure ZnS nanocrystals, significant blue shift of the absorption spectrum due to alloy, and/or shape irregularity of the resulting core/shell nanocrystals were commonly encountered.28,40−42
Synthesis of the Mn:ZnSe/ZnS core/shell doped nanocrystals (Mn:ZnSe/ZnS d-dots) was realized through a modified SILAR (successive ion layer adsorption reaction) epitaxial scheme using elemental S and Zn stearate as the precursors.43 We varied fatty acids with different chain length and chain structure, and no obvious improvement was observed (data not shown). It was found to be critical to add S into the Mn:ZnSe d-dots solution as the initial step. Furthermore, the S to Zn molar ratio added in consecutive steps in one growth cycle must be S rich, usually with the S:Zn ratio being >1.2. If either the initial step was with the Zn precursor or the S to Zn ratio in one growth cycle was close to 1, appearance of pure ZnS nanocrystals and insufficient shell growth would be inevitable (Figure S6). Presumably, excess S on the surface of the nanocrystals would promote the surface passivation from relatively strong carboxylate ligands to the weak fatty amine ones. The fast ligand dynamics of fatty amines44 would subsequently enhance probability of surface epitaxy of ZnS.
The technical strategies described in the above paragraph would make both epitaxial growth and self-nucleation be extremely fast. This would accelerate formation of pure ZnS nanocrystals as well. In addition, when epitaxy was too fast, size distribution of the resulting core/shell nanocrystals would become broadened upon shell growth (Figure S6). To address these problems, a significant amount of free fatty acid (either 0.5–1 mL of oleic acid or an equivalent amount of stearic acid in 6 mL of reaction mixture) was added. Free fatty acid could reduce the reactivity of metal carboxylate to suppress the extremely fast growth and dissolve newly formed tiny ZnS nanocrystals.
With control of all these key parameters, the resulting Mn:ZnSe/ZnS d-dots retained the size and shape distribution of the Mn:ZnSe d-dots with a desirable thickness of the ZnS shell, i.e., ∼7 monolayers (Figure 2c).
Photochemical stability of the d-dots in Figure 2a is one obvious requirement for applications of unique emission properties of d-dots. Epitaxial growth of a relatively thick ZnS shell, i.e., ∼7 monolayers, was found to be sufficient to boost the stability of the d-dots against other types of challenging environments. Stability of Mn:ZnSe/ZnS d-dots was studied by comparing with that of CdSe/CdS core/shell nanocrystals with 5 monolayers of CdS shell. This specific reference emitted at a similar PL peak position with PL quantum yield of its exciton emission greater than 90% (see details in Figure S7). The PL decay dynamics of intrinsic emission of the CdSe/CdS core/shell nanocrystals was single-exponential.
One targeted application field of d-dots would be PL lifetime multiplexing for biomedical systems. The homogeneity and stability of the PL decay dynamics of the emitters in aqueous solution with different pH are basic requirements for such potential applications, considering the large variation of pH values in biological systems (e.g., lysosome ∼5, cytosol 7.0–7.4, human blood 6.8–7.5, etc.).45 Mn:ZnSe/ZnS d-dots were converted to be soluble in aqueous solution by exchanging the original ligands with mercaptopropanoic acid. After the ligand exchange, the PL quantum yield, peak position, and PL decay dynamics all remained unaltered for the d-dots. Even after dispersion of the resulting water-soluble d-dots in buffer solutions with various pH values, their PL properties were identical to those in the organic solvents (Figure 3a). In comparison, ligand exchange of the CdSe/CdS core/shell nanocrystals and dispersion in the buffer solutions with various pH values changed their emission properties noticeably, which was found to be consistent with the literature.46
Figure 3.
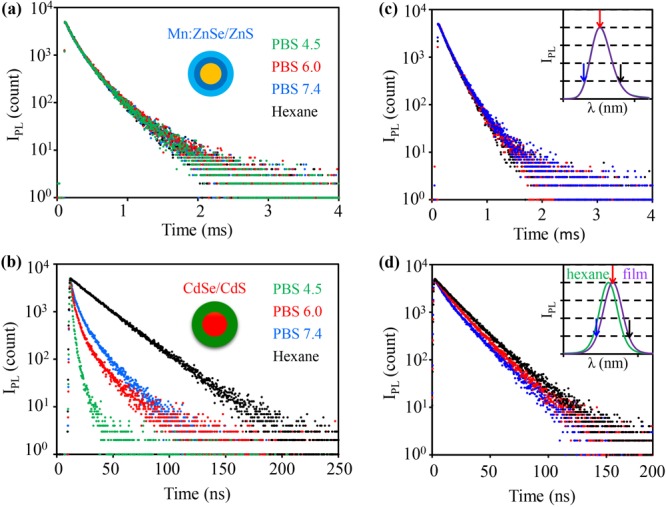
PL decay curves of (a)Mn:ZnSe/ZnS d-dots and (b) CdSe/CdS core/shell nanocrystals before phase transfer in hexane (black) and after phase transfer in PBS with pH 4.5 (green), 6.0 (red), and 7.4 (blue). (c) PL decay curves of Mn:ZnSe/ZnS d-dots and (d) CdSe/CdS core/shell nanocrystals in solid films at different wavelengths with the PL spectra in solution (green) and film (purple) as the insets. The color-coded arrows represented the emission wavelengths for recording PL decay curves.
Both steady-state PL and transient PL of the dopant emission of Mn:ZnSe/ZnS d-dots in solid thin films—necessary for LEDs,47 solid-sate lighting,48 luminescent solar concentrators,49,50 document security,51,52 and information storage53—were studied. The PL spectrum of Mn:ZnSe/ZnS d-dots in thin solid film was practically identical to that in the original solution (Figure 3c (inset)), and their PL decay dynamics did not show any wavelength dependence in the solid film (Figure 3c). Conversely, for the reference CdSe/CdS core/shell nanocrystals, significant red shift of the PL spectra was evidenced (Figure 3d, inset), and their PL decay dynamics differed from one wavelength to another in the solid film. Moreover, compared to that in solution, apparent sink (rise) features could be observed for the PL decay curves of the CdSe/CdS core/shell nanocrystals in the solid film (Figure 3d). These changes of the PL properties for intrinsic semiconductor nanocrystals should be mainly caused by Förster resonance energy transfer, which is efficient for the exciton emission of undoped nanocrystals at high concentrations and/or in the solid state.54 In comparison, for d-dots, Förster resonance energy transfer is eliminated by the large Stokes shift (see Figure 1a).
High excitation power results in high emission intensity, which would be beneficial for certain applications. PL lifetime multiplexing is in this category, because PL lifetime multiplexing relies on transient emission, instead of steady-state emission used in PL color multiplexing. The PL decay dynamics and lifetime of the undoped host semiconductor nanocrystals—ZnSe/ZnS core/shell nanocrystals—were confirmed to be closely related to excitation power (Figure S8), which is consistent with the appearance of bi- and multiexciton emission under high excitation power reported in the literature.55 In comparison, under the same excitation conditions, the PL decay dynamics of the Mn:ZnSe/ZnS d-dots apparently did not change by increasing the excitation power up to tens of photons absorbed per pulse for each dot (Figure S8). Moreover, local heating is often a side effect of high power excitation. Results revealed that the PL decay dynamics of the dopant emission in d-dots was found to be much more stable than that of the exciton emission of the undoped semiconductor nanocrystals against local heating Figure S9). High thermal stability was observed previously in solution and was attributed to the inner atomic emission of the dopant PL.56
With a microsecond excitation source, Figure 4a records a three-dimensional plot of the transient PL of a mixture of a common protein (bull serum albumin, BSA) and two types of water-soluble Mn:ZnSe/ZnS d-dots, namely, one peaked at 605 nm with 110 μs lifetime and the other peaked at 595 nm with 840 μs lifetime. BSA possesses very broad fluorescence with its PL peak at 475 nm and lifetime <1 μs, which was applied to mimic biological autoluminescence. Figure 4a reveals that, as time evolved, the emission contour and peak position changed substantially. Qualitatively, the PL peak was at ∼475 nm at very short decay time and swiftly shifted to a peak near 600 nm. This means that the bright and stable d-dots described in this work would be suited for time-gated imaging. Time-gated imaging, namely, discarding the first snapshot of luminescence (within 10 μs in Figure 4), takes advantage of the lifetime difference of background emission—usually from biological substances in a range of a few nanoseconds—and the labels. Intrinsic semiconductor nanocrystals were demonstrated applicable for time-gated imaging in some cases because their exciton PL lifetime—several tens of nanoseconds—is somewhat longer than that of typical biological autoluminescence.57 Evidently, time-gated imaging could be better realized by d-dots.21 The larger the lifetime difference between the labels and the background luminescence, the less signal loss of the labels.
Figure 4.
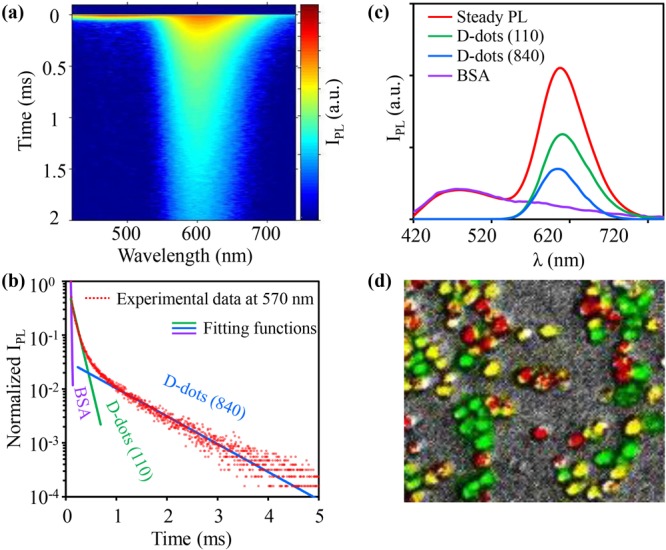
(a) Time resolved emission spectrum of the solution composed of BSA and two types of d-dots. Colors from red to blue represent the logarithmic PL intensity from high to low. (b) PL decay curve at 570 nm with fitting functions. (c) PL spectra of the solution and three reconstructed components. (d) In vitro image (false color) of a mixture of three groups of cells labeled with d-dots with three different PL lifetimes.
Time-gated imaging does not require the PL decay dynamics to be single-exponential though stability and brightness of the emission are important. However, single-exponential decay dynamics is necessary for PL lifetime multiplexing. The PL decay dynamics shown in Figure 1c is substantially single-exponential in comparison with that of the d-dots with pure dopant emission synthesized by the established methods (see Figure S3). In Figure 1c, some of the samples reached single-exponential but some of the samples were not “perfect” single-exponential. In the literature, “perfect” single-exponential requires the goodness-of-fit (χR2) to be 1.0–1.3.58 Within 3–4 orders of magnitude of intensity (usually 5000 counts), the goodness-of-fit (χR2) for representative samples in Figure 1 was reproducibly in the range between 1.2 and 2.5. Experimental results (see below) revealed that the extremely large time dimension of the tunable PL lifetime (∼50–900 μs) made the degree of single-exponential PL decay for all samples be sufficient for PL lifetime multiplexing.
By fixing the emission wavelength in Figure 4a, one could obtain a regular PL decay curve of the mixture at the given emission wavelength. Figure 4b is such an example, which was extracted from Figure 4a by fixing the emission wavelength at 575 nm. Assuming single-exponential PL decay for all three emitters, eq 1 can be applied to fit the results at a given wavelength.
| 1 |
The terms on the right side depends on the number of emissive components in the system. For the system in Figure 4a, there were three components. Here t is the decay time and Iλ, Iλ1 e−t/τ1, Iλ2 e−t/τ2, Iλ3 e−t/τ3 are respectively the PL intensity of total, Component 1, Component 2, and Component 3 at the given wavelength (λ). The single-exponential PL decay lifetime values of three components are τ1, τ2, and τ3, respectively. For λ equal to 575 nm, three lifetime components obtained from the fitting were in excellent accordance with the lifetimes of bull serum albumin (<10 μs, limited by the time resolution of the optical system) and two d-dots (Figure 4b). Applying eq 1 to each wavelength, one would obtain three sets of PL intensity and wavelength, which would result in three separate spectra for three components. Figures 4c and S10 show that the entire emission spectrum reconstructed for each component matched the spectrum of the corresponding pure substance nicely, which quantitatively confirmed suitability of the d-dots emitters for PL lifetime multiplexing.
In imaging applications of PL lifetime multiplexing, PL decay dynamics needs to be deconvoluted in real space, instead of in wavelength dimension discussed above. Three types of Mn:ZnSe/ZnS d-dots with their nearly single-exponential decay lifetime being 220, 550, and 840 μs were separately loaded into three batches of RAW264.7 mouse macrophage cells. After subsequent mixing of three batches of cells, a series of digital fluorescence pictures were taken under a microscope coupled with a pulse laser. The PL signals of the pictures were deconvoluted using eq 2.
| 2 |
The difference between eqs 1 and 2 is the terms for PL intensity. Here, I(x,y), I1(x,y) e−t/τ1, I2(x,y) e−t/τ2, and I3(x,y) e−t/τ3 are respectively the PL intensity of total, Component 1, Component 2, and Component 3 at a given pixel in the pictures with its spatial position as (x, y). Figure 4d shows the false color image of three batches of cells with good spatial resolution.
Results further verified that the representative d-dots were sufficiently single-exponential to resolve imaging information with spatial overlapping. Figure 5a is a steady-state fluorescence image of an oval overlapped with two letters “M” and “n”, in which each of three objects was printed with one specific type of Mn:ZnSe/ZnS d-dots with nearly single-exponential decay dynamics. Upon exciting the same pattern with a pulsed laser, a series of digital pictures were taken with 67 μs intervals (see three sample pictures in Figure 4b). To obtain the encoded information within one exposure, the average number of photons absorbed per d-dot per pulse was about 14, which took advantage of the power insensitivity of PL decay dynamics of the Mn:ZnSe/ZnS d-dots (Figure S8). Using eq 2, one could retract the original information as shown in Figure 5c in false colors.
Figure 5.
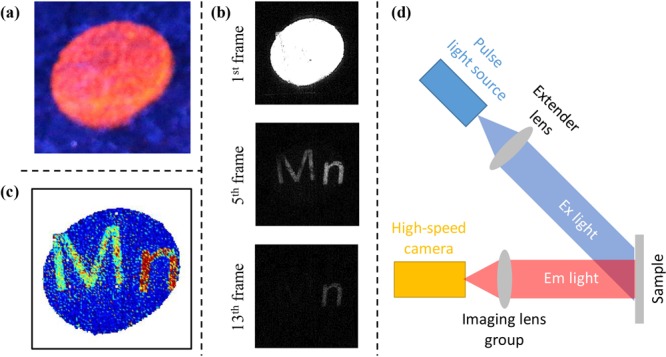
(a) Digital photograph of a pattern encoded with three types of d-dots under continuous irradiation of a 365 nm UV lamp. (b) Digital photographs of the 1st, 5th, and 13th frames captured by high-speed camera at different delay times after one pulse of laser excitation. (c) Reconstructed pattern in false color. (d) Scheme of imaging system of PL lifetime multiplexing using the d-dots described in this work.
The submillisecond lifetime, nearly single-exponential PL decay dynamics, broad and intense absorption, and high PL quantum yield enabled greatly simplified instrumentation for PL lifetime multiplexing (Figure 5d). It can be as simple as a pulsed light source with ∼10 μs pulse width, a commercial high-speed camera with ∼100 μs frame rate, and some lens. It is worth mentioning that the exciton emission of intrinsic semiconductor nanocrystals was applied in conjugation with organic dyes in PL lifetime multiplexing.59 However, because of their sensitivity to the environment (Figure 3) and nanosecond lifetime, it would be difficult for intrinsic semiconductor nanocrystals to improve the traditional schemes of PL lifetime multiplexing significantly. For the same reasons, intrinsic semiconductor nanocrystals would not be compatible with the scheme shown in Figure 5d.
PL color multiplexing and PL lifetime multiplexing imply powerful and unique solutions for data storage,53 document security,51,52 and biomedical detection/imaging.60,61 Working in time dimension, PL lifetime multiplexing renders rich coding channels with low sensitivity to excitation and emission intensity.22 However, the nanosecond PL lifetime of common emitters—usually organic dyes—made PL lifetime multiplexing tightly bound with time-correlated single photon counting (TCSPC), whose point-by-point scanning feature dictates the lifetime imaging to be time-consuming and instrumentation intensive. Though time-gated and wide-field imaging techniques shorten detection time, they require highly stable excitation and sophisticated detection scheme, given the short lifetime of common emitters. Up-conversion nanoparticles62 offer a significantly broad lifetime window, i.e., in the microsecond range, but their low absorption coefficient and low PL quantum yield could be challenging for applications with simple scheme shown in Figure 5d.
The magnetic properties at excited states could be different from those at the ground state, at least for the Mn–Mn dimers.13,63,64 Interestingly, it was reported that the PL decay dynamics of d-dots seemed to be closely related to the magnetic properties at excited states of d-dots.13 Single-exponential PL decay dynamics in a large time window imply nearly monodisperse magnetic coupling and magnetoluminescence in a given sample described in this work. These monodisperse properties should depend on the Mn2+ ion concentration of a given sample. Magnetic properties and magnetoluminescence of these unique emitters are under study for exploring applications in quantum computer and many other spin-based information technologies.65
In conclusion, the dopant emission of Mn:ZnSe/ZnS d-dots could be synthesized not only with high PL quantum yield and desirable excitation properties but also with nearly single-exponential decay dynamics. Furthermore, their single-exponential PL decay lifetime was reproducibly controlled yet tunable in a large time window, approximately between 50 and 900 μs. By epitaxial growth of thick ZnS shell, their PL brightness and decay dynamics were both robust against challenging environments. In comparison to exciton emission of intrinsic semiconductor nanocrystals, these PL properties were found to be unique and would enable interesting applications, such as various schemes of PL lifetime multiplexing, which would be difficult for intrinsic semiconductor nanocrystals. Demonstration of these unique emitters implies that d-dots can be complementary to intrinsic semiconductor nanocrystals to potentially cover a large range of emissive materials in advanced technologies.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants 21303159 and 21233005), Zhejiang Provincial Natural Science Foundation (Grant LQ12H18001), and Fundamental Research Fund for the Central Universities (Grant 2014FZA3006).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00327.
Experimental details and figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brus L. Electronic Wave-Functions in Semiconductor Clusters: Experiment and Theory. J. Phys. Chem. 1986, 90, 2555–2560. 10.1021/j100403a003. [DOI] [Google Scholar]
- Bhargava R. N.; Gallagher D.; Hong X.; Nurmikko A. Optical-Properties of Manganese-Doped Nanocrystals of Zns. Phys. Rev. Lett. 1994, 72, 416–419. 10.1103/PhysRevLett.72.416. [DOI] [PubMed] [Google Scholar]
- Pradhan N.; Goorskey D.; Thessing J.; Peng X. G. An alternative of CdSe nanocrystal emitters: Pure and tunable impurity emissions in ZnSe nanocrystals. J. Am. Chem. Soc. 2005, 127, 17586–17587. 10.1021/ja055557z. [DOI] [PubMed] [Google Scholar]
- Norris D. J.; Efros A. L.; Erwin S. C. Doped nanocrystals. Science 2008, 319, 1776–1779. 10.1126/science.1143802. [DOI] [PubMed] [Google Scholar]
- Pradhan N.; Sarma D. D. Advances in Light-Emitting Doped Semiconductor Nanocrystals. J. Phys. Chem. Lett. 2011, 2, 2818–2826. 10.1021/jz201132s. [DOI] [Google Scholar]
- Yang Y. A.; Chen O.; Angerhofer A.; Cao Y. C. Radial-Position-Controlled Doping of CdS/ZnS Core/Shell Nanocrystals: Surface Effects and Position-Dependent Properties. Chem. - Eur. J. 2009, 15, 3186–3197. 10.1002/chem.200802295. [DOI] [PubMed] [Google Scholar]
- Suyver J. F.; Wuister S. F.; Kelly J. J.; Meijerink A. Luminescence of nanocrystalline ZnSe: Mn2+. Phys. Chem. Chem. Phys. 2000, 2, 5445–5448. 10.1039/b006950g. [DOI] [Google Scholar]
- Beaulac R.; Archer P. I.; Gamelin D. R. Luminescence in colloidal Mn2+-doped semiconductor nanocrystals. J. Solid State Chem. 2008, 181, 1582–1589. 10.1016/j.jssc.2008.05.001. [DOI] [Google Scholar]
- Berezin M. Y.; Achilefu S. Fluorescence Lifetime Measurements and Biological Imaging. Chem. Rev. 2010, 110, 2641–2684. 10.1021/cr900343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. J.; Yuan X.; Ikezawa M.; Jing P. T.; Liu X. Y.; Zheng Z. H.; Kong X. G.; Zhao J. L.; Masumoto Y. Efficient Photoluminescence of Mn2+ Ions in MnS/ZnS Core/Shell Quantum Dots. J. Phys. Chem. C 2009, 113, 16969–16974. 10.1021/jp906390y. [DOI] [Google Scholar]
- Eilers J.; Groeneveld E.; de Mello Donegá C. D.; Meijerink A. Optical Properties of Mn-Doped ZnTe Magic Size Nanocrystals. J. Phys. Chem. Lett. 2012, 3, 1663–1667. 10.1021/jz300300g. [DOI] [PubMed] [Google Scholar]
- Yang B. P.; Shen X. C.; Zhang H. C.; Cui Y. P.; Zhang J. Y. Luminescent and Magnetic Properties in Semiconductor Nanocrystals with Radial-Position-Controlled Mn2+ Doping. J. Phys. Chem. C 2013, 117, 15829–15834. 10.1021/jp402129k. [DOI] [Google Scholar]
- Bradshaw L. R.; May J. W.; Dempsey J. L.; Li X. S.; Gamelin D. R. Ferromagnetic excited-state Mn2+ dimers in Zn1-xMnxSe quantum dots observed by time-resolved magnetophotoluminescence. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 89, 115312. 10.1103/PhysRevB.89.115312. [DOI] [Google Scholar]
- Beaulac R.; Archer P. I.; Liu X. Y.; Lee S.; Salley G. M.; Dobrowolska M.; Furdyna J. K.; Gamelin D. R. Spin-polarizable excitonic luminescence in colloidal Mn2+-doped CdSe quantum dots. Nano Lett. 2008, 8, 1197–1201. 10.1021/nl080195p. [DOI] [PubMed] [Google Scholar]
- Pandey A.; Brovelli S.; Viswanatha R.; Li L.; Pietryga J. M.; Klimov V. I.; Crooker S. A. Long-lived photoinduced magnetization in copper-doped ZnSe-CdSe core-shell nanocrystals. Nat. Nanotechnol. 2012, 7, 792–797. 10.1038/nnano.2012.210. [DOI] [PubMed] [Google Scholar]
- Xie R.; Peng X. Synthesis of Cu-doped InP nanocrystals (d-dots) with ZnSe diffusion barrier as efficient and color-tunable NIR emitters. J. Am. Chem. Soc. 2009, 131, 10645–10651. 10.1021/ja903558r. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Karan N. S.; Pradhan N. Ultrasmall Color-Tunable Copper-Doped Ternary Semiconductor Nanocrystal Emitters. Angew. Chem., Int. Ed. 2011, 50, 6065–6069. 10.1002/anie.201101572. [DOI] [PubMed] [Google Scholar]
- Jana S.; Srivastava B. B.; Jana S.; Bose R.; Pradhan N. Multifunctional Doped Semiconductor Nanocrystals. J. Phys. Chem. Lett. 2012, 3, 2535–2540. 10.1021/jz3010877. [DOI] [PubMed] [Google Scholar]
- Vlaskin V. A.; Janssen N.; van Rijssel J.; Beaulac R.; Gamelin D. R. Tunable Dual Emission in Doped Semiconductor Nanocrystals. Nano Lett. 2010, 10, 3670–3674. 10.1021/nl102135k. [DOI] [PubMed] [Google Scholar]
- Hsia C. H.; Wuttig A.; Yang H. An Accessible Approach to Preparing Water-Soluble Mn2+-Doped (CdSSe)ZnS (Core)Shell Nanocrystals for Ratiometric Temperature Sensing. ACS Nano 2011, 5, 9511–9522. 10.1021/nn2025622. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Chen Y.; Jiang L. P.; Geng J.; Zhang J. R.; Zhu J. J. Manganese-Doped ZnSe Quantum Dots as a Probe for Time-Resolved Fluorescence Detection of 5-Fluorouracil. Anal. Chem. 2011, 83, 9076–9081. 10.1021/ac202101u. [DOI] [PubMed] [Google Scholar]
- Ebrecht R.; Don Paul C.; Wouters F. S. Fluorescence lifetime imaging microscopy in the medical sciences. Protoplasma 2014, 251, 293–305. 10.1007/s00709-013-0598-4. [DOI] [PubMed] [Google Scholar]
- Han M. Y.; Gao X. H.; Su J. Z.; Nie S. Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules. Nat. Biotechnol. 2001, 19, 631–635. 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- Resch-Genger U.; Grabolle M.; Cavaliere-Jaricot S.; Nitschke R.; Nann T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- Gan C. L.; Zhang Y. P.; Battaglia D.; Peng X. G.; Xiao M. Fluorescence lifetime of Mn-doped ZnSe quantum dots with size dependence. Appl. Phys. Lett. 2008, 92, 241111. 10.1063/1.2945274. [DOI] [Google Scholar]
- Kneip M. K.; Yakovlev D. R.; Bayer M.; Maksimov A. A.; Tartakovskii I. I.; Keller D.; Ossau W.; Molenkamp L. W.; Waag A. Spin-lattice relaxation of Mn ions in ZnMnSe/ZnBeSe quantum wells measured under pulsed photoexcitation. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 73, 045305. 10.1103/PhysRevB.73.045305. [DOI] [Google Scholar]
- Chen O.; Shelby D. E.; Yang Y. A.; Zhuang J. Q.; Wang T.; Niu C. G.; Omenetto N.; Cao Y. C. Excitation-Intensity-Dependent Color-Tunable Dual Emissions from Manganese-Doped CdS/ZnS Core/Shell Nanocrystals. Angew. Chem., Int. Ed. 2010, 49, 10132–10135. 10.1002/anie.201004926. [DOI] [PubMed] [Google Scholar]
- Zeng R. S.; Zhang T. T.; Dai G. Z.; Zou B. S. Highly Emissive, Color-Tunable, Phosphine-Free Mn:ZnSe/ZnS Core/Shell and Mn:ZnSeS Shell-Alloyed Doped Nanocrystals. J. Phys. Chem. C 2011, 115, 3005–3010. 10.1021/jp111288h. [DOI] [Google Scholar]
- Peng X. G. Band Gap and Composition Engineering on a Nanocrystal (BCEN) in Solution. Acc. Chem. Res. 2010, 43, 1387–1395. 10.1021/ar100025m. [DOI] [PubMed] [Google Scholar]
- Zu L. J.; Wills A. W.; Kennedy T. A.; Glaser E. R.; Norris D. J. Effect of Different Manganese Precursors on the Doping Efficiency in ZnSe Nanocrystals. J. Phys. Chem. C 2010, 114, 21969–21975. 10.1021/jp106594n. [DOI] [Google Scholar]
- Erwin S. C.; Zu L.; Haftel M. I.; Efros A. L.; Kennedy T. A.; Norris D. J. Doping semiconductor nanocrystals. Nature 2005, 436, 91–94. 10.1038/nature03832. [DOI] [PubMed] [Google Scholar]
- Chen D. A.; Viswanatha R.; Ong G. L.; Xie R. G.; Balasubramaninan M.; Peng X. G. Temperature Dependence of ″Elementary Processes″ in Doping Semiconductor Nanocrystals. J. Am. Chem. Soc. 2009, 131, 9333–9339. 10.1021/ja9018644. [DOI] [PubMed] [Google Scholar]
- Nag A.; Chakraborty S.; Sarma D. D. To dope Mn2+ in a semiconducting nanocrystal. J. Am. Chem. Soc. 2008, 130, 10605–10611. 10.1021/ja801249z. [DOI] [PubMed] [Google Scholar]
- Pu C. D.; Zhou J. H.; Lai R. C.; Niu Y.; Nan W. N.; Peng X. G. Highly reactive, flexible yet green Se precursor for metal selenide nanocrystals: Se-octadecene suspension (Se-SUS). Nano Res. 2013, 6, 652–670. 10.1007/s12274-013-0341-7. [DOI] [Google Scholar]
- Yang Y. A.; Chen O.; Angerhofer A.; Cao Y. C. On Doping CdS/ZnS Core/Shell Nanocrystals with Mn. J. Am. Chem. Soc. 2008, 130, 15649–15661. 10.1021/ja805736k. [DOI] [PubMed] [Google Scholar]
- Vlaskin V. A.; Barrows C. J.; Erickson C. S.; Gamelin D. R. Nanocrystal Diffusion Doping. J. Am. Chem. Soc. 2013, 135, 14380–14389. 10.1021/ja4072207. [DOI] [PubMed] [Google Scholar]
- Schimpf A. M.; Ochsenbein S. T.; Gamelin D. R. Surface Contributions to Mn2+ Spin Dynamics in Colloidal Doped Quantum Dots. J. Phys. Chem. Lett. 2015, 6, 457–463. 10.1021/jz5026678. [DOI] [PubMed] [Google Scholar]
- Ithurria S.; Guyot-Sionnest P.; Mahler B.; Dubertret B. Mn2+ as a radial pressure gauge in colloidal core/shell nanocrystals. Phys. Rev. Lett. 2007, 99, 265501. 10.1103/PhysRevLett.99.265501. [DOI] [PubMed] [Google Scholar]
- Crabtree D. F. Luminescence, Optical-Absorption, and ESR of ZnS-Se: Mn. Phys. Status Solidi A 1974, 22, 543–552. 10.1002/pssa.2210220220. [DOI] [Google Scholar]
- Chen H. S.; Lo B.; Hwang J. Y.; Chang G. Y.; Chen C. M.; Tasi S. J.; Wang S. J. J. Colloidal ZnSe, ZnSe/ZnS, and ZnSe/ZnSeS quantum dots synthesized from ZnO. J. Phys. Chem. B 2004, 108, 17119–17123. 10.1021/jp047035w. [DOI] [Google Scholar]
- Jana S.; Srivastava B. B.; Acharya S.; Santra P. K.; Jana N. R.; Sarma D. D.; Pradhan N. Prevention of photooxidation in blue-green emitting Cu doped ZnSe nanocrystals. Chem. Commun. 2010, 46, 2853–2855. 10.1039/b925980e. [DOI] [PubMed] [Google Scholar]
- Jana S.; Srivastava B. B.; Pradhan N. Correlation of Dopant States and Host Bandgap in Dual-Doped Semiconductor Nanocrystals. J. Phys. Chem. Lett. 2011, 2, 1747–1752. 10.1021/jz200673q. [DOI] [Google Scholar]
- Li J. J.; Wang Y. A.; Guo W. Z.; Keay J. C.; Mishima T. D.; Johnson M. B.; Peng X. G. Large-scale synthesis of nearly monodisperse CdSe/CdS core/shell nanocrystals using air-stable reagents via successive ion layer adsorption and reaction. J. Am. Chem. Soc. 2003, 125, 12567–12575. 10.1021/ja0363563. [DOI] [PubMed] [Google Scholar]
- Pradhan N.; Reifsnyder D.; Xie R. G.; Aldana J.; Peng X. G. Surface ligand dynamics in growth of nanocrystals. J. Am. Chem. Soc. 2007, 129, 9500–9509. 10.1021/ja0725089. [DOI] [PubMed] [Google Scholar]
- Boron W. F.; Boulpaep E. L.. Medical Physiology: A Cellular and Molecular Approach, 1st ed.; Elsevier: 2004. [Google Scholar]
- Chen O.; Zhao J.; Chauhan V. P.; Cui J.; Wong C.; Harris D. K.; Wei H.; Han H. S.; Fukumura D.; Jain R. K.; Bawendi M. G. Compact high-quality CdSe-CdS core-shell nanocrystals with narrow emission linewidths and suppressed blinking. Nat. Mater. 2013, 12, 445–451. 10.1038/nmat3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki Y.; Supran G. J.; Bawendi M. G.; Bulovic V. Emergence of colloidal quantum-dot light-emitting technologies. Nat. Photonics 2013, 7, 13–23. 10.1038/nphoton.2012.328. [DOI] [Google Scholar]
- Schubert E. F.; Kim J. K. Solid-state light sources getting smart. Science 2005, 308, 1274–1278. 10.1126/science.1108712. [DOI] [PubMed] [Google Scholar]
- Debije M. G.; Verbunt P. P. C. Thirty Years of Luminescent Solar Concentrator Research: Solar Energy for the Built Environment. Adv. Energy Mater. 2012, 2, 12–35. 10.1002/aenm.201100554. [DOI] [Google Scholar]
- Meinardi F.; Colombo A.; Velizhanin K. A.; Simonutti R.; Lorenzon M.; Beverina L.; Viswanatha R.; Klimov V. I.; Brovelli S. Large-area luminescent solar concentrators based on ’Stokes-shift-engineered’ nanocrystals in a mass-polymerized PMMA matrix. Nat. Photonics 2014, 8, 392–399. 10.1038/nphoton.2014.54. [DOI] [Google Scholar]
- Lu Y. Q.; Zhao J. B.; Zhang R.; Liu Y. J.; Liu D. M.; Goldys E. M.; Yang X. S.; Xi P.; Sunna A.; Lu J.; Shi Y.; Leif R. C.; Huo Y. J.; Shen J.; Piper J. A.; Robinson J. P.; Jin D. Y. Tunable lifetime multiplexing using luminescent nanocrystals. Nat. Photonics 2014, 8, 32–36. 10.1038/nphoton.2013.322. [DOI] [Google Scholar]
- Kim W. J.; Nyk M.; Prasad P. N. Color-coded multilayer photopatterned microstructures using lanthanide (III) ion co-doped NaYF4 nanoparticles with upconversion luminescence for possible applications in security. Nanotechnology 2009, 20, 185301. 10.1088/0957-4484/20/18/185301. [DOI] [PubMed] [Google Scholar]
- Zijlstra P.; Chon J. W. M.; Gu M. Five-dimensional optical recording mediated by surface plasmons in gold nanorods. Nature 2009, 459, 410–413. 10.1038/nature08053. [DOI] [PubMed] [Google Scholar]
- Crooker S. A.; Hollingsworth J. A.; Tretiak S.; Klimov V. I. Spectrally resolved dynamics of energy transfer in quantum-dot assemblies: Towards engineered energy flows in artificial materials. Phys. Rev. Lett. 2002, 89, 186802. 10.1103/PhysRevLett.89.186802. [DOI] [PubMed] [Google Scholar]
- Klimov V. I.; Mikhailovsky A. A.; McBranch D. W.; Leatherdale C. A.; Bawendi M. G. Quantization of multiparticle Auger rates in semiconductor quantum dots. Science 2000, 287, 1011–1013. 10.1126/science.287.5455.1011. [DOI] [PubMed] [Google Scholar]
- Pradhan N.; Peng X. G. Efficient and color-tunable Mn-doped ZnSe nanocrystal emitters: Control of optical performance via greener synthetic chemistry. J. Am. Chem. Soc. 2007, 129, 3339–3347. 10.1021/ja068360v. [DOI] [PubMed] [Google Scholar]
- Dahan M.; Laurence T.; Pinaud F.; Chemla D. S.; Alivisatos A. P.; Sauer M.; Weiss S. Time-gated biological imaging by use of colloidal quantum dots. Opt. Lett. 2001, 26, 825–827. 10.1364/OL.26.000825. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Baltimore, 2006. [Google Scholar]
- Grabolle M.; Kapusta P.; Nann T.; Shu X.; Ziegler J.; Resch-Genger U. Fluorescence Lifetime Multiplexing with Nanocrystals and Organic Labels. Anal. Chem. 2009, 81, 7807–7813. 10.1021/ac900934a. [DOI] [PubMed] [Google Scholar]
- De Rosa S. C.; Brenchley J. M.; Roederer M. Beyond six colors: A new era in flow cytometry. Nat. Med. 2003, 9, 112–117. 10.1038/nm0103-112. [DOI] [PubMed] [Google Scholar]
- Li Y. G.; Cu Y. T. H.; Luo D. Multiplexed detection of pathogen DNA with DNA-based fluorescence nanobarcodes. Nat. Biotechnol. 2005, 23, 885–889. 10.1038/nbt1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J. C.; van Veggel F. C. J. M. Absolute quantum yield measurements of colloidal NaYF4: Er3+, Yb3+ upconverting nanoparticles. Nanoscale 2010, 2, 1417–1419. 10.1039/c0nr00253d. [DOI] [PubMed] [Google Scholar]
- Mcclure D. S. Optical Spectra of Exchange Coupled Mn++ Ion Pairs in ZnS: MnS. J. Chem. Phys. 1963, 39, 2850–2855. 10.1063/1.1734115. [DOI] [Google Scholar]
- Busse W.; Gumlich H. E.; Meissner B.; Theis D. Time Resolved Spectroscopy of ZnS: Mn by Dye-Laser Technique. J. Lumin. 1976, 12–13, 693–700. 10.1016/0022-2313(76)90163-0. [DOI] [Google Scholar]
- Wolf S. A.; Awschalom D. D.; Buhrman R. A.; Daughton J. M.; von Molnar S.; Roukes M. L.; Chtchelkanova A. Y.; Treger D. M. Spintronics: A spin-based electronics vision for the future. Science 2001, 294, 1488–1495. 10.1126/science.1065389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.