

Abstract
Fetal Alcohol Syndrome (FAS) is a common birth defect in many societies. Affected individuals have neurodevelopmental disabilities and a distinctive craniofacial dysmorphology. These latter deficits originate during early development from the ethanol-mediated apoptotic depletion of cranial facial progenitors, a population known as the neural crest. We showed previously that this apoptosis is caused because acute ethanol exposure activates a G protein-dependent intracellular calcium within cranial neural crest progenitors, and this calcium transient initiates the cell death. The dysregulated signals that reside downstream of ethanol’s calcium transient and effect neural crest death are unknown. Here we show that ethanol’s repression of the transcriptional effector β-catenin causes the neural crest losses. Clinically-relevant ethanol concentrations (22–78 mM) rapidly deplete nuclear β-catenin from neural crest progenitors, with accompanying losses of β-catenin transcriptional activity and downstream genes that govern neural crest induction, expansion and survival. Using forced expression studies we show that β-catenin loss of function (via dominant-negative TCF) recapitulates ethanol’s effects on neural crest apoptosis, whereas β-catenin gain-of-function in ethanol’s presence preserves neural crest survival. Blockade of ethanol’s calcium transient using Bapta-AM normalizes β-catenin activity and prevents the neural crest losses, whereas ionomycin treatment is sufficient to destabilize β-catenin. We propose that ethanol’s repression of β-catenin causes the neural crest losses in this model of FAS. β-Catenin is a novel target for ethanol’s teratogenicity. β-Catenin/Wnt signals participate in many developmental events and its rapid and persistent dysregulation by ethanol may explain why the latter is such a potent teratogen.
Keywords: ethanol, neural crest, apoptosis, intracellular calcium, β-catenin, Wnt signaling
Introduction
Fetal Alcohol Spectrum Disorders (FASD) is the most common non-genetic birth defect in western nations and a leading cause of neurodevelopmental disability in children. The most severe form, Fetal Alcohol Syndrome, affects 0.33–2.2 per 1000 live births with substantially higher rates (8–89.2/1000) in populations with appreciable ethanol abuse (Clarren et al. 2001; May et al. 2007). FAS is characterized by neurodevelopmental deficits, growth retardation, and a distinctive craniofacial appearance including micrognathia, maxillary hypoplasia, flattened midface, and deficits of the overlying soft tissue (Smith and Debelak-Kragtorp, 2006). These facial structures are derived from the neural crest, a pluripotent stem cell population that originates early during development within the dorsal neuroectoderm (LeDouarin and Kalcheim, 1999). As the elevated neural folds fuse, neural crest cells emigrate into the cranial mesenchyme where they differentiate to form the craniofacial bone, cartilage and connective tissue, and portions of the cranial ganglia. Animal studies show that the distinctive facial hypoplasia of FAS results, in part, from the apoptotic deletion of premigratory and early migratory cranial neural crest in response to ethanol exposure (Cartwright et al. 1998; Dunty et al. 2006; Su et al. 2001). This cell death is apoptotic because it is sensitive to caspase inhibition and is identified using TUNEL techniques (Cartwright et al. 1998; Dunty et al. 2006. Because ethanol lacks a specific receptor, the mechanism by which ethanol initiates apoptosis in this cell population is incompletely understood.
Using a chick embryo model in which neural crest development is well understood, we found that ethanol exposure at any time during gastrulation (stage 4, neural crest specification), neurulation (stage 6, neural crest induction) headfold formation (stage 8, neural crest expansion) and the onset of neural crest migration (stage 10, 10 somites) produces significant cell death within neural crest. Interestingly, regardless of when the ethanol is administered within that window, the apoptosis itself does not occur immediately and instead largely occurs at stage 12/13- (16–18 somites), a time that coincides with the endogenous cell death normally observed within a subset of hindbrain neural crest (Cartwright and Smith, 1995; Cartwright et al. 1998). This suggested that ethanol disrupted one or more events crucial to the early induction and expansion of these premigratory neural crest populations. To elucidate this mechanism we investigated the early cellular signaling events that are altered by acute ethanol exposure. Those studies revealed that clinically relevant ethanol concentrations cause the rapid mobilization of intracellular calcium (Cai+2) stores within the premigratory neural crest progenitors that reside in the dorsal neural folds (Debelak-Kragtorp et al. 2003). We found that this Cai+2 transient originates from ethanol’s activation of G protein-coupled signaling via Gαi2/3 and Gβγ (Garic-Stankovic et al. 2005, 2006), which in turn activates phospholipase Cβ and phosphoinositide-mediated release of Cai+2 stores. Chelators of intracellular calcium prevent both the ethanol-induced Cai+2 rise and the resulting cell death (Debelak-Kragtorp et al. 2003), thus normalizing neural crest survival. Why this ethanol-induced Cai+2 transient initiates neural crest apoptosis is unknown.
One potential target of ethanol’s Cai+2 transient is non-canonical Wnt/Ca signaling and its central effector β-catenin. β-catenin is a bifunctional protein that is crucial for neural crest development. In one role, β-catenin interacts with E-cadherin and the actin cytoskeleton and contributes to cell adhesions (Lein et al. 2006). In its other function, β-catenin is a transcriptional effector within the canonical/non-canonical Wnt signaling pathway (reviewed in MacDonald et al. 2009). Wnt/β-Catenin/TCF signals have multiple, essential roles in neural crest development (reviewed in Yanfeng et al. 2003). In the early embryo, Wnt/β-catenin contributes to cranial neural crest induction and expansion, and Wnt/β-catenin loss-of-function produces widespread neural crest apoptosis and craniofacial deficiencies (Brault et al. 2001; Ikeya et al. 1997). β-catenin modulates neural crest migration (De Melker et al. 2004), and, later in development, promotes neural crest differentiation into melanocyte and sensory neuron fates (Dorsky et al. 1998; Lee et al. 2004). Many of these actions are attributed to β-catenin’s transcriptional activity.
Two intersecting pathways act at the posttranslational level to regulate β-catenin’s stability and thus its transcriptional activity. In the canonical Wnt pathway, secreted Wnt glycoproteins interact with their cell surface Frizzled receptors to inhibit the protein kinase GSK3β (MacDonald et al. 2009). In the presence of Wnt/Frizzled signals, β-catenin interacts with T cell transcription factor/lymphoid enhancer factor (TCF/LEF) proteins to initiate transcription. When Wnt signal is absent, GSK3β is no longer silenced and it phosphorylates β-catenin and targets the protein for ubiquitination and proteosomal destruction. In the noncanonical Wnt pathway, Cai+2 signals originating from G protein activity similarly initiate the phosphorylation and destabilization of β-catenin, albeit in a GSK3β-independent manner, and also deplete nuclear β-catenin and terminate its transcriptional activity (Kohn and Moon, 2005). Because β-catenin is essential for neural crest survival and can be destabilized by intracellular calcium, we hypothesized that ethanol and its Cai+2 transient might dysregulate β-catenin and its transcriptional activity, and these changes might in turn be responsible for the neural crest losses. Here we identify the dysregulation of transcriptionally active β-catenin as a key target of ethanol’s developmental pathogenicity. Ethanol’s suppression of β-catenin/TCF signaling mediates the neural crest losses in this avian model of FAS.
Methods
Embryos and Ethanol Treatment
Fertile white leghorn chick eggs (W98, Hyline, Spencer, IA; SexLink, Sunnyside Farms, Beaver Dam, WI) were incubated to the desired developmental stage (Hamburger and Hamilton, 1951); all studies compared embryos that were precisely matched in somite number and developmental stage. For in ovo studies, saline or 0.43 mmol ethanol in isotonic saline was injected into the egg yolk center at stage 8/9; this produces a peak embryonic ethanol concentration of 50–60 mM for 1–1.5 hr (Debelak and Smith, 2000). Ex vivo studies incubated stage 8/9 embryos 2hr in Tyrode’s buffer ± 52 mM ethanol; this ethanol concentration causes half-maximal calcium release in these embryos (Garic-Stankovic et al. 2006). Some embryos were pretreated with Bapta-AM (1 mM, 15 min) or ionomycin (50 μM, 5 min) prior to ethanol challenge. Although developmental stages from gastrulation (neural crest induction) through cranial neural tube formation (early neural crest migration) are equally sensitive to ethanol-induced apoptosis (Cartwright and Smith, 1995; Cartwright et al. 1998), studies herein use ethanol treatment at stage 8/9 (neural crest expansion) for uniformity.
Calcium Imaging
Ratiometric visualization and quantitation of intracellular calcium release using Fura2 was performed exactly as described previously (Garic-Stankovic et al. 2009).
Cell Death Analysis
Cell death was visualized at HH12/13- using LysoTracker Red (0.5 μM, Invitrogen), which we and others have shown detects apoptotic death in the early embryo FAS model (Cartwright et al. 1998; Dunty et al. 2006). Neural crest cells were visualized using immunostain for the neural crest markers Slug (#27568, 1/1000, AbCam, Cambridge MA) or Sox9 (1/1000, Abcam) followed by Alexa488-conjugated secondary antibody. Experiments evaluated 15–20 embryos per treatment and were performed in at least triplicate. To quantify neural crest death, we counted the total number of slug+ (Sox9+) and LTR+slug+ (Sox9+) cells residing in the dorsal neural roof and adjacent lateral quadrant, in transverse sections through r4 and r6. We enumerated 6–10 sections per embryo and 3–4 embryos per treatment, counting >100 slug+ cells per embryo and normalized to the number of sections counted.
β-Catenin Immunostain
HH8 (3–4 somites) embryos were challenged 2hr with saline or ethanol and then were fixed in Dent’s solution and immunostained for β-catenin with antibody #15B8 (1/1000, Novus Biologicals, Littleton CO) and Alexa 488-conjugated secondary antibody. DAPI counterstain visualized nuclei.
Western Analysis
Protein isolated from somite-matched HH8/9 embryos was separated on a 7.5% SDS acrylamide gel, analyzing one dissected head per lane. Proteins were transferred using semi-dry electrophoresis to a PVDF membrane (Immobilon-P, Millipore, Billerica MA). Primary antibodies were directed against β-catenin (15B8, 1:50,000), phospho-β-catenin (#9561, 1/1000, Cell Signaling, Danvers MA) and GAPDH (#G8795, 1:50,000, Sigma). Secondary antibodies conjugated to horseradish peroxidase (1:50,000, Southern Biotech, Birmingham AL) were detected using the SuperSignal West Pico chemillumination kit (Pierce, Rockford IL). Membranes were exposed to x-ray film and scanned densitometrically (OptiQuant Acquisition and Analysis v4.0, Packard Instruments, Palo Alto CA). Experiments analyzed three embryos per treatment and were performed in at least triplicate.
Real-Time PCR
RNA was isolated from a pool of 5–6 stage-matched crania, dissected at the boundary between somite 2 and somite 3, using the Ambion Melt Total Nucleic Acid Isolation kit. Primer sequences are presented in Supplemental Table 1. Real-time PCR was performed as described (Flentke et al. 2004) except cDNA was synthesized using random hexamers and ImProm-II reverse transcriptase (Promega, Madison WI) according to the manufacturer. Amplification was performed using an ABI Prism 7000 (Applied Biosystems, Foster City CA) and the ABI SYBR Green PCR Master Mix. Expression was normalized to GAPDH content. Mean relative expression change was calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001). Samples were run in triplicate and each gene was analyzed in 3–5 separate experiments.
Electroporation and Cell Death Assays
cDNA encoding eGFP and the parent expression vector pCAGGS (Niwa et al. 2001) were kind gifts of T. Suzuki. The β-catenin construct was obtained from Addgene (#13435, Cambridge MA; Lee et al. 2001). The ΔTCF construct (Addgene #14019; Kardon et al. 2003) lacks the β-catenin binding domain (amino acids 2–50) and thus has dominant-negative activity. Constructs were used at a 3:1 ratio (1.2:0.4 μg/μl) of experimental:eGFP cDNA. DNA (2 μl in Fast Green) was injected into the lumen of the HH9 posterior hindbrain, and embryos were immediately electroporated (20V/100 msec on, 999.9 msec off, 5 pulses; CUY-21, Protech International) using 1 mm platinum electrodes with a fixed 4 mm gap. Eggs were injected 3hr later with saline or 0.43 mmol ethanol.
At HH12/13-, 20 hr later, cell death and neural crest numbers were visualized as described above using LysoTracker Red and slug immunostain. The electroporation efficiency was visualized using eGFP fluorescence; embryos lacking eGFP in r4/5/6 were discarded from analysis. An experiment evaluated 15–20 embryos per treatment and was performed in at least triplicate. We counted the total number of slug+ (Sox9+) and LTR+slug+ (Sox9+) cells residing in the dorsal neural roof and adjacent lateral quadrant, in transverse sections through r4 and r6. We enumerated 6–10 sections per embryo and 3–4 embryos per treatment, counting > 100 slug+ cells per embryo and normalized to the number of sections counted.
β-Catenin/TCF Luciferase Reporter Assay
To measure endogenous transcriptional activity of the β-catenin/TCF complex, embryos were electroporated as above with the β-catenin/TCF reporter construct pBARL or pfuBARL (5 μg/μl; kind gift of R. Moon) + CMV-Renilla vector (0.06 μg/μl; Promega) in dilute Methyl Green, because redox-active dyes inhibit the catalytic FAD in luciferase (Kurfurst et al. 1982). pBARL contains twelve functional β-catenin/TCF response elements upstream of luciferase, and pfuBARL contains mutated β-catenin/TCF sites that eliminate transcriptional activity (Biechele and Moon 2008). For the forced expression studies, β-catenin or ΔTCF were co-electroporated with pBARL and CMV-Renilla at 5:2:0.06 μg/μl. Eggs were injected 3hr later ± 0.43 mmol ethanol and the dissected crania were isolated 21 hr thereafter. Enzyme activity was quantified using a modification of the Dual Luciferase Reporter Assay System (Promega). Individual crania were ultrasonic homogenized (Branson) on ice using the Passive Lysis Buffer (Promega) and spin-clarified. The assay was otherwise run per manufacturer’s suggestions. Signal was quantified using an EG&G Berthold Lumat LB9507 luminometer. Luciferase values were normalized to Renilla activity. Extracts were assayed in duplicate with 6–8 embryos per treatment; experiments were run in at least duplicate. Purified luciferase and Renilla enzymes were from Promega.
Statistics
Normally distributed data were subjected either to unpaired t-test or to one-way ANOVA and the appropriate post-hoc analysis as indicated, using SigmaStat v.2.0 (Systat Software, Point Richmond, CA). Data not normally distributed were analyzed using Kruskal-Wallis one-way ANOVA on ranks and the nonparametric version of Dunn’s post hoc analysis, again using SigmaStat. P<0.05 was the criterion for significance. Results are presented as mean ± SEM unless otherwise indicated.
Results
Transient Ethanol Exposure Causes Neural Crest Cell Death
As we showed previously (Debelak-Kragtorp et al. 2003; Garic-Stankovic et al. 2005) transient exposure of early somite-stage embryos (2–5 somites) to 50–60 mM ethanol caused a significant rise in Cai+2 levels within the neural folds and including the neural crest progenitors (compare signal at asterisks, Fig. 1A–C). Eighteen hr thereafter this Cai+2 transient caused significant cell death within neuronal populations within the midbrain and hindbrain rhombomeres (compare signal at arrows, Fig. 1D, E). The ethanol-induced cell death coincided temporally and spatially with the well characterized programmed apoptosis within the subset of neural crest progenitors that reside within these regions (Fig. 1D; Ellies et al. 2002). We found that many of the affected cells expressed the transcriptional repressor slug, which demarcates premigratory and early migratory neural crest at this developmental stage (compare green and red signal at arrows, Fig. 1F, G). Their dorsal hindbrain position further endorsed their neural crest identity (LeDouarin and Kalcheim 1999). Few dead cells were observed in equivalent regions of control hindbrain (Fig. 1F).
Figure 1.

Ethanol exposure causes neural crest cell death. (A) Diagram of 3-somite embryo showing boxed headfold region imaged for intracellular calcium. (B, C) Fura-2 imaging of intracellular calcium in a 3-somite embryo following challenge with saline (B) and 52 mM ethanol (C). Asterisk indicates dorsal neural fold enriched in neural crest progenitors. (D, E) Cell death is visualized in 17-somite embryos using LysoTracker Red (LTR, white dots). (D) Saline (Sal) control reveals programmed cell death at the dorsal midline of the midbrain (mb) and hindbrain rhombomeres r1/2, r3 and r5 (arrows). (E) Ethanol treatment (52 mM) substantially increases the number of LTR+ cells within the midbrain and hindbrain (compare signal at arrows). (F, G) Transverse histological sections through rhombomere 4 of 17-somite embryos immunostained for Slug and cell death (LTR). Dorsal is at the top. Saline-treated r4 (F) contains a substantial slug+ neural crest population (green signal at arrow) with few apoptotic neural crest (red signal). In contrast, the ethanol-treated r4 (G) has many apoptotic neural crest cells, shown in yellow (green-red overlay, arrows). OV, otic vesicle.
Ethanol Selectively Reduces Nuclear β-Catenin
To identify the downstream targets of ethanol’s calcium transient that are responsible for this cell death, we examined the distribution of β-catenin, which is a known calcium target. Prior to neural tube closure, neural crest progenitors inhabit the dorsal aspect of the elevating neural folds (boxed region, Fig. 2A; LeDouarin and Kalcheim 1999). Cells within the dorsal neuroepithelium, including neural crest progenitors, were substantially enriched in β-catenin protein, as compared with the underlying ventral populations. Within the neural crest progenitors, β-catenin immunostaining was observed in the nucleus (arrows in enlargement, Fig. 2A); signal was also observed along the cell border and likely reflected its association with cadherin (Lein et al. 2006). This distribution pattern was consistent with previous reports for β-catenin expression within premigratory cranial neural crest (Brault et al. 2001; Maretto et al. 2003; Yanfeng et al. 2003). We found that brief (2hr) exposure to 52 mM ethanol substantially reduced β-catenin protein levels within the neural folds, an effect that was especially pronounced within dorsal populations including the neural crest (Fig. 2A, B). Within this population, high magnification revealed that the β-catenin losses were most apparent within the nuclear compartment and might also affect the cytoskeletal-affiliated levels (insert, Fig. 2B).
Figure 2.

Acute ethanol treatment rapidly depletes transcriptionally active β-catenin from neural crest. (A, B) Transverse sections through presumptive hindbrain of 3-somite embryo treated 2hr ± 52 mM ethanol, dorsal at top, and stained for β-catenin protein (green) and nuclei (blue). Control hindbrain (A) shows strong β-catenin expression within dorsal regions including dorsolateral populations enriched in neural crest progenitors (boxed region). In contrast, ethanol-treated hindbrain (B) has significant β-catenin loss in the nuclei of dorsal populations including premigratory neural crest (arrows). Insets show magnified views of boxed regions; arrows indicate nuclear signal. m, midline; nc, neural crest; nf, neural folds. Scale bar equals 50 μm. Exposure time was constant between images. (C) Western blot analysis confirms the dose-dependent loss of β-catenin content following 2hr ethanol exposure. β-catenin content is normalized to GAPDH. One-way ANOVA with Holm-Sidak post-hoc analysis, * p < 0.01, ** p ≤ 0.001 vs. 0 mM ethanol. Mean ± SEM for 3–5 independent experiments. (D) Representative western blot shows the decline in β-catenin content with increasing ethanol concentration. Because each lane contains the total cranial protein from a single embryo, there is loading variation between lanes that is normalized against GAPDH content. Upper arrow, intact β-catenin; lower arrow, proteolytic fragments of β-catenin. (E) Western blot showing the increased phospho-β-catenin content and lower molecular weight protein fragments following 2hr challenge with 52 mM ethanol.
Western blot analysis confirmed the histochemical findings and demonstrated that ethanol exposure significantly reduced β-catenin levels in a dose-dependent manner (Fig. 2C, D). At higher ethanol concentrations, we detected lower molecular weight signals that suggested the presence of proteolytic fragments, a finding consistent with the proteosomal degradation mechanism commonly used to regulate nuclear β-catenin content (MacDonald et al. 2009). This was confirmed by western analysis for phospho-β-catenin, which revealed the presence of multiple, lower molecular weight bands consistent with a phosphorylation and proteolysis mechanism for β-catenin depletion (Fig. 2E).
β-catenin exerts its cellular activity, in part, through its interaction with transcriptional effectors of the TCF/LEF family. To ascertain whether the ethanol-induced β-catenin loss affected its transcriptionally active form as part of the β-catenin/TCF/LEF complex (hereafter β-catenin/TCF), we electroporated the early hindbrain in vivo with the luciferase reporter construct pBARL, which has 12 TCF/LEF binding sites in its promoter region and thus is selectively induced by β-catenin/TCF (Biechele and Moon, 2008); the negative control pfuBARL contains nonfunctional mutant sites. At this developmental stage the dorsal neural progenitors including neural crest are the predominant source of nuclear β-catenin and β-catenin-mediated reporter activity within this tissue. Ethanol treatment significantly reduced pBARL but not pfuBARL reporter activity in a dose-dependent manner (Fig. 3A). Ethanol inhibited pBARL transcription at doses as low as 0.11 mmol/egg, indicating that its activity against this transcriptional pathway was quite potent. Ethanol did not inhibit purified luciferase or Renilla enzymes. In this in ovo exposure model, embryonic ethanol concentrations return to baseline (≤ 9 mM) by 3 hr following ethanol addition (Cartwright et al. 1998;Debelak and Smith 2000). Luciferase activity was quantified 21 hr after ethanol administration, indicating that ethanol’s reduction of pBARL transcriptional activity was long lasting.
Figure 3.

Ethanol significantly reduced β-catenin/TCF transcriptional activity. (A) β-Catenin/Tcf transcriptional activity measured using the pBARL luciferase reporter. Left side: Significant decreases were found at all ethanol concentrations tested. Middle: No luciferase activity was obtained using the pfuBARL reporter that contains mutant Tcf binding sequences. Right side: Recombinant luciferase and Renilla luciferase were not inhibited by 52 mM ethanol. Kruskal-Wallis one-way ANOVA on ranks with Dunn’s post hoc analysis, * p<0.05 vs. 0 mmol ethanol. Mean ± SEM of 4–6 experiments with 6–8 dissected crania/treatment analyzed individually. (B) Treatment with 52 mM ethanol does not alter short-term expression of canonical Wnt effectors in neural crest, as quantified using real-time PCR. Within-treatment values were normalized against GAPDH expression. Transcript levels in ethanol-treated neural crest were then normalized against the saline-treated levels. Shown is mean ± SD of 3–5 experiments using 25–30 pooled HH8/9 crania assayed in triplicate.
Although nuclear β-catenin is typically regulated at the level of protein phosphorylation and turnover, one explanation for its rapid loss could be that ethanol dysregulated one or more of the Wnts and accessory proteins that act upstream of β-catenin to govern its nuclear stability. Neural crest expresses several of these during this developmental period, including the positive effectors Wnt1, Wnt5a, Wnt6, Wnt9a, and Wnt11, and the negative effectors APC, GSK3β and sFRP2. We found that ethanol treatment did not alter the short-term expression of these Wnt effectors within the 2hr time period of nuclear β-catenin loss (Fig. 3B). Ethanol treatment also did not affect the transcript levels encoding β-catenin itself (92 ± 7% of control levels). Thus the nuclear β-catenin reductions caused by ethanol exposure were not attributed to transcriptional changes in either β-catenin or the upstream Wnt signals that stabilize the nuclear protein.
Ethanol Reduces β-Catenin’s Transcriptional Activity
The loss of β-catenin suggested that transcription of β-catenin/TCF target genes would be impaired following ethanol challenge. We evaluated the expression of neural crest genes that are known transcriptional targets of β-catenin/TCF, including FoxD3, Slug, and Wnt6 (Garcia-Castro et al. 2002; Kos et al. 2001; Taneyhill and Bronner-Fraser 2005; Tribulo et al. 2004; Yanfeng et al. 2003). Six hr after 52 mM ethanol challenge, the cranial neural crest had significantly reduced mRNA levels of three β-catenin targets, FoxD3, Slug, and Wnt6 (Fig. 4A). Ethanol treatment did not affect the expression of Bmp4, Tgfβ2 and Snail2, neural crest genes whose transcription is β-catenin-independent. Thus the ethanol-induced loss of β-catenin was functionally relevant and was accompanied by significant and selective reductions in β-catenin/TCF-mediated transcriptional activity.
Figure 4.

Ethanol selectively represses transcriptional targets of β-catenin/TCF signaling in neural crest. (A) Transcript levels in HH9 neural crest 6hr after ethanol (52 mM) challenge, normalized to saline controls (100%). Ethanol selectively reduced the expression of β-catenin/TCF-dependent neural crest genes Wnt6, FoxD3 and Slug, but not the neural crest genes Tgfβ3, Bmp4 or Snail. Mean ± SD of 5–6 pooled crania assayed in triplicate. * p <0.05 using unpaired t-test. (B) Time course analysis revealed that ethanol’s reduction of FoxD3 (⋄) and Wnt6 (Δ) was developmentally persistent and the reduction in slug (○) was transient. Expression of the β-catenin-independent genes Snail (▲), Tgfβ2 (■) and Bmp4 (◆) were largely unaffected until the onset of neural crest apoptosis (stage 12/13). Shown is mean expression with SEMs omitted for clarity; these averaged 10–15% of mean values. Values were normalized to GAPDH and expressed relative to saline controls (100%).
Given the persistent reduction of β-catenin reporter activity following ethanol challenge, we investigated whether there was a similar, lasting reduction in endogenous β-catenin/TCF-mediated transcriptional activity within neural crest. β-catenin has distinct actions in pre-migratory versus post-migratory neural crest (De Melker et al. 2004; Yanfeng et al. 2003). Because neural crest sensitivity to ethanol is greatest during the pre-migratory period (Cartwright et al. 1998), we evaluated the consequences of ethanol challenge to gene expression during neural crest induction through the onset of their migration. We found that ethanol’s repression of β-catenin/TCF-mediated signaling was long lasting, with respect to the developmental time scale. Ethanol exposure rapidly and significantly reduced the expression of Wnt-dependent transcripts as compared with controls (Fig. 4B). Importantly, FoxD3 and Wnt6, which are key contributors to neural crest establishment (Garcia-Castro et al. 2002; Kos et al. 2001), were significantly repressed throughout the premigratory induction period even though ethanol levels had dropped to ≤ 9 mM (Debelak and Smith 2000). This repression was selective because expression of the β-catenin/TCF-independent neural crest transcripts, Snail, Tgfβ3, and Bmp4, was largely unaffected by ethanol treatment. Both sets of transcripts, β-catenin/TCF-dependent and -independent, were significantly reduced at HH12/13, when ethanol-induced cell death occurs. We conclude that ethanol caused a specific and selective repression of β-catenin/TCF-mediated transcriptional activity and adversely affected the expression of genes critical for neural crest development. Moreover, this repression persisted throughout premigratory neural crest morphogenesis.
β-Catenin Loss Initiates Neural Crest Apoptosis
Because one role of β-catenin/TCF activity in early neural crest is to support cell survival (Brault et al. 2001; Ikeya et al. 1997), we hypothesized that the loss of this activity was responsible for the ethanol-induced cell death. We tested this by quantifying neural crest survival following the forced expression in ovo of either β-catenin, which stabilizes β-catenin/TCF signaling (Lee et al. 2001), or dominant-negative TCF (ΔTCF), which represses β-catenin/TCF activity (Kardon et al. 2003). Expression of the parent eGFP reporter construct controlled for non-specific effects. We verified the constructs’ effects upon β-catenin/TCF-dependent transcription using co-transfection of the pBARL reporter.
In the presence of eGFP expression, ethanol again significantly suppressed TCF/LEF reporter activity as compared with saline-treated controls (p < 0.05; Fig. 5A). eGFP expression itself did not alter the effects of ethanol or saline upon neural crest. Levels of cell death were comparable with that in untransfected neural crest as determined visually (compare Fig. 6A, B and Suppl. Fig. 1A, D vs. Fig. 1F,G) and by quantifying the slug+ populations. Ethanol treatment significantly increased the number of apoptotic neural crest (Fig. 5B, p < 0.05; compare red signal in Fig. 6A, B) and reduced neural crest numbers (Fig. 5C, p = 0.025; compare green signal in Fig. 5A, B) in the eGFP controls.
Figure 5.

β-catenin/TCF misexpression alters ethanol-induced cell death and neural crest survival. (A) β-catenin/TCF transcriptional activity in neural crest expressing β-catenin/TCF constructs. Ethanol treatment significantly reduces β-catenin/TCF transcriptional activity and this is normalized by forced expression of β-catenin, whereas dominant-negative ΔTCF inhibits β-catenin/TCF activity in both treatment groups. Activities were normalized to eGFP-saline controls. Mean ± SEM of 3–4 experiments. *p<0.05 vs. saline-eGFP. (B) Quantitation of LTR+-slug+ neural crest following the indicated treatments. β-catenin significantly reduced the number of apoptotic neural crest in ethanol treated hindbrain, whereas ΔTCF increased neural crest death to levels caused by ethanol. Mean ± SEM of 3–4 embryos per group. *p<0.05 vs. Saline-eGFP; † p<0.05 vs. Ethanol-eGFP. (C) Quantitation of slug+ neural crest cells following the indicated treatments. Forced expression of β-catenin significantly increased the number of slug+ neural crest in both ethanol and saline-treated hindbrain, whereas ΔTCF lowered those numbers to that caused by ethanol. Mean ± SEM of 3–4 embryos per group. * p<0.025 vs. Saline-eGFP; † p=0.006 vs. Ethanol-eGFP. (A, B) were analyzed using Kruskal-Wallis one-way ANOVA with Dunn’s post hoc analysis. (C) was analyzed using one-way ANOVA and Student-Newman-Kuels post-hoc analysis
Figure 6.
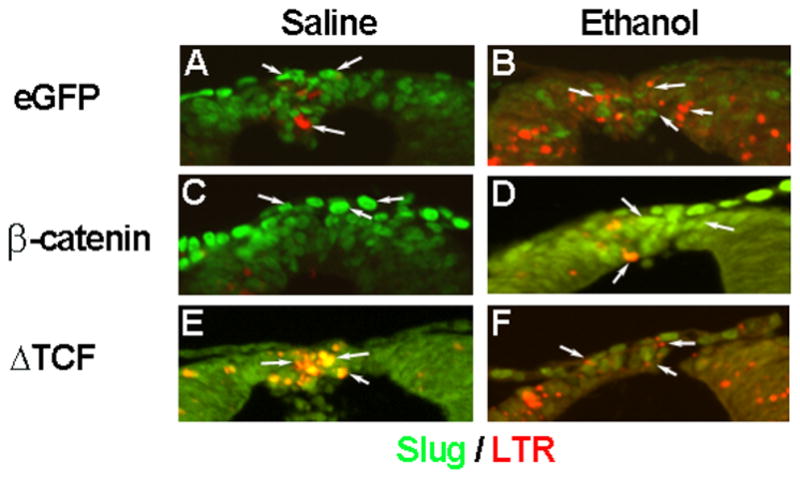
Forced expression of β-catenin and ΔTCF alters ethanol-induced cell death and neural crest numbers. Shown are transverse sections of the r4 dorsal roof, using representative embryos analyzed in Figure 4. Arrows highlight individual slug+ cells (green). (A, B) Ethanol (B) appreciably increased the number of apoptotic slug+ neural crest as compared with saline controls (A). (C, D) Forced expression of β-catenin increased the number of slug+ neural crest and decreased the number of apoptotic slug+ cells, following saline (C) and ethanol treatment (D). (E, F) In contrast, ΔTCF substantially decreased the number of slug+ cells and increased the number of apoptotic cells within r4 following both saline (E) and ethanol treatment (F).
As predicted, forced expression of β-catenin significantly enhanced pBARL reporter activity in saline-treated embryos (p < 0.05, Fig. 5A), showing that the construct was transcriptionally active. It also normalized the reporter activity following ethanol-treatment although not to the level observed in saline-treatment plus β-catenin. β-catenin over-expression also prevented ethanol-induced cell death in neural crest. Ethanol-treated hindbrain with forced β-catenin expression had significantly fewer apoptotic neural crest cells (Fig. 5B; p < 0.05) and significantly more slug+ neural crest (Fig. 5C; p = 0.006), as compared with eGFP controls (compare red and green signal in Fig. 6B vs. D; compare white signal in Suppl. Fig. 1D vs. E). Interestingly, β-catenin over-expression in the saline controls similarly reduced neural crest apoptosis (Fig. 5B, p=0.05) and elevated slug+ cell numbers (Fig. 5C, p=0.001), suggesting that β-catenin had pro-survival effects upon the neural crest subpopulations that were normally fated to undergo programmed cell death.
Conversely, if the β-catenin transcriptional loss was responsible for the ethanol-induced apoptosis, then targeted inactivation of β-catenin/TCF activity should adversely affect neural crest survival. The ΔTCF construct lacks the β-catenin binding site but still binds DNA via its C-terminal HMG box and thus is a dominant-negative repressor of β-catenin/TCF transcriptional activity (Kardon et al. 2003). Forced ΔTCF expression strongly suppressed pBARL reporter activity regardless of ethanol treatment status (Fig. 5A, p< 0.05). In both treatment groups ΔTCF substantially and significantly increased neural crest cell death (p < 0.005, Figs. 5B, 6E,F, Suppl. Fig. 1) and significantly reduced neural crest numbers to the levels caused by ethanol treatment (p< 0.05, Fig. 5C). In the ethanol-treated embryos the effects of ethanol and ΔTCF were not additive and did not significantly differ. This suggested that ethanol and ΔTCF operated through a shared mechanism to adversely affect neural crest survival.
Ethanol’s Cai+2 Transient Mediates the β-Catenin Loss
Intracellular calcium transients originating from GPCR activity have been shown in several model systems to destabilize β-catenin, increasing its proteolytic removal and terminating its transcriptional activity (Kohn and Moon 2005; Schneider et al. 2008). We tested whether the ethanol-induced Cai+2 transient was responsible for the β-catenin loss. Embryos were treated with the intracellular calcium chelator Bapta-AM prior to ethanol challenge. We showed previously that Bapta-AM prevents both ethanol’s Cai+2 transient and the neural crest losses (Debelak et al. 2001; Garic-Stankovic et al. 2005). Ethanol-exposed embryos treated with Bapta-AM had normal β-catenin expression within the dorsal neural folds including premigratory neural crest (Fig. 7C) as compared with ethanol-only (Fig. 7B). Bapta also altered neural fold morphology in a manner consistent with the requirement for calcium in neural fold elevation (Ferreira and Hilfer 1993). In contrast, cranial neural folds exposed to the calcium ionophore ionomycin were substantially depleted in β-catenin to levels comparable to ethanol (Fig. 7D), thus indicating that the β-catenin in these populations was sensitive to calcium-mediated destabilization. The Bapta-mediated restoration of β-catenin levels in ethanol-treated populations was accompanied by normalized β-catenin transcriptional activity, as determined using the pBARL reporter assay (Fig. 8A). Evaluation of Sox9+ neural crest populations revealed that Bapta-AM pretreatment also prevented the neural crest cell death (Fig. 8B) and rescued neural crest survival (Fig. 8C) in ethanol-treated embryos. Taken together these data indicate that ethanol’s Cai+2 transient mediated the loss of β-catenin and its cell survival effects in this cell population.
Figure 7.

Neural fold β-catenin content is altered by ethanol-induced Cai+2 signals. Embryos having 3 somites were challenged with saline, ethanol and/or the indicated calcium effector, and β-catenin protein (green, arrows) was visualized 2hr thereafter. Righthand panels show a 40× magnification of each right dorsal neural fold where premigratory neural crest resides. (A) Saline-treated embryo has robust β-catenin distribution in the dorsal neural folds (arrows). (B) 52 mM ethanol substantially reduces β-catenin content in the dorsal neural folds. (C) Embryo pretreatment with the Cai+2 chelator Bapta-AM prior to 52 mM ethanol challenge prevents β-catenin loss. (D) Treatment of otherwise normal embryos with the calcium ionophore Ionomycin is sufficient to deplete β-catenin from dorsal neural folds.
Figure 8.

Cai+2 effectors modulate β-catenin transcriptional activity and neural crest survival. (A) β-catenin/TCF transcriptional activity in neural crest treated with Bapta-AM ± 52 mM ethanol, measured using embryonic crania transfected with the pBARL luciferase reporter. Bapta pretreatment reverses the ethanol-mediated suppression of β-catenin/TCF transcriptional activity. Results are normalized to saline controls. Mean ± SEM of 3 experiments. (B) Quantitation of Sox9+ neural crest cells following the indicated treatments. Pretreatment with Bapta-AM normalized the number of Sox9+ neural crest in ethanol-treated hindbrain. Mean ± SEM of 3–4 embryos per group. (C) Quantitation of LTR+Sox9+ neural crest following the indicated treatments. Pretreatment with Bapta-AM significantly reduced the number of apoptotic neural crest in ethanol-treated hindbrain to normal levels. Mean ± SEM of 3–4 embryos per group. (A, B) were analyzed using Kruskal-Wallis one-way ANOVA with Dunn’s post hoc analysis. (C) was analyzed using one-way ANOVA and Student-Newman-Kuels post-hoc analysis. *p<0.005 vs. saline, + p<0.05 vs. ethanol, ++ p<0.01 vs. ethanol.
Discussion
Ethanol exposure during early embryogenesis causes significant cell death within the neural crest and these losses contribute to the multiple neurochristopathies that partly characterize FAS and FASD. The mechanism by which ethanol causes their apoptosis is uncertain. We showed previously that a Cai+2 transient originating from Gαi2/3 signaling initiates their apoptosis (Debelak-Kragtorp et al. 2003; Garic-Stankovic et al. 2005, 2006). Here we report that a major target of this Cai+2 transient is the transcriptional effector β-catenin and the loss of its activity mediates the neural crest losses. The ethanol-mediated destabilization of β-catenin loss occurred rapidly and at clinically relevant ethanol concentrations. It was accompanied by a persistent reduction in its transcriptional activity and the loss of downstream signals essential for neural crest development and survival including slug, FoxD3 and Wnt6. Forced expression of β-catenin in the presence of ethanol, as well as blockade of the Cai+2 transient, prevented the neural crest losses. Ethanol’s destabilization of β-catenin/TCF and its downstream products explains why the β-catenin-enriched premigratory neural crest is sensitive to ethanol during this developmental period. This is the first report that ethanol directly dysregulates β-catenin signal transduction and its transcriptional activity in any embryonic cell population.
The direct suppression of β-catenin/TCF transcriptional activity by ethanol is a novel mechanism of ethanol’s neurotoxicity and has not been previously considered as a neural target in acute ethanol exposure. Several studies report changes in Wnt/β-catenin in response to ethanol treatment but most of these investigated chronic ethanol exposure and thus may reflect compensatory cellular changes in response to ethanol challenge. The data are strongest for bone, in which chronic ethanol treatment substantially reduces Wnt signaling and the expression of Wnt effectors (Chen et al. 2010; Himes et al. 2008; Yeh et al. 2008). This may be secondary due to the commensurate loss of BMP activity, although acute ethanol can also rapidly deplete nuclear β-catenin from osteoblasts (Chen et al. 2010), a finding consistent with our own studies with neural crest progenitors. For neurons the findings are contradictory, with chronic ethanol exposure reducing the total β-catenin content of cultured hippocampal neurons (Singh et al. 2009) but elevating β-catenin in the frontal cortex of chronic alcoholics (Al-Housseini et al. 2008). These neuronal studies did not distinguish between the cytoskeletal and nuclear pools and thus the functional consequences of those changes are uncertain. Directly bearing on our work, Chen et al. (2010) recently reported that 50 mM ethanol caused the rapid depletion of ectopic nuclear β-catenin from in vitro osteoblasts and a commensurate reduction of β-catenin signaling, using a TopFlash reporter similar to ours. Ethanol’s ability to rapidly repress Wnt/β-catenin signaling in two distinct cell lineages suggests this action may represent a broader consequence of acute ethanol exposure than has been previously considered. Wnt signaling is highly conserved and is critical for numerous cellular processes including cell growth and proliferation, cell fate specification, differentiation, positional identity, and cellular adhesion. It is important not only in neuronal lineages but for a wide range of embryonic, fetal, and adult tissues. Our data suggest that ethanol’s suppression of canonical Wnt signaling contributes to the dysmorphic effects of ethanol, and may also contribute to ethanol’s effects upon fetal development and through adulthood. Thus β-catenin/TCF signals are thus a novel, direct target of ethanol exposure.
These data also show that this loss of β-Catenin/TCF signaling underlies ethanol’s toxicity and, specifically, it caused the neural crest losses in this FAS model. The forced expression of β-catenin rescued the ethanol-treated embryos and prevented the neural crest apoptosis. Conversely, β-catenin loss-of-function via ΔTCF overexpression was minimally sufficient to cause the apoptosis. These findings are consistent with genetic models of β-catenin loss-of-function, which show widespread cranial neural crest apoptosis at E9–10.5 and craniofacial hypoplasia (Brault et al. 2001). The pattern and timing of neural crest death in the loss-of-function genetic model parallels that observed here and in murine FAS models (Cartwright et al. 1998; Dunty et al. 2006). The results are consistent with the proposed function of β-catenin to provide critical signals that enhance neural crest survival and prevent their apoptosis. A possible effector of this apoptosis is the transcriptional repressor slug (also known as snail2), which is a downstream target of β-catenin. Slug has been shown to inhibit neural crest apoptosis by controlling the expression of apoptosis signals such as caspases and XR11 (Bcl2; Tribulo et al 2004). We found that Slug expression was strongly suppressed in our embryos just 6 hr after ethanol exposure. This loss of slug might contribute to the neural crest apoptosis observed in this FAS model.
Our data show that ethanol also causes neural crest hypocellularity by suppressing signals that govern neural crest specification and expansion, events supported in part by β-catenin (Brault et al. 2001; Dorsky et al. 1998; Garcia-Castro et al. 2002; Ikeya et al. 1997; Yanfeng et al. 2003). β-catenin induces several key effectors of early neural crest development including FoxD3, Wnt6, and slug (Garcia-Castro et al. 2002; Kos et al. 2001; Taneyhill and Bronner-Fraser 2005; Tribulo et al. 2004). Ethanol treatment sharply reduced the expression of these effectors, and such losses would impair these early developmental events and reduce the number of neural crest progenitors available for craniofacial development. This hypothesis is further supported by our demonstration that forced β-catenin expression was sufficient to increase the number of cranial neural crest cells in both the ethanol and control groups. We propose that the combination of increased apoptosis and reduced neural crest expansion reduces the neural crest pool size and contributes to the neurochristopathies that are observed in FAS. These data indicate that β-catenin/TCF transcriptional activity is an important regulator of premigratory neural crest survival. β-Catenin also has important roles in later neural crest development, through its interactions with cadherin to promote neural crest migration (de Melker et al. 2004; De Calisto et al. 2005) and though its transcriptional stimulation of melanocyte and sensory neuron specification (Dorsky et al. 1998; Lee et al. 2004). Ethanol’s dysregulation of β-catenin may not be limited to the premigratory neural crest period and could potentially explain the impaired neural crest migration (Rovasio and Battiato, 1995, 2002) and sensory deficits associated with prenatal alcohol exposure. It will be important to determine if the effects upon β-catenin reported here are stage and cell type dependent or instead reflect a broader impact of ethanol upon β-catenin activity.
Regulation of β-catenin’s transcriptional activity is largely post-translational, through the phosphorylation of amino-terminal serines that signals the protein’s ubiquitination and proteolytic degradation (reviewed in MacDonald et al. 2009; Sugimura and Li 2010). Signaling via the canonical Wnt pathway prevents this degradation and thus promotes the activity of the β-catenin/Tcf transcriptional complex. However, we found that ethanol exposure did not appreciably alter the expression of many of the canonical Wnt effectors known to be present in neural crest, suggesting that a putative loss of canonical Wnt signaling was unlikely to account for the β-catenin transcriptional losses. Instead, we found that the Cai+2 transient evoked by ethanol was responsible for the β-catenin loss. Calcium is an established negative regulator of β-catenin stability and transcriptional activity as a component of the noncanonical Wnt/Ca pathway and its downstream effectors including CaMKII, protein kinase C, calpain, and calcineurin-NFAT (reviewed in Kohn and Moon, 2005; Sugimura and Li 2010). These first three proteins act by enhancing β-catenin phosphorylation, ubiquitination and/or proteolytic destruction, whereas NFAT appears to affect β-catenin levels at the transcription level. We found that ethanol significantly increased the neural crest content of phospho-β-catenin and its proteolytic fragments, suggesting that ethanol/Cai+2 acted through one or more of these calcium-activated kinases or proteases. Insight into this question may come from our recent demonstration that a major downstream target of the calcium transient in these cells is the activation of CaMKII; suppression of CaMKII prevents the ethanol-induced cell death, whereas its constitutive activation promotes neural crest losses (Garic et al., in press). CaMKII is a known effector of the Wnt/Ca pathway through its activation by GPCR signaling (Kohn and Moon 2005). This suggests that ethanol may have reduced β-catenin activity through its stimulation of GPCR-mediated Wnt/Ca signaling, or by interacting with a GPCR that mimics or converges upon Wnt/Ca pathways. Ethanol frequently alters cellular activity through its ability to bind hydrophillic proteins, many of which are GPCRs (Mihic et al. 1997). Studies are underway to investigate whether CaMKII activation contributes to the β-catenin destabilization documented here.
Two other potential modulators of canonical Wnt signaling merit consideration. GSK3β is a potent and negative effector of β-catenin stability through its ability to phosphorylate β-catenin and target it for ubiquitination and proteolysis (MacDonald et al. 2009). Importantly, GSK3β activity can be significantly enhanced by acute ethanol exposure and its activation contributes to ethanol’s neurotoxicity, as shown for Drosophila sensory neurons and mouse cerebral cortex (French and Heberlein 2009; Liu et al. 2009). Although GSK3β activation is thought to be calcium independent, we cannot rule out its possible contribution in this model. Interestingly, caspase proteases have also been shown to cleave β-catenin and inhibit canonical Wnt signaling (Abdul-Ghani et al. 2011; Steinhausen et al. 2000; Van de Craen et al. 1999). The cell death studied here is caspase-dependent (Cartwright et al. 1998); although the precise timing of caspase activation in this model is not known, such an action could also account for the β-catenin losses. Studies are underway to establish which of these pathways mediates the β-catenin losses caused by acute ethanol exposure and its Cai+2 transient in this neural crest model
In summary we show that the Cai+2 transient triggered by clinically relevant ethanol concentrations suppress β-catenin and its transcriptional activity within the developing neural crest. This repression mediates the subsequent cranial neural crest losses observed in this FAS model. β-catenin/Wnt signals also govern additional developmental events known to be disrupted by gestational ethanol exposure including cardiac and limb morphogenesis, and neurodevelopment events such as neurogenesis, dendrite formation and synaptogenesis (Gao et al. 2007; Mao et al. 2009; Miller et al. 2006; Serrano et al. 2010; Zechner et al. 2003). We suggest that ethanol’s dysregulation of β-catenin signaling could underlie the seemingly diverse pathological outcomes in FAS.
Supplementary Material
Ethanol-induced apoptosis and neural crest numbers are altered by β-catenin and TCF transfection in hindbrain. Shown are dorsal views of the intact HH12+ (17s) hindbrain, transfected as indicated and challenged 3hr thereafter with saline or 52 mM ethanol. Panels A–F depict apoptotic cells visualized with LysoTracker Red (LTR, white dots). Panels A′-F′ show green fluorescent signals overlaid on bright-field images of the same embryos, showing expression of the eGFP internal control in the right lateral hindbrain and migrating neural crest. Lines indicate the neural crest migrating from rhombomeres 2 (r2), r4 and r6. (A–C) Saline-treated embryos transfected with (A) eGFP, (B) β-catenin + eGFP, or (C) ΔTCF + eGFP. (D–F) Embryos challenged with 52 mM ethanol and transfected with (D) eGFP, (E) β-catenin + eGFP, or (F) ΔTCF + eGFP. Ethanol treatment significantly enhanced the apoptosis level in eGFP-treated hindbrain (compare abundance of white dots in A vs. D). In contrast, β-catenin overexpression strongly reduced the apoptosis levels in both saline and ethanol-treated hindbrain (compare abundance of white dots, B vs. A, E vs. D). ΔTCF overexpression elevated apoptosis levels in both saline-treated and ethanol-treated hindbrain (compare abundance of white dots, C vs. A, F vs. D). In embryos transfected with ΔTCF + eGFP, although eGFP expression extended along the hindbrain’s rostrocaudal length, the eGFP signal per unit area was consistently lower than for embryos receiving eGFP-only and β-catenin + eGFP. This may reflect the increased apoptosis of the ΔTCF-transfected cells. Abbreviations: ov, otic vesicle; r, rhombomere.
Figure 9.
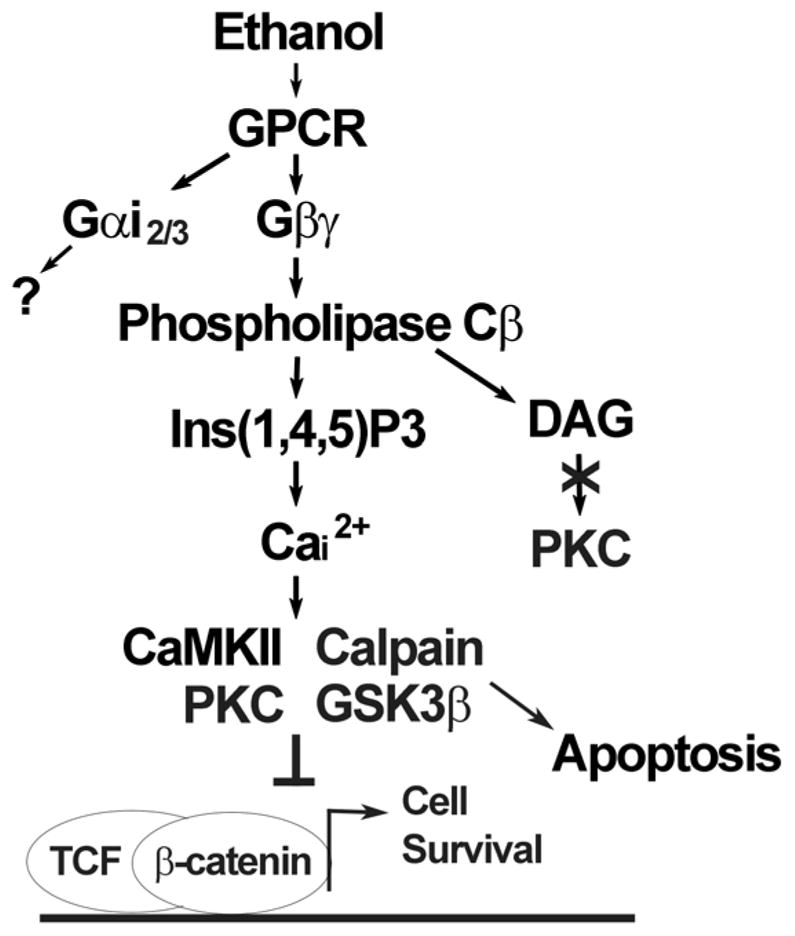
Ethanol signaling pathway that initiates neural crest apoptosis. Shown is a summary of published studies (Debelak-Kragtorp et al. 2003; Garic-Stankovic et al. 2005, 2006) and results herein. Ethanol interacts with a G-protein-coupled receptor (GPCR) of unknown identity to activate Gαi2/3 and Gβγ. Within seconds, the latter stimulates Phospholipase Cβ-mediated synthesis of inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) and calcium release predominantly from intracellular stores. The mobilized calcium interacts with one of several possible effectors, including CaMKII, Protein Kinase C or calpain to destabilize β-catenin and terminate its transcriptional stimulation of the TCF/LEF complex. Alternately, ethanol may destabilize β-catenin through its potential stimulation of GSK3b. The loss of β-catenin’s transcriptional activity promotes apoptosis whereas its maintenance promotes the survival of neural crest and neuronal precursors. “?” indicates that the contribution of this signal to ethanol-mediated cell death is unknown at this time.
Acknowledgments
Supported by NIH Merit Award R37 AA11085 and R21 AA17287 to S.M.S.
References
- Al-Housseini AM, Sivanandam TM, Bradbury EL, Tannenberg RK, Dodd PR, Gu Q. Up-regulation of beta-catenin levels in superior frontal cortex of chronic alcoholics. Alcohol Clin Exp Res. 2008;32:1080–1090. doi: 10.1111/j.1530-0277.2008.00670.x. [DOI] [PubMed] [Google Scholar]
- Biechele TL, Moon RT. Assaying β-catenin/TCF transcription with β-catenin/TCF transcriptional-based reporter constructs. Methods Mol Biol. 2008;468:99–110. doi: 10.1007/978-1-59745-249-6_8. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the β-catenin gene by Wnt-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Tessmer LL, Smith SM. Ethanol-induced neural crest apoptosis is coincident with their endogenous death, but is mechanistically distinct. Alcohol Clin Exp Res. 1998;22:142–149. [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Badger TM, Ronis MJ. A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/beta-catenin signaling. J Bone Miner Res. 2010;25:1117–27. doi: 10.1002/jbmr.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarren SK, Randels SP, Sanderson M, Fineman RM. Screening for fetal alcohol syndrome in primary schools: a feasibility study. Teratology. 2001;63:3–10. doi: 10.1002/1096-9926(200101)63:1<3::AID-TERA1001>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Debelak KA, Smith SM. Avian genetic background modulates the neural crest apoptosis induced by ethanol exposure. Alcohol Clin Exp Res. 2000;24:307–314. [PubMed] [Google Scholar]
- Debelak-Kragtorp KA, Armant DR, Smith SM. Ethanol-induced cephalic apoptosis requires phospholipase C-dependent intracellular calcium signaling. Alcohol Clin Exp Res. 2003;27:515–523. doi: 10.1097/01.ALC.0000056615.34253.A8. [DOI] [PubMed] [Google Scholar]
- De Calisto J, Araya C, Marchant L, Riaz CF, Mayor R. Essential role of non-canonical Wnt signaling in neural crest migration. Develoment. 2005;132:2587–2597. doi: 10.1242/dev.01857. [DOI] [PubMed] [Google Scholar]
- De Melker AA, Desban N, Duband JL. Cellular localization and signaling activity of β-catenin in migrating neural crest cells. Dev Dyn. 2004;230:708–726. doi: 10.1002/dvdy.20091. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signaling pathway. Nature. 1998;396:370–373. doi: 10.1038/24620. [DOI] [PubMed] [Google Scholar]
- Dunty WC, Chen SY, Zucker RM, Dehart DB, Sulik KK. Neural crest and alcohol exposure. Alcohol Clin Exp Res. 2006;25:1523–1535. [PubMed] [Google Scholar]
- Ellies DL, Tucker AS, Lumsden A. Apoptosis of premigratory neural crest cells in rhombomeres 3 and 5: consequences for patterning of the branchial region. Dev Biol. 2002;251:118–128. doi: 10.1006/dbio.2002.0815. [DOI] [PubMed] [Google Scholar]
- Ferreira MC, Hilfer SR. Calcium regulation of neural fold formation: visualization of the actin cytoskeleton in living chick embryos. Dev Biol. 1993;159:427–440. doi: 10.1006/dbio.1993.1253. [DOI] [PubMed] [Google Scholar]
- Flentke GR, Baker MW, Docterman KE, Power S, Lough J, Smith SM. Dev Dyn. 2004;229:886–898. doi: 10.1002/dvdy.10489. [DOI] [PubMed] [Google Scholar]
- French RL, Heberlein U. Glycogen synthase kinase-3/Shaggy mediates ethanol-induced excitotoxic cell death of Drosophila olfactory neurons. Proc Natl Acad Sci USA. 2009;106:20924–20929. doi: 10.1073/pnas.0910813106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Arlotta P, Macklis JD, Chen J. Conditional knock-out of β-catenin in postnatal-born dentate gyrus granule neurons results in dendritic malformation. J Neurosci. 2007;27:14317–14325. doi: 10.1523/JNEUROSCI.3206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Castro MI, Marcelle C, Bronner-Fraser M. Ectodermal Wnt function as a neural crest inducer. Science. 2002;297:848–851. doi: 10.1126/science.1070824. [DOI] [PubMed] [Google Scholar]
- Garic A, Flentke GR, Amberger E, Hernandez M, Smith SM. CaMKII Activation Is a Novel Effector of Alcohol’s Neurotoxicity in Neural Crest Stem/Progenitor Cells. J Neurochem. doi: 10.1111/j.1471-4159.2011.07273.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garic-Stankovic A, Hernandez MA, Flentke GR, Debelak-Kragtorp KA, Armant DR, Smith SM. Ethanol triggers neural crest apoptosis thru the selective activation of a pertussis toxin-sensitive G-protein and a phospholipase Cβ-dependent Ca2+ transient. Alcohol Clin Exp Res. 2005;29:1237–1246. doi: 10.1097/01.alc.0000172460.05756.d9. [DOI] [PubMed] [Google Scholar]
- Garic-Stankovic A, Hernandez M, Flentke GR, Smith SM. Alcohol Clin Exp Res. 2006;30:552–559. doi: 10.1111/j.1530-0277.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Himes R, Wezeman FH, Callaci JJ. Identification of novel bone-specific molecular targets of binge alcohol and ibandronate by transcriptome analysis. Alcohol Clin Exp Res. 2008;32:1167–80. doi: 10.1111/j.1530-0277.2008.00736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeya M, Lee SMK, Johnson JE, McMahon AP, Takada S. Wnt signaling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–970. doi: 10.1038/40146. [DOI] [PubMed] [Google Scholar]
- Kardon G, Harfe BD, Tabin CJ. A Tcf-4-positive mesodermal population provides a prepattern for vertebrate limb muscle patterning. Dev Cell. 2003;5:937–944. doi: 10.1016/s1534-5807(03)00360-5. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Moon RT. Wnt and calcium signaling: β-catenin-independent pathways. Cell Calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Kos R, Reedy MV, Johnson RL, Erickson CA. The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development. 2001;128:1467–1479. doi: 10.1242/dev.128.8.1467. [DOI] [PubMed] [Google Scholar]
- Kurfurst M, Ghisla S, Presswood R, Hastings JW. Structure and catalytic inactivity of the bacterial luciferase neutral flavin radical. Eur J Biochem. 1982;123:355–361. doi: 10.1111/j.1432-1033.1982.tb19775.x. [DOI] [PubMed] [Google Scholar]
- Le Douarin LM, Kalcheim C. The Neural Crest. 2. Cambridge University Press; Cambridge: 1999. [Google Scholar]
- Lee HY, Kleber M, Hari L, Brault V, Suter U, Taketo MM, Kemler R, Sommer L. Instructive role of Wnt/β-catenin in sensory fate specification in neural crest stem cells. Science. 2004;303:1020–1023. doi: 10.1126/science.1091611. [DOI] [PubMed] [Google Scholar]
- Lee E, Salic A, Kirschner MW. Physiological regulation of β-catenin stability by Tcf3 and CK1ε. J Cell Biol. 2001;154:983–993. doi: 10.1083/jcb.200102074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Vasioukhin V. Cadherin-catenin proteins in vertebrate development. Curr Opin Cell Biol. 2006;18:499–506. doi: 10.1016/j.ceb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen G, Ma C, Bower KA, Xu M, Fan Z, Shi X, Ke ZJ, Luo J. Overexpression of glycogen synthase kinase 3β sensitizes neuronal cells to ethanol toxicity. J Neurosci Res. 2009;87:2793–2802. doi: 10.1002/jnr.22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-[Delta][Delta]CT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in Schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/β-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci USA. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP, Marais AS, Adnams CM, Hoyme HE, Jones KL, Robinson LK, Khaole NC, Snell C, Kalberg WO, Hendricks L, Brooke L, Stellavato C, Viljoen DL. The epidemiology of fetal alcohol syndrome and partial FAS in a South African community. Drug Alcohol Depend. 2007;88:259–271. doi: 10.1016/j.drugalcdep.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende M, Christophorou NAD, Streit A. Specific and effective gene knock-down in early chick embryos using morpholinos but not pRFPRNAi vectors. Mech Dev. 2008;125:947–962. doi: 10.1016/j.mod.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Grennblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Miller MW. Brain Development: Normal Processes and the Effects of Alcohol and Nicotine. Oxford University Press; New York: 2006. [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Rovasio RA, Battiato NL. Role of early migratory neural crest cells in developmental anomalies induced by ethanol. Int J Dev Biol. 1995;39:421–422. [PubMed] [Google Scholar]
- Rovasio RA, Battiato NL. Ethanol induces morphological and dynamic changes on in vivo and in vitro neural crest cells. Alcohol Clin Exp Res. 2002;26:1286–1298. doi: 10.1097/01.ALC.0000026102.73486.65. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Kume S, Amasaki Y, Mikoshiba K. The Wnt/calcium pathway activates NF-AT and promotes ventral cell fate in Xenopus embryos. Nature. 2002;417:295–299. doi: 10.1038/417295a. [DOI] [PubMed] [Google Scholar]
- Schneider I, Houston DW, Rebagliati MR, Slusarski DC. Calcium fluxes in dorsal forerunner cells antagonize b-catenin and alter left-right patterning. Development. 2008;135:75–84. doi: 10.1242/dev.004713. [DOI] [PubMed] [Google Scholar]
- Serrano M, Han M, Brinez P, Linask KK. Fetal alcohol syndrome: cardiac birth defects in mice and prevention with folate. Am J Obstet Gynecol. 2010;203:75.e7–75.e15. doi: 10.1016/j.ajog.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Singh AK, Gupta S, Jiang Y, Younus M, Ramzan M. In vitro neurogenesis from neural progenitor cells isolated from the hippocampus region of the brain of adult rats exposed to ethanol during early development through their alcohol-drinking mothers. Alc Alcohol. 2009;44:185–198. doi: 10.1093/alcalc/agn109. [DOI] [PubMed] [Google Scholar]
- Smith SM, Debelak-Kragtorp KA. Neural crest and alcohol exposure. In: Miller MW, editor. The Developing Brain: Lessons Learned from Alcohol and Nicotine Exposures. Oxford University Press; New York: 2006. pp. 279–294. [Google Scholar]
- Su B, Debelak KA, Tessmer LA, Cartwright MM, Smith SM. Genetic influences on craniofacial outcome in an avian model of prenatal alcohol exposure. Alcohol Clin Exp Res. 2001;25:60–69. [PubMed] [Google Scholar]
- Sugimura R, Li L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Res C. 2010;90:243–256. doi: 10.1002/bdrc.20195. [DOI] [PubMed] [Google Scholar]
- Taneyhill LA, Bronner-Fraser M. Dynamic alterations in gene expression after Wnt-mediated induction of avian neural crest. Mol Biol Cell. 2005;16:5283–5293. doi: 10.1091/mbc.E05-03-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribulo C, Aybar MJ, Sanchez SS, Mayor R. A balance between the anti-apoptotic activity of Slug and the apoptotic activity of msx1 is required for the proper development of neural crest. Dev Biol. 2004;275:325–342. doi: 10.1016/j.ydbio.2004.07.041. [DOI] [PubMed] [Google Scholar]
- Yanfeng W, Saint-Jeannet JP, Klein PS. Wnt-frizzled signaling in the induction and differentiation of the neural crest. BioEssays. 2003;25:317–325. doi: 10.1002/bies.10255. [DOI] [PubMed] [Google Scholar]
- Yeh CH, Chang JK, Wang YH, Ho ML, Wang GJ. Ethanol may suppress Wnt/beta-catenin signaling on human bone marrow stroma cells: a preliminary study. Clin Orthop Relat Res. 2008;466:1047–53. doi: 10.1007/s11999-008-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner D, Fujita Y, Hulsken J, Muller T, Walther I, Taketo MM, Crenshaw EB, Birchmeier W, Birchmeier C. β-catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. 2003;258:406–418. doi: 10.1016/s0012-1606(03)00123-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ethanol-induced apoptosis and neural crest numbers are altered by β-catenin and TCF transfection in hindbrain. Shown are dorsal views of the intact HH12+ (17s) hindbrain, transfected as indicated and challenged 3hr thereafter with saline or 52 mM ethanol. Panels A–F depict apoptotic cells visualized with LysoTracker Red (LTR, white dots). Panels A′-F′ show green fluorescent signals overlaid on bright-field images of the same embryos, showing expression of the eGFP internal control in the right lateral hindbrain and migrating neural crest. Lines indicate the neural crest migrating from rhombomeres 2 (r2), r4 and r6. (A–C) Saline-treated embryos transfected with (A) eGFP, (B) β-catenin + eGFP, or (C) ΔTCF + eGFP. (D–F) Embryos challenged with 52 mM ethanol and transfected with (D) eGFP, (E) β-catenin + eGFP, or (F) ΔTCF + eGFP. Ethanol treatment significantly enhanced the apoptosis level in eGFP-treated hindbrain (compare abundance of white dots in A vs. D). In contrast, β-catenin overexpression strongly reduced the apoptosis levels in both saline and ethanol-treated hindbrain (compare abundance of white dots, B vs. A, E vs. D). ΔTCF overexpression elevated apoptosis levels in both saline-treated and ethanol-treated hindbrain (compare abundance of white dots, C vs. A, F vs. D). In embryos transfected with ΔTCF + eGFP, although eGFP expression extended along the hindbrain’s rostrocaudal length, the eGFP signal per unit area was consistently lower than for embryos receiving eGFP-only and β-catenin + eGFP. This may reflect the increased apoptosis of the ΔTCF-transfected cells. Abbreviations: ov, otic vesicle; r, rhombomere.