

Abstract
Cardiovascular disease is the leading cause of death and debility in women in the USA, and cardiac arrhythmias are a major concern. Voltage-gated potassium (Kv) channels along with the binding partners; Kvβ subunits are major regulators of the action potential (AP) shape and duration (APD). The regulation of Kv channels by the Kvβ1 subunit is unknown in female hearts. In the present study, we hypothesized that the Kvβ1 subunit is an important regulator of female cardiac physiology. To test this hypothesis, we ablated (knocked out; KO) the KCNAB1 isoform 1 (Kvβ1.1) subunit in mice and evaluated cardiac function and electrical activity by using ECG, monophasic action potential recordings and echocardiography. Our results showed that the female Kvβ1.1 KO mice developed cardiac hypertrophy, and the hearts were structurally different, with enlargement and increased area. The electrical derangements caused by Kvβ1.1 KO in female mice included long QTc and QRS intervals along with increased APD (APD20–90% repolarization). The male Kvβ1.1 KO mice did not develop cardiac hypertrophy, but they showed long QTc and prolonged APD. Molecular analysis showed that several genes that support cardiac hypertrophy were significantly altered in Kvβ1.1 KO female hearts. In particular, myosin heavy chain αexpression was significantly elevated in Kvβ1.1 KO mouse heart. Using a small interfering RNA strategy, we identified that knockdown of Kvβ1 increases myosin heavy chain αexpression in H9C2 cells. Collectively, changes in molecular and cell signalling pathways clearly point towards a distinct electrical and structural remodelling consistent with cardiac hypertrophy in the Kvβ1.1 KO female mice.
Introduction
Cardiovascular disease is a leading cause of death for women in the USA. In 2009 in the USA, ~33,000 females died from high blood pressure and heart failure (Go et al. 2013). Sex differences have been reported in cardiovascular disease, in which females are at lower risk for atrial fibrillation, but they present with higher incidences of torsades de pointes and are therefore at greater risk for sudden death (Makkar et al. 1993; Benjamin et al. 1994; Feinberg et al. 1995). In addition, females demonstrate more severe ischaemic heart disease, including stress-induced cardiomyopathy, plaque erosion and microvascular dysfunction (Crea et al. 2015). Health disparities in females are highly prevalent, and the treatment modalities are based on male-driven parameters (Daniels & Maisel, 2015; Yahagi et al. 2015).
One prominent sex disparity is in heart-rate-corrected QT (QTc) interval and action potential (AP) duration (APD), indicating dramatic sex-specific differences in the cardiac repolarization phase. This phenomenon is well documented in women taking anti-arrhythmic drugs, who demonstrate a greater risk of drug-induced long QT syndrome (Makkar et al. 1993; Lehmann et al. 1996; Li et al. 2013; Tadros et al. 2014). The prolonged QTc and APD suggest that there is less repolarization reserve in females (Yang et al. 1997; Gowd & Thompson, 2012). Cardiac sex differences noted in humans have also been demonstrated in animal studies, which show an increase in QT duration and an increase in the incidence of early after-depolarization in female rabbits after anti-arrhythmic drug exposure (Ebert et al. 1998; Pham et al. 2001; Odening et al. 2012). The repolarization reserve in the heart is constituted by three main potassium currents (IKr, IKs and IK1), which work in synchrony to allow the return of membrane potentials to the resting state and therefore play a vital role in cardiovascular function and disease (Schmitt et al. 2014). Mice demonstrate similar repolarization reserves to humans, and these reserves are constituted by distinct potassium currents (IKslow1, IKslow2 and Iss); although slightly different in kinetics, the currents show similar contributions to action potentials (Nerbonne, 2014). Disruption of the repolarization reserve can lead to cardiovascular disease, including arrhythmic events (Olson et al. 2006; Chapalamadugu et al. 2015).
Given that voltage-gated potassium (Kv) channels are pivotal to the repolarization reserve, understanding how Kv channel accessory subunits can alter this reserve may provide novel mechanistic insights. The voltage-gated potassium channel subunit Kvβ1 belongs to the aldo-keto reductase superfamily, which is found abundantly in the heart. The Kvβ subunit demonstrates a unique ability to bind and regulate many Kv channels, including Kv4 and Kv1 (Aimond et al. 2005; Pongs & Schwarz, 2010; David et al. 2012; Fischer et al. 2014).
Previous research on Kvβ1 knockout (KO) mice demonstrated reduced K+ channel inactivation (Aimond et al. 2005) and after-hyperpolarization as well as increased neuronal excitability in the brain (Giese et al. 1998; Murphy et al. 2004). Genomic research has begun to shed light on the importance of Kvβ1 (human gene name KCNAB1) in cardiovascular health. In 2008, a patient who suffered sudden cardiac death demonstrated a deletion in KCNAB1 (Banerjee et al. 2008). In addition, recent genomic studies identified KCNAB1 as a gene of interest for genetic association with blood pressure (BP) and a causal variant in humans with hypertension (Chiu et al. 2014; McCarthy et al. 2014).
In this present work, we demonstrate that 12- to 14-week-old Kvβ1.1 KO mice are physiologically altered compared with wild-type (WT) control animals. This is the first report to delineate the physiological role of Kvβ1.1 in female murine hearts. In addition, we used physiological comparison between males and females to identify electrophysiological and vascular differences in order to understand the roles of Kvβ1.1.
Methods
Ethical approval of animals
The Kvβ1.1 KO (global; Giese et al. 1998) and C57BL6/NJ (WT) female and male mice were obtained from Jackson Laboratories (stock number 007747; Bar Harbor, ME, USA). Mice were used at 12–14 weeks of age (20 female mice with average weights of 19–21 g and 15 male mice with average weights of 26–28 g, of each strain). All animal protocols and use were approved by the Institutional Animal Care and Use Committee at the University of South Florida (Tampa, FL, USA), which is consistent with the practices approved by US National Institutes of Health guidelines. The investigators understand the ethical principles under which the journal operates and that this work complies with the journal’s animal ethics checklist. All the mice had continuous access to food and water, ad libitum. Mice were injected with heparin (360 USP; Sigma) and killed with Somnasol [Pentobarbital Sodium, 50 mg (kg body weight)−1, Henry Schein Animal Health, Dublin, OH, USA] by I.P. injection, and heart tissue was excised after thoracotomy, snap frozen in liquid N2 and stored at −80°C until further use.
Cell (H9C2) culture and small interfering RNA
The H9C2 cells were purchased from ATCC (Manassas, VA, USA), and were cultured in an incubator (Thermo Fisher Scientific, Waltham, MA, USA) in air enriched with 5% CO2 using standard Dulbecco’s modified Eagle’s medium (Invitrogen, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Invitrogen), penicillin and streptomycin (10 mg ml−1 of each) antibiotics (Invitrogen). For small interfering RNA (siRNA) transfection experiments, the cells were transfected with 50 nM of either scrambled inhibitor or Kvβ1.1 siRNA (catalogue no. EMU086161; Sigma Aldrich, St. Louis, MO, USA) at 70% confluence using siLentFectTM lipid reagent (BioRad Hercules, CA, USA). Cells were observed for signs of toxicity under a microscope every 24 h. No detectable cell loss or change in cell morphology was observed in either group. Total RNA was extracted 72 h post-siRNA treatment using an Exiqon RNA isolation kit as described in the Quantitative real-time PCR (qRT-PCR) section.
Histochemistry
The hearts of female Kvβ1.1 KO and WT mice were frozen after removal by thoracotomy and then sectioned (25 μm thick) using a cryostat (Microm HM505 E, Walldorf, Germany). Sections were stained with Haematoxylin and Eosin for histological examination (Panguluri et al. 2013b).
Echocardiography
Serial transaortic echocardiography (Visualsonic Vevo 770TM, 30 MHz linear signal transducer; Toronto, Ontario, Canada) was carried out under general anaesthesia with 2–3% isoflurane in oxygen. The mice were depilated as required for imaging and placed on a 37°C heated platform throughout the imaging procedure. Echo measurements were taken from at least three different cardiac cycles for each mouse. M-Mode imaging from the short axis of the left ventricle (LV), using the papillary muscles for reference, was used to obtain measurements of LV posterior and anterior wall (LVPW and LVAW) thicknesses as well as LV internal dimensions in diastole and systole (LVIDd and LVIDs). Fractional shortening (FS%) and ejection fraction (EF%) were calculated as previously described (Panguluri et al. 2013b). For measurment of systolic flow, pulse wave (PW) Doppler was used to image the ascending aortic arch as well as the pulmonary artery in long axis to obtain mean flow velocity and the velocity–time integral of both the aortic arch and the pulmonary artery.
Electrocardiography
Electrocardiographic recordings were obtained from mice under general anaesthesia with 4–5% isoflurane in oxygen using surface probes in lead II configuration. The ECGs were acquired for a total duration of 15 min, with 1 min recordings obtained at 5 min intervals. Heart rate (HR) was measured while ECG signals were obtained. Signal was acquired at a rate of 1kHz by using a PowerLab system operated with LabChart 7.2 software (AD Instruments, Colorado Springs, CO, USA), and data were analysed offline using the ECG module of LabChart 7.2 software, as reported elsewhere (Berul et al. 1996; Chapalamadugu et al. 2015). The intervals (in milliseconds) of R–R, P–R, QRS and J–T were measured. The QT interval was measured from the start of the Q peak to the point where the T wave returns to the isoelectric baseline (TP baseline), and QTc interval was obtained using the following formula: QTc = QT/(RR/100)½ (Mitchell et al. 1998; El Gebeily et al. 2015).
Blood pressure measurements
A non-invasive tail-cuff method was used to measure BP and HR in conscious mice. Mice were placed in plastic restrainers and placed on a water heater at 37°C for 10 min. A pressure transducer was placed on the tail of the mice. Mice were allowed to habituate to this procedure for 5 days before experiments were performed. Blood pressure and HR values were recorded using a Model CODA Standard, one-animal non-invasive blood pressure system (Kent Scientific, Torrington, CT, USA). Final measurements were averaged from 10 consecutive readings obtained from each mouse.
Monophasic action potentials
Monophasic action potentials were recorded from ex vivo KO and WT female mouse hearts. Mice were injected with 1 mg heparin (180 USP; Sigma) and killed with Somnasol [Pentobarbital Sodium, 50 mg (kg body weight)−1, Henry Schein Animal Health] by I.P. injection. Hearts were isolated through a bilateral thoracotomy and retrograde perfusion with Krebs–Hanseleit buffer (mM: 119 NaCl, 25 NaHCO3, 4 KCl, 1.2 KH2PO4, 1 MgCl2, 1.8 CaCl2, 10 D-glucose and 2 sodium pyruvate, pH 7.4) at a constant flow rate of ~2.2 ml min−1, maintained at 37°C (Chapalamadugu et al. 2015). Monophasic action potentials were recorded from the LV epicardial surface using contact electrode (Harvard Apparatus, Holliston, MA, USA). Hearts were stabilized for 15 min, and AP data were acquired using an eight-channel PowerLab system (AD Instruments).
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from the ventricular apex of Kvβ1.1 KO and WT female hearts using an Exiqon miRCURY RNA Isolation kit (Exiqon, Woburn, MA, USA) according to the manufacturer’s protocol. Complimentary DNA (cDNA) from total RNA was synthesized, and quantitative real-time PCR (qRT-PCR) analysis was performed to measure the mRNA expression of hypertrophic markers, including Myosin Heavy chain alpha (MHCα), Myosin Heavy Chain Beta (MHCβ), GATA-Binding Protein 6 (GATA6), Bone Morphogenetic Protein 10 (BMP10) and Phosphatidylinositol-4,5-Bisphosphate 3-Kinase (PI3K). All the cDNA synthesis and qRT-PCR procedures were performed as described previously (Panguluri et al. 2013b). The expression of mouse HPRT transcript was used as an endogenous reference. Data were expressed as the mean fold change (±SEM; n = 3).
Western blotting
Protein lysates for Western blotting were prepared from both KO and WT female mouse hearts. Left ventricles were homogenized, and procedures were performed as described previously (Panguluri et al. 2013a,b). The supernatant was collected and stored at −80°C. Equivalent amounts of protein were loaded and separated by 4–20% gradient SDS polyacrylamide gels (Bio-Rad Laboratories). Proteins were detected with a dilution of primary antibody; MHCβ (MAB1548) and MHCα (AB50967) at 1:200 and 1:1000, respectively. Target protein band densities were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and normalized with Ponceau S-stained total protein band densities.
Pull-down assay
To identify the interaction between Kvβ1.1 and MHCα in the heart, we conducted a pull-down assay using whole ventricular tissue lysate. Briefly, 5 μg of Ddf4-dependent protein kinase (DDK)-tagged Kvβ1.1 plasmid (Origene, Rockville, MD, USA) was transiently expressed (72 h) in Cos-7 cells grown to 90% confluence in a 10 cm plate. Total cellular protein was extracted from Kvβ1.1–DDK-expressing Cos-7 cells and mouse ventricles by homogenization using extraction buffer containing (mM): 50 Tris, pH 7.4, 150 NaCl, 5 EDTA and 1% Nonidet P40 (NP40; ThermoScientific, Waltham, MA, USA) supplemented with 10 mM DDT, 1:100 protease inhibitor (Sigma-Aldrich, St Louis, MO) and 1:100 protease inhibitor (Sigma). Tissue lysate was then centrifuged at 10,000g for 10 min at 4°C, and the supernatant was collected. Protein quantification was performed using a Pierce 660 assay (Thermo Fisher Scientific, Waltham, MA, USA). Two hundred micrograms of Kvβ1.1–DDK Cos-7 lysate was incubated with anti-DDK agarose beads (Origene) for 3 h at 4°C, and 500 μg of precleared ventricular tissue lysate was then added and incubated overnight at 4°C. Bound proteins were then eluted, and immunoblot analysis was conducted using MHCα antibody.
Pathway analysis
Differentially expressed genes from qRT-PCR data were selected for network analysis using Ingenuity Pathway Analysis software (Ingenuity Systems, Inc., Redwood City, CA, USA). Based on the existing literature, Ingenuity Pathway Analysis identified the networks from its library of canonical pathways that were most significant to the data set. The significance of the association between the data set and the pathway network was measured by a ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway.
Statistical analysis
Student’s paired t test was used to identify significant pairwise comparisons for all parameters between WT and KO mice. Statistical analyses were performed with Sigma Plot (version 11.0, Sigma-Aldrich, San Jose, CA, USA) and MS Excel. Data were expressed as means ± SEM, and P values ≤0.05 were considered statistically significant.
Results
Kvβ1.1 knockout females demonstrated altered cardiac structure and systolic function
The Kvβ1.1 KO female mouse hearts demonstrated a significant increase in heart weight compared with WT control hearts (Fig. 1A). The Kvβ1.1 KO male mouse hearts, however, demonstrated no significant heart weight difference when compared with WT male control hearts (Fig. 1B). Histological sections of female hearts showed an overall increase in the size of the KO hearts compared with WT control hearts (Fig. 1C and D).
Figure 1. Cardiac structural analysis by morphometry.

A, female heart weight (HW) normalized with tibia length (TL) from wild-type (WT) and Kvβ1.1 knockout (KO) hearts; bar graph shows means ± SEM (n = 10), and *P < 0.05. B, male HW normalized with TL from WT and KO hearts; bar graph shows means ± SEM (n = 15). C, cross-sectional image of the heart from WT and KO female mice. Heart sections were 25 μm thick and stained with Haematoxylin and Eosin; left ventricle (Lv) and right ventricle (Rv) are labelled. D, cross-sectional area of hearts measured with ImageJ software, and means ± SEM plotted using a bar graph (n = 4), and *P < 0.05.
Echocardiographic measurements confirmed the overall cardiac size increase, because measurements of LV mass showed a significant increase in KO females compared with WT control animals (122 ± 7 versus 103 ± 4 mg; Fig. 2C). M-Mode short-axis measurements demonstrated a significant increase in the LVID at both systole and diastole, in addition to the volume pumped at both systole and diastole (Fig. 2A and B). Furthermore, the stroke volume was significantly higher in KO females compared with WT control females (41 ± 1.8 versus 35 ± 1.7 μl; Fig. 2D). However, no differences were noted in LV wall thickness (including anterior and posterior) or functional indices, including ejection fraction and fractional shortening, between the female KO mice and WT control females (Tables 1 and 2).
Figure 2. Female cardiac measurements by echocardiography.

A, B-mode short-axis image (left panel) of the left ventricle (LV) with papillary muscles (P) visible, and M-mode image (right panel) of the interior of the LV with left ventricular internal dimension at systole and diastole (LVIDs and LVIDd) along with left ventricular anterior and posterior wall at diastole (LVAWd and LVPWd). B, LVIDs and LVIDd dimensions measured in WT and KO hearts. Bar graph shows means ± SEM (n = 13), and *P < 0.05. C, LV mass estimated using M-mode images; bar graph represents means ± SEM (n = 13), and *P < 0.05. D, stroke volume (SV) per beat obtained from M-mode images; bar graph shows means ± SEM (n = 13), and *P < 0.05.
Table 1.
Echocardiographic M-mode measurements
| Female Mice | LVAWs (mm) | LVAWd (mm) | LVPWs (mm) | LVPWd (mm) |
|---|---|---|---|---|
| Kvβ1.1−/− | 1.3 ± 0.05 | 0.88 ± 0.04 | 1.1 ± 0.03 | 0.8 ± 0.01 |
| Wild-type | 1.3 ± 0.05 | 0.87 ± 0.04 | 1.1 ± 0.06 | 0.8 ± 0.03 |
| P Value | n.s. | n.s. | n.s. | n.s. |
LVAWs, Left ventricular anterior wall systolic; LVAWd, Left ventricular anterior wall diastolic; LVPWs, Left ventricular posterior wall systolic; LVPWd, Left ventricular posterior wall diastolic.
Table 2.
Echocardiographic M-mode measurements
| Female Mice | V(s) (μl) | V(d) (μl) | EF% | FS% | CO (ml min−1) |
|---|---|---|---|---|---|
| Kvβ1.1−/− | 27.7 ± 1.6 | 69 ± 3.1 | 60 ± 1.2 | 31.6 ± 0.8 | 18 ± 0.9 |
| Wild-type | 21.4 ± 2.2 | 57 ± 2.8 | 63 ± 2 | 34 ± 2 | 15.8 ± 0.8 |
| P Value | 0.03 | 0.008 | n.s. | n.s. | 0.07 |
V(s), Volume systolic; V(d), Volume diastolic; EF%, Ejection fraction; FS%, Fractional shortening; CO, Cardiac output.
Arterial and blood pressure differences in Kvβ1.1 KO female mice
Blood pressure measurements in female KO mice demonstrated significant elevation at both systole and diastole compared with female WT control females (Fig. 3A and B). Male KO mice showed only a small and statistically non-significant elevation in blood pressure when compared with WT control males (Fig. 3D and E). Heart rates between KO and WT in both sexes indicated no significant difference (Fig. 3C and F).
Figure 3. Blood pressure measurements.

A–C, female systolic blood pressure (SBP; A), female diastolic blood pressure (DBP; B) and female heart rate (HR; C) measurements from WT and KO hearts; bar graphs show means ± SEM (n = 8 mice), and *P < 0.05. D–F, male SBP (D), male DBP (E) and male HR (F) measurements from WT and KO hearts; bar graphs show means ± SEM (n = 8 mice).
Pulse wave Doppler imaging was used to assess the ascending aorta and pulmonary artery to measure systolic flow parameters of the left and right side of the heart. Female KO mice demonstrated an increase in mean gradient pressure as well as velocity–time integral (Fig. 4A–C) in the ascending aorta when compared with WT mice, which is indicative of increased LV pressure in female KO mice. A small increase in velocity–time integral was also observed in the pulmonary artery, but this was not significantly different from WT control animals (Fig. 4D).
Figure 4. Female haemodynamic changes.

A, B-mode image of the ascending aorta (Ao Arch) and pulse wave (PW) Doppler image of the blood flow in the ascending aorta (top panel), and B-mode image of the pulmonary artery (Pul Artery) and PW Doppler image of the blood flow in the pulmonary artery (bottom panel). B–D, Aortic mean gradient pressure (in millimetres of mercury (B), aortic velocity–time integral (VTI) taken from PW Doppler imaging (C) and pulmonary VTI taken from PW Doppler imaging (D), in WT and KO mice; bar graphs show means ± SEM (n = 8), and *P < 0.05.
Kvβ1.1 knockout prolonged QTc interval in both male and female mice
As shown in Fig. 5A, the averaged traces showed sign-ificantly longer QTc interval (51 ± 1.8 ms) in KO female mice when compared with WT females (45 ± 2.1 ms; Fig. 5B). In addition, the QRS interval was significantly prolonged in KO females compared with WT females (Fig. 5C). Male Kvβ1.1 KO mice showed significantly longer QTc intervals (Fig. 5D), but the QRS interval did not change significantly (Fig. 5E). These data suggest that the repolarization phase in Kvβ1.1 KO mice is altered compared with WT control animals. Additional ECG measurements, including P wave duration, demonstrated slight significance in KO females (Tables 3 and 4).
Figure 5. Electrocardiographic recordings.

A, averaged trace of lead II ECG recording from WT (black) and KO (red) hearts showing QTc duration from female mice. B–E, female QTc interval (B) female QRS duration (C), male QTc interval (D) and male QRS duration (E), in WT and KO mice; bar graphs represent means ± SEM (n = 10), and *P < 0.05.
Table 3.
Electrocardiographic measurements (females)
| Female mice | RR Interval (ms) | PR Interval (ms) | P Duration (ms) | QT Interval (ms) | JT Interval (ms) |
|---|---|---|---|---|---|
| Kvβ1.1−/− | 148 ± 3 | 43 ± 1.6 | 12 ± 0.7 | 20 ± 0.7 | 10 ± 0.7 |
| Wild-type | 144 ± 3 | 40 ± 0.7 | 10 ± 0.5 | 17 ± 0.7 | 9.5 ± 0.8 |
| P Value | n.s. | n.s. | 0.04 | 0.02 | n.s. |
Table 4.
Electrocardiographic measurements (males)
| Male mice | RR Interval (ms) | PR Interval (ms) | P Duration (ms) | QT Interval (ms) | JT Interval (ms) |
|---|---|---|---|---|---|
| Kvβ1.1−/− | 144 ± 4 | 42 ± 0.5 | 9.8 ± 0.6 | 22 ± 0.6 | 14 ± 0.7 |
| Wild-type | 140 ± 2.3 | 53 ± 4.5 | 12 ± 0.8 | 20 ± 0.5 | 11 ± 0.5 |
| P Value | n.s. | 0.04 | n.s. | 0.007 | 0.002 |
Prolonged monophasic action potentials in Kvβ1.1 KO hearts
Monophasic action potential traces were recorded from the left ventricular epicardial surface of hearts using ex vivo perfusion in females (Fig. 6A) and males (Fig. 6C). Analysis of the traces from female KO hearts revealed a significantly prolonged APD from 20–90% repolarization levels; APD90 (57 ± 1.8 versus 49 ± 2.5 ms), when compared with WT (Fig. 6B). However, male KO hearts demonstrated significantly prolonged APDs from 50 to 90% repolarization levels only (Fig. 6D). Nevertheless, these data suggest that Kvβ1.1 KO hearts demonstrate prolonged APDs.
Figure 6. Ventricular repolarization changes.

A, averaged recordings of the monophasic action potential in WT (black) and KO (red) hearts from females. B, female action potential durations (in milliseconds) at 20, 50, 70 and 90% repolarization in WT and KO mice; bar graph shows means ± SEM (n = 6), and *P < 0.05. C, averaged recordings of monophasic action potential in WT (black) and KO (red) hearts (n = 3) from males. D, male action potential durations (in milliseconds) at 20, 50, 70 and 90% repolarization in WT and KO mice; bar graph shows means ± SEM (n = 9), and *P < 0.05. Monophasic action potential recordings were obtained during perfusion at 37°C.
Differential mRNA expression of key hypertrophic markers and Kv channels
Given that we observed significant differences in the structural, haemodynamic and electrical indices in KO female mouse hearts compared with WT, we examined the expression levels of various genes that have previously been linked to these phenotypes such as, MHCα(6), MHCβ(7), PI3K, GATA4, GATA6 and BMP10. Data from the present study revealed that mRNA levels of MHCα were significantly increased in KO female mouse hearts compared with WT control females, whereas no change in MHCβ transcripts was noted (Fig. 7A). The mRNA levels of GATA4, GATA6 and BMP10 were significantly increased, whereas PI3K decreased in KO female hearts compared with WT female hearts (Fig. 7A).
Figure 7. Female cardiac real-time PCR expression, protein and protein–protein interaction analysis.

A, expression of myosin heavy chain (MHC) isoforms α and β, PI3K, GATA4, GATA6 and BMP10 (known cardiac specific hypertrophy markers); bar graph shows means ± SEM (n = 3), and *P < 0.05. Genes were normalized with a housekeeping gene (HPRT). B, Western blot images of MHCα and MHCβ from WT and KO left ventricular homogenate. C, bar graph shows means ± SEM (n = 3), and *P < 0.05; bands were normalized with Ponceau S-stained full lanes. D, Western blot image of KO left ventricle (Lv) and Cos-7 cells transfected with Kvβ1.1–DDK plasmid (β1.1DDK; lane 1) and Cos-7 cells transfected with β1.1DDK alone (lane 2). Lane 1 was incubated overnight with DDK-coated agarose beads. Myosin heavy chain α (MHCα) primary antibody (1:200 dilution) was incubated overnight with blot, and a 225 kDa band was noted in lane 1, with a limited band seen in lane 2. E, PCR expression of MHCα, GATA4 and GATA6 72 h after Kvβ1.1 small interfering RNA treatment in H9C2 cells (rat cardiomyoblasts); bar graph shows means ± SEM (n = 3), and *P < 0.05. †P < 0.07.
Differential expression of myosin isoform proteins
Gene expression changes noted in MHCα (Fig. 7A) were confirmed by using Western blot, which showed a significant increase in MHCα expression within the LV in KO female mice compared with WT female control mice (Fig. 7B and C). Western blot analysis also revealed a small increase (not significant) in MHCβ expression in the LV KO heart compared with WT mice.
MHCα interacts with Kvβ1.1 subunit
To evaluate the protein–protein interaction, we performed a pull-down assay to identify the association between Kvβ1 and MHCα. As shown in Fig. 7D, lane 1 identifies MHCα as the protein that was pulled using the DDK–Kvβ1 affinity assay. However, no relevant protein was identified in lane 2 over a similar molecular weight range. Overall, these data demonstrate that Kvβ1 protein interacts with MHCα in the mouse heart.
Kvβ1.1 knockdown caused MHCα upregulation
We evaluated the co-regulation of Kvβ1 and MHCα by using siRNA knockdown in H9C2 cells (rat cardiomyoblasts). Inhibiting the expression of Kvβ1.1 in H9C2 cells clearly led to higher expression of MHCα in the cardiac cells. As shown in Fig. 7E, we identified the regulation of key genes, including GATA4, GATA6 and MHCα, altered in the Kvβ1.1 knockdown group compared with the scrambled siRNA. These data confirm that Kvβ1 knockdown modulates the expression of the genes that were altered in the Kvβ1.1 KO mouse model.
Expression of key Kv channels and Kvβ subunits
The mRNA expression of key Kv channels and Kvβ subunits was not significantly altered (Fig. 8A and B). Comparison of Kvβ1.1 levels between male and female WT mouse hearts showed significantly increased expression in females (Fig. 8C).
Figure 8. Real-time PCR expression analysis.

A, female expression of voltage-gated potassium channels involved in cardiac repolarization, including Kv1.4, Kv1.5, Kv2.1, Kv4.2, Kv4.3 and Kv10.2; bar graph shows means ± SEM (n = 3), and *P < 0.05. Genes were normalized with a housekeeping gene (18S). B, expression of voltage-gated potassium channel subunits Kvβ1.2, Kvβ2 and KCHIP2; bar graph shows means ± SEM (n = 3), and *P < 0.05. C, expression of voltage-gated potassium channel subunit Kvβ1.1 in wild-type male (WTM) versus wild-type female (WTF) mouse heart; bar graph shows means ± SEM (n = 3), and *P < 0.05. Genes were normalized with a housekeeping gene (HPRT).
Discussion
In the present study, we report the physiological role of Kvβ1.1 in the murine heart. Our morphometric and echocardiographic assessment clearly demonstrated that KO female mice had significantly enlarged hearts with altered cardiac function compared with their WT controls. Male KO mice, however, demonstrated no alteration in heart size in comparison, emphasizing a sex-specific difference in Kvβ1.1 KO female mice. The ECG and monophasic action potential analysis identified prolongation in QTc and APDs, demonstrating that the repolarization reserve was depleted in both male and female Kvβ1.1 KO mice. At the molecular level, we identified novel protein–protein interactions between Kvβ1 and MHCα and confirmed that MHCα expression can be specifically modulated by Kvβ1 knockdown.
Electrical remodelling in Kvβ1.1 KO mice
Prolonged repolarization indices, such as QTc and APD, in KO mice suggest that Kvβ1.1 is necessary for Kv channel activity and therefore vital to the repolarization reserve. It is well known that Kv channels, such as Kv1.x and Kv4.x, are major contributors to the repolarization reserve in the heart. Alterations in Kv channels in the heart can lead to arrhythmic events and altered cardiac metabolism (Chapalamadugu et al. 2015; Ohanyan et al. 2015). Several studies have demonstrated that Kvβ subunits bind to and modulate the activities of Kv1.x and Kv4.x channels (Pongs & Schwarz, 2010). Heterologous expression studies have shown that different splice isoforms of Kvβ1 can bind to and confer inactivation of both slowly and rapidly inactivating Kv1.x channels (Rettig et al. 1994; Tipparaju et al. 2007, 2012). It has been shown that Kvβ1.1 binds to Kv4.2 and Kv4.3 in mouse ventricles, and deletion of Kvβ1 leads to decreased Ito,f densities in male mice (Aimond et al. 2005). Consistent with this evidence, our present data show prolonged APD and QTc in the KO group, suggesting that Kvβ1.1 is an essential contributor to cardiac repolarization. Repolarization defects were noted in KO female hearts, which also showed significant hypertrophy, suggesting differential structural remodelling in females.
Vascular alterations in Kvβ1.1 KO mice
Power Doppler analysis indicates that increased vascular resistance may be the most likely cause of the observed hypertrophy in KO female mice, because both the mean aortic gradient pressure and the aortic velocity–time integral were significantly higher in KO females compared with WT control females. Blood pressure recordings demonstrated a significant increase in KO females when compared with WT female control mice. Knockout males, however, demonstrated a small and statistically insignificant elevation in blood pressure. An increase in blood pressure can result in pressure overload on the heart, leading to the development of left ventricular hypertrophy that can progress further to hypertensive heart disease (Drazner, 2011). Echocardiographic analysis also supports this idea, because female KO mice have an increased LV internal diameter at diastole and systole, indicating LV dilatation (Du et al. 2000), and increased left ventricular mass, which is corroborated by the larger cross-sectional area observed using stained tissue sections, which collectively suggests that female KO mouse hearts are hypertrophic (Panguluri et al. 2013b; Chapalamadugu et al. 2015). Lack of any significant change in ejection fraction, which reflects a fractional change in the LV end-diastolic volume, indicates no differences in the fractional LV output at each cycle. Despite this, we observed an increased aortic blood flow rate, and KO females presented with higher blood pressure than WT control females, which could, at least in part, contribute to the development of cardiac hypertrophy.
Effect of Kvβ1.1 on cardiac MHCα expression
Cardiac remodelling involves changes in the expression of key genes involved in regulation of the electrical function of the heart. Therefore, we assessed mRNA and/or protein expression of key genes in the heart. We found that myosin isoform expression was significantly altered in the heart of KO females. The significantly larger elevation of MHCα (fast isoform) expression, at both mRNA and protein levels, clearly indicates features of a hypertrophic response in the KO female heart. Furthermore, mRNA expression of Kvβ1.1 in the heart demonstrated a significant increase in female compared with male WT mice, indicating that Kvβ1.1 may play a significant cardiac-specific role in females. To develop an overall understanding of the Kvβ1.1 gene at the molecular level, we used the Ingenuity Pathway Analysis and provided experimental data as the input for predicting possible pathways that are involved in cardiac remodelling. Based on this analysis, the final targets for cardiac hypertrophy in murine heart are GATA4, GATA6 and MHCα (MYH6), which are significantly altered in KO female hearts (Fig. 9). The transcription factors GATA4 and GATA6 have previously been demonstrated to have a profound effect on MHCα and MHCβ expression (Molkentin et al. 1994; Zhu et al. 2005; Maitra et al. 2009; Hu et al. 2010). Furthermore, protein kinase A and protein kinase C, which are known targets affected by cardiac hypertrophy, demonstrate significant interactions with GATA4, which is one of the key transcription factor that activates MHCα (MYH6; Wang et al. 2005; Sharma et al. 2007). Although it remains unclear how Kvβ1.1 or absence of Kvβ1.1 alters the expression of GATA4, GATA6 and MHCα (MYH6), the protein–protein interaction between Kvβ1.1 and MHCα in conjunction with elevated MHCα expression in Kvβ1.1 KO mouse hearts and Kvβ1.1 siRNA-treated H9C2 cells strongly suggests a potent inhibitory role of Kvβ1.1 in MHCα regulation. Collectively, the expression data and network analysis suggests that in female KO mice there is an upregulation of the hypertrophic pathway that involves altered expression of myosin heavy chain genes as well as key transcription factors, including GATA4.
Figure 9. Gene network analysis.
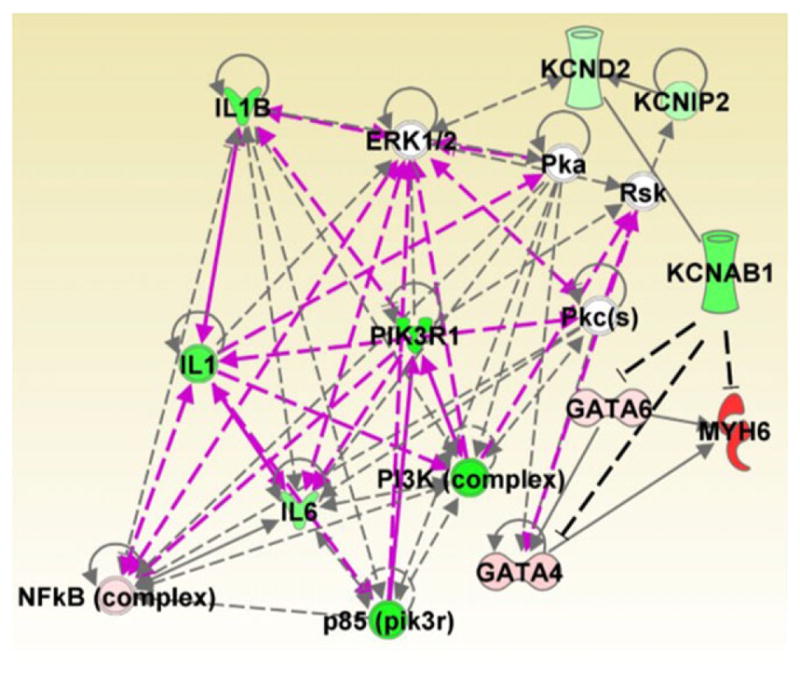
The top two networks identified by Ingenuity Pathway Analysis based on qPCR-expression data were merged. Potential interactions between KCNAB1 and MYH6 (MHCα) or GATA factors were incorporated into the analysis and indicated by dashed black lines. Relative gene expression differences between the WT and KO groups are depicted by a colour gradient from green to red; green represents higher expression for WT, whereas red represents KO. ERK1/2, extracellular signal-regulated protein kinases 1 and 2; IL1, Interleukin-1; IL1B, Interleukin 1-Beta; IL6, Interleukin-6; KCND2, Kv4.2; KCNIP2, Kv Channel Interacting Protein 2; KCNAB1, Kv Beta 1.1; MYH6, Myosin heavy chain alpha; NFkB, Nuclear Factor Kappa-B; PI3K, Phosphoinositide-3-Kinase; PIK3R1, Phosphoinositide-3-Kinase Regulatory Subunit 1; Pka, cAMP-dependent Protein Kinase, Pkc, Protein Kinase C; RsK, Ribosomal Protein S6 Kinase.
Study limitations
In the present study, mice with global knockout of the Kvβ1.1 gene were used for experimentation. Our study demonstrated significant cardiac structural and haemodynamic differences in the female KO mice. However, given that Kvβ1.1 KO female mice have high blood pressure, it is likely that vascular changes may be involved in causing cardiac hypertrophy. Future studies are necessary to identify the vascular component and how deletion of Kvβ1.1 affects the female mice.
Conclusion
In conclusion, we identified structural, electrical and haemodynamic differences in Kvβ1.1 KO in murine hearts. This is the first study to demonstrate that deletion of Kvβ1.1 leads to increased blood pressure, electrical changes and cardiac hypertrophy in female mice. We identified that the male mice failed to develop cardiac hypertrophy and high blood pressure despite altered electrical activity. Overall, the female mouse hearts depicted distinct physiological changes upon deletion of the Kvβ1.1 gene compared with male mice. At a molecular level, the female hearts confirmed the major hallmarks for cardiac hypertrophy, such as MHCα and its binding to Kvβ1.1. Therefore, the present study provides fundamental new information on the role of Kvβ1.1 in female murine hearts and its relationship to cardiovascular physiology.
New Findings.
What is the central question of this study?
The goal of this study was to evaluate sex differences and the role of the potassium channel ß1 (Kvß1) subunit in the heart.
What is the main finding and its importance?
Genetic ablation of Kvβ1.1 in females led to cardiac hypertrophy characterized by increased heart size, prolonged monophasic action potentials, elevated blood pressure and increased myosin heavy chain α (MHCα) expression. In contrast, male mice showed only electrical changes. Kvβ1.1 binds the MHCα isoform at the protein level, and small interfering RNA targeted knockdown of Kvβ1.1 upregulated MHCα.
Acknowledgments
Funding
We acknowledge funding support from National Institutes of Health HL102171 (to S.M.T.).
The authors wish to thank the College of Pharmacy and College of Medicine, Department of Molecular Medicine of the University of South Florida for its support and assistance.
Footnotes
Competing interests
None declared.
Author contributions
S.M.T. was responsible for conception and design of the research. J.T., K.C.C., T.P. and S.L.B. performed the experiments. J.T., K.C.C., P.J.K., A.B. and S.M.T. performed data analysis and interpretation. J.T. and K.C.C. drafted the manuscript. S.M.T. edited and revised the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
References
- Aimond F, Kwak SP, Rhodes KJ, Nerbonne JM. Accessory Kvβ1 subunits differentially modulate the functional expression of voltage-gated K+ channels in mouse ventricular myocytes. Circ Res. 2005;96:451–458. doi: 10.1161/01.RES.0000156890.25876.63. [DOI] [PubMed] [Google Scholar]
- Banerjee B, Peiris DN, Koo SH, Chui P, Lee EJ, Hande MP. Genomic imbalances in key ion channel genes and telomere shortening in sudden cardiac death victims. Cytogenet Genome Res. 2008;122:350–355. doi: 10.1159/000167822. [DOI] [PubMed] [Google Scholar]
- Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271:840–844. [PubMed] [Google Scholar]
- Berul CI, Aronovitz MJ, Wang PJ, Mendelsohn ME. In vivo cardiac electrophysiology studies in the mouse. Circulation. 1996;94:2641–2648. doi: 10.1161/01.cir.94.10.2641. [DOI] [PubMed] [Google Scholar]
- Chapalamadugu KC, Panguluri SK, Bennett ES, Kolliputi N, Tipparaju SM. High level of oxygen treatment causes cardiotoxicity with arrhythmias and redox modulation. Toxicol Appl Pharmacol. 2015;282:100–107. doi: 10.1016/j.taap.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YF, Chung RH, Lee CY, Kao HY, Hou L, Hsu FC. Identification of rare variants for hypertension with incorporation of linkage information. BMC Proc. 2014;8:S109. doi: 10.1186/1753-6561-8-S1-S109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crea F, Battipaglia I, Andreotti F. Sex differences in mechanisms, presentation and management of ischaemic heart disease. Atherosclerosis. 2015;241:157–168. doi: 10.1016/j.atherosclerosis.2015.04.802. [DOI] [PubMed] [Google Scholar]
- Daniels LB, Maisel AS. Cardiovascular biomarkers and sex: the case for women. Nat Rev Cardiol. 2015;12:588–596. doi: 10.1038/nrcardio.2015.105. [DOI] [PubMed] [Google Scholar]
- David M, Macías A, Moreno C, Prieto Á, Martíinez-Mármol R, Vicente R, González T, Felipe A, Tamkun MM, Valenzuela C. Protein kinase C (PKC) activity regulates functional effects of Kvβ1.3 subunit on KV1.5 channels: identification of a cardiac Kv1.5 channelosome. J Biol Chem. 2012;287:21416–21428. doi: 10.1074/jbc.M111.328278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic receptors in the heart. Cardiovasc Res. 2000;48:448–454. doi: 10.1016/s0008-6363(00)00187-5. [DOI] [PubMed] [Google Scholar]
- Ebert SN, Liu XK, Woosley RL. Female gender as a risk factor for drug-induced cardiac arrhythmias: evaluation of clinical and experimental evidence. J Womens Health. 1998;7:547–557. doi: 10.1089/jwh.1998.7.547. [DOI] [PubMed] [Google Scholar]
- El Gebeily G, El Khoury N, Mathieu S, Brouillette J, Fiset C. Estrogen regulation of the transient outward K+ current involves estrogen receptor α in mouse heart. J Mol Cell Cardiol. 2015;86:85–94. doi: 10.1016/j.yjmcc.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–473. [PubMed] [Google Scholar]
- Fischer F, Vonderlin N, Seyler C, Zitron E, Korkmaz S, Szabó G, Thomas D, Katus HA, Scholz EP. Isoenzyme-specific regulation of cardiac Kv1.5/Kvβ1.2 ion channel complex by protein kinase C: central role of PKCβII. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:469–476. doi: 10.1007/s00210-014-0965-5. [DOI] [PubMed] [Google Scholar]
- Giese KP, Storm JF, Reuter D, Fedorov NB, Shao LR, Leicher T, Pongs O, Silva AJ. Reduced K+ channel inactivation, spike broadening, and after-hyperpolarization in Kvβ1.1-deficient mice with impaired learning. Learn Mem. 1998;5:257–273. [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- Gowd BM, Thompson PD. Effect of female sex on cardiac arrhythmias. Cardiol Rev. 2012;20:297–303. doi: 10.1097/CRD.0b013e318259294b. [DOI] [PubMed] [Google Scholar]
- Hu DL, Chen FK, Liu YQ, Sheng YH, Yang R, Kong XQ, Cao KJ, Gu HT, Qian LM. GATA-4 promotes the differentiation of P19 cells into cardiac myocytes. Int J Mol Med. 2010;26:365–372. [PubMed] [Google Scholar]
- Lehmann MH, Hardy S, Archibald D, quart B, MacNeil DJ. Sex difference in risk of torsade de pointes with d,l-sotalol. Circulation. 1996;94:2535–2541. doi: 10.1161/01.cir.94.10.2535. [DOI] [PubMed] [Google Scholar]
- Li G, Cheng G, Wu J, Zhou X, Liu P, Sun C. Drug-induced long QT syndrome in women. Adv Ther. 2013;30:793–802. doi: 10.1007/s12325-013-0056-x. [DOI] [PubMed] [Google Scholar]
- McCarthy NS, Vangjeli C, Cavalleri GL, Delanty N, Shianna KV, Surendran P, O’Brien E, Munroe PB, Masca N, Tomaszewski M, Samani NJ, Stanton AV. Two further blood pressure loci identified in ion channel genes with a gene-centric approach. Circ Cardiovasc Genet. 2014;7:873–879. doi: 10.1161/CIRCGENETICS.113.000190. [DOI] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol. 2009;326:368–377. doi: 10.1016/j.ydbio.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol Heart Circ Physiol. 1998;274:H747–H751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Kalvakolanu DV, Markham BE. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the α-myosin heavy-chain gene. Mol Cell Biol. 1994;14:4947–4957. doi: 10.1128/mcb.14.7.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GG, Fedorov NB, Giese KP, Ohno M, Friedman E, Chen R, Silva AJ. Increased neuronal excitability, synaptic plasticity, and learning in aged Kvβ1.1 knockout mice. Curr Biol. 2004;14:1907–1915. doi: 10.1016/j.cub.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM. Mouse models of arrhythmogenic cardiovascular disease: challenges and opportunities. Curr Opin Pharmacol. 2014;15:107–114. doi: 10.1016/j.coph.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, Schofield L, Centracchio J, Zehender M, Peng X, Brunner M, Koren G. Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while progesterone is protective. Heart Rhythm. 2012;9:823–832. doi: 10.1016/j.hrthm.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanyan V, Yin L, Bardakjian R, Kolz C, Enrick M, Hakobyan T, Kmetz JG, Bratz I, Luli J, Nagane M, Khan N, Hou H, Kuppusamy P, Graham J, Fu FS, Janota D, Oyewumi MO, Logan SJ, Lindner JR, Chilian WM. Requisite role of Kv1.5 channels in coronary metabolic dilation. Circ Res. 2015;117:612–621. doi: 10.1161/CIRCRESAHA.115.306642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- Panguluri SK, Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Tipparaju SM. MicroRNA-301a mediated regulation of Kv4.2 in diabetes: identification of key modulators. PLoS ONE. 2013a;8:e60545. doi: 10.1371/journal.pone.0060545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panguluri SK, Tur J, Fukumoto J, Deng W, Sneed KB, Kolliputi N, Bennett ES, Tipparaju SM. Hyperoxia-induced hypertrophy and ion channel remodeling in left ventricle. Am J Physiol Heart Circ Physiol. 2013b;304:H1651–H1661. doi: 10.1152/ajpheart.00474.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TV, Sosunov EA, Gainullin RZ, Danilo P, Jr, Rosen MR. Impact of sex and gonadal steroids on prolongation of ventricular repolarization and arrhythmias induced by IK-blocking drugs. Circulation. 2001;103:2207–2212. doi: 10.1161/01.cir.103.17.2207. [DOI] [PubMed] [Google Scholar]
- Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Schmitt N, Grunnet M, Olesen SP. Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol Rev. 2014;94:609–653. doi: 10.1152/physrev.00022.2013. [DOI] [PubMed] [Google Scholar]
- Sharma A, Masri J, Jo OD, Bernath A, Martin J, Funk A, Gera J. Protein kinase C regulates internal initiation of translation of the GATA-4 mRNA following vasopressin-induced hypertrophy of cardiac myocytes. J Biol Chem. 2007;282:9505–9516. doi: 10.1074/jbc.M608874200. [DOI] [PubMed] [Google Scholar]
- Tadros R, Ton AT, Fiset C, Nattel S. Sex differences in cardiac electrophysiology and clinical arrhythmias: epidemiology, therapeutics, and mechanisms. Can J Cardiol. 2014;30:783–792. doi: 10.1016/j.cjca.2014.03.032. [DOI] [PubMed] [Google Scholar]
- Tipparaju SM, Li XP, Kilfoil PJ, Xue B, Uversky VN, Bhatnagar A, Barski OA. Interactions between the C-terminus of Kv1.5 and Kvβ regulate pyridine nucleotide-dependent changes in channel gating. Pflugers Arch. 2012;463:799–818. doi: 10.1007/s00424-012-1093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipparaju SM, Liu S-Q, Barski OA, Bhatnagar A. NADPH binding to β-subunit regulates inactivation of voltage-gated K+ channels. Biochem Biophys Res Commun. 2007;359:269–276. doi: 10.1016/j.bbrc.2007.05.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Paradis P, Aries A, Komati H, Lefebvre C, Wang H, Nemer M. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Mol Cell Biol. 2005;25:9829–9844. doi: 10.1128/MCB.25.22.9829-9844.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahagi K, Davis HR, Arbustini E, Virmani R. Sex differences in coronary artery disease: pathological observations. Atherosclerosis. 2015;239:260–267. doi: 10.1016/j.atherosclerosis.2015.01.017. [DOI] [PubMed] [Google Scholar]
- Yang H, Elko P, Fromm BS, Baga JJ, Pires LA, Schuger CD, Steinman RT, Lehmann MH. Maximal ascending and descending slopes of the T wave in men and women. J Electrocardiol. 1997;30:267–276. doi: 10.1016/s0022-0736(97)80038-6. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhu S, Liu D, Yu Z, Yang Y, van der Giet M, Tepel M. GATA4-mediated cardiac hypertrophy induced by d-myo-inositol 1,4,5-tris-phosphate. Biochem Biophys Res Commun. 2005;338:1236–1240. doi: 10.1016/j.bbrc.2005.10.086. [DOI] [PubMed] [Google Scholar]