

Abstract

Controlling the assembly of soft and deformable molecular aggregates into mesoscale structures is essential for understanding and developing a broad range of processes including rare earth extraction and cleaning of water, as well as for developing materials with unique properties. By combined synchrotron small- and wide-angle X-ray scattering with large-scale atomistic molecular dynamics simulations we analyze here a metalloamphiphile–oil solution that organizes on multiple length scales. The molecules associate into aggregates, and aggregates flocculate into meso-ordered phases. Our study demonstrates that dipolar interactions, centered on the amphiphile headgroup, bridge ionic aggregate cores and drive aggregate flocculation. By identifying specific intermolecular interactions that drive mesoscale ordering in solution, we bridge two different length scales that are classically addressed separately. Our results highlight the importance of individual intermolecular interactions in driving mesoscale ordering.
Short abstract
Combining synchrotron X-ray scattering with atomistic simulations reveals intermolecular dipolar interactions that drive mesoscopic assemble of metal-centered aggregates in a metalloamphiphile phase.
Introduction
Controlling the assembly of matter across length scales using well-defined building blocks is essential for the development of advanced materials and processes.1,2 Numerous mechanisms have been proposed that describe the growth and behavior of materials through the nano- and mesoscales, including nanoparticle aggregation3 and interfacial self-assembly.4 However, the various associative processes that drive the organization of matter into meso-ordered domains are often separated by orders of magnitude in space and time from molecular-level processes. For example, sub-nanometer processes are understood in terms of molecular building blocks that aggregate into supramolecular particles (e.g., colloids) through interactions that take place on the picosecond time scale, whereas nano- to micrometer processes are often understood in terms of flocculating particulate units with dynamics on the nanosecond time scale. The gulf between the mesoscopic and molecular domains is the reason why supramolecular chemistry is considered as a separate discipline from the science of complex fluids, and bridging these length and time scales requires new approaches. Herein we study an amphiphilic molecular solution that is driven toward mesoscopic ordering, giving new insight into the interactions that drive both molecular self-assembly and cooperative long-range organizations.
There is a growing consensus that molecular solutions bearing high concentrations of metal ions display nano/mesoscale behaviors, in that clusters of ions and solvent molecules interact collectively through intercluster interactions (ICIs).5 Such systems belong to a broad category of material known as nonideal solutions. The concept of nonideal behavior in solution has been known for more than a century and can lead to perturbations in the thermodynamic properties, as well as phase behaviors often associated with complex fluids (e.g., viscosity changes, phase transitions, etc.). For example, dissolving metal salts into water has an effect beyond the local H-bonded network structure, where ordered “icelike” regions can be seeded around some ions and these local regions can manifest mesoscopic (collective) phenomena such as viscosity changes, which arise through ICIs.5 The effect is exaggerated when the solvent and solute differ significantly in polarity. Pronounced nano- and mesoscale structuring is exhibited by solutions of metal ions in nonpolar solvents. An important example of this kind of system is metalloamphiphile solutions, which are hybrid materials that combine the activity of a coordination complex with the surfactant property of an amphiphile. This renders metalloamphiphiles highly susceptible to aggregation in solution, causing the metal complex to localize into higher-ordered architectures,6,7 offering a route to concentrate metal ions locally with useful properties for applications such as catalysis, sensing, and separations.8,9
Mesoscale behavior has been probed with particular vigor in metalloamphiphile solutions that are used in metal refining applications, where they underpin a multibillion dollar hydrometallurgy industry.10 In these systems, high concentrations of metal ions are transported from an aqueous phase into hydrocarbon oil via complexation with an amphiphilic ligand, the result being a highly concentrated metalloamphiphile–oil solution. This solution is notoriously prone to mesoscopic ordering, which leads to problematic phase changes that must be avoided.11 Due to the commercial importance of this particular issue, the ICIs that lead to mesoscale behaviors in metalloamphiphile–oil solutions of relevance to metal refining have received a great deal of attention.11,12 The mesoscale structural dynamics are understood using a model known as “Baxter sticky spheres” (Figure 1), which treats the metalloamphiphile aggregates as spherical particles with surface adhesion that is approximated by a narrow attractive potential well.13 The origin of the attractive potential has previously been assigned to van der Waals interactions between groups of atoms within the aggregate cores in accordance with Hamaker theory,14 despite this being inconsistent with the narrow delta function approximated by the Baxter model.
Figure 1.

Concept of the Baxter sticky sphere model as applied to metalloamphiphile solutions.15 The metalloamphiphile aggregate is treated as a solid nanoscale particle with the hydrophilic coordination complex (purple) inside a solid spherical core (red) surrounded by the lipophilic functionality endowed by the organic amphiphile ligands (headgroups in orange, tails in gray). Particle aggregates interact through a narrow attractive well U(r) (dashed line, eq 4 in the Supporting Information), which is an example of ICI that causes inelastic collisions between molecular aggregates that drive mesoscopic assembly.
The specific example of aggregating metalloamphiphile–oil solutions (such as those used in metal refining applications) illustrates a general dichotomy that is presented by describing an aggregating molecular solution system—which is inherently dominated by molecular-level dynamics—using a simple sticky-particle model. Without a molecular-based understanding of the interactions that drive mesoscopic assembly in aggregating solution systems, predictive control of the organization of molecular aggregates into mesoscale architectures remains out of our grasp. This problem is universal, from natural processes to technological applications such as in the synthesis of meso-ordered functional materials.
The paucity in understanding of molecular-level mechanisms that drive nano- to mesoscale structuring in metalloamphiphile solutions (and nonideal solutions in general) derives from the scarcity of appropriate experimental techniques. These are extremely limited by the disorder and dynamics of solution systems. Direct imaging methods including X-ray crystallography or electron microscopy are inappropriate because they require measurements to be performed on solid substrates that lack the crucial molecular-level dynamics that define solutions. Indeed, as exemplified in our recent publication, the solution structure of even simple metal salts can be very different from the solid state.16 Further complications arise from the structural continuum that characterizes nonideal solutions, stretching from the molecular to the mesoscale. The interacting units (i.e., molecules) that drive assembly are in the sub-nanometer size range, and these control both the formation of nanoscale clusters and interactions between them (i.e., mesoscale). This requires techniques that encapsulate length scales from the angstrom to hundreds of nanometers. Most laboratory-based optical techniques cannot span these size domains and have added complications in analyzing such concentrated systems. In simple aqueous solutions, vibrational spectroscopy such as Raman scattering can probe interactions between the clustering water molecules provided they have bands that are easily distinguishable with shifts that depend predictively on clustering. Unfortunately, this technique is of limited use for more complex solutions because the self-assembling molecular units (in our case hydrocarbons, amides, nitrates, metal ions, etc.) often have numerous overlapping bands that are difficult to deconvolute and shift in an unpredictable manner. Perhaps the most powerful technique we can bring to bear on aggregating metalloamphiphile solutions is synchrotron small- and wide-angle X-ray scattering (SWAXS). The small wavelength (high energy) of X-rays available at synchrotrons, as well as the high flux, provide scattering data that can be used to probe the molecular-to-meso length scales. However, interpretations of SWAXS data are wrought with ambiguity arising from the complicated interactions between molecules and clusters.
Bridging the spatial and chronological scales that separate the molecular (i.e., molecular self-assembly) from the mesoscopic (i.e., interactions between self-assembled nanoscale clusters) requires an understanding of the collective dynamics of systems involving tens to hundreds of thousands of atoms with processes that occur on picosecond to microsecond time frames. Advances in computer technology have allowed large-scale atomistic molecular dynamics (MD) simulations to be performed on amphiphile systems.17−19 We recently used atomistic MD simulations to demonstrate how the presence of metal ions in amphiphile–oil solution changes the structure of molecular aggregates.20,21 Although these studies show a general qualitative agreement with the morphology of aggregates suggested by analysis of experimental SAXS data, there was no quantitative comparison of simulation with experiment. Last year, Ferru et al. demonstrated the generation of SAXS data from amphiphile–oil solutions,22 providing a new approach to bridge experiment with simulation. However, this particular method did not accurately reproduce SAXS collected from amphiphile–oil solutions bearing metal ions (i.e., metalloamphiphiles).23 Despite the recently acquired ability to simulate large-scale aggregating solution systems, there is lacking a study that fully bridges MD simulations with experimental measurement. This is needed to uncover the intermolecular interactions controlling the mesoscopic assembly predicted by particle modeling of SAXS data (e.g., the Baxter model).
Here we present a study that fully bridges atomistic MD simulations of a metalloamphiphile solution with particle-based analysis of experimental SWAXS data, leading to a molecular-level understanding of the mesoscale dynamics. Initially, we adopt a new approach to generating X-ray scattering data from atomistic MD simulations of metalloamphiphile solutions, providing experimentally reflective SWAXS from these metal-bearing systems. We then analyze the atomistic MD simulations to provide a quantitative measurement of average aggregate morphology that reflects real-space functions generated from experimental SWAXS. Finally, we generate parameters from the simulation that converge with those derived from the classical Baxter model interpretation of experimental SWAXS. This crucial step allows us to use the simulations to isolate specific intermolecular interactions that drive the attractive force between nanoscale clusters. This gives original insight onto intermolecular interactions that drive mesoscale ordering in nonideal solutions. Such interactions may account for a variety of emergent behaviors (e.g., viscosity changes and phase transitions) exhibited in a broad variety of nonideal solution systems.
Results and Discussion
Development of Metalloamphiphile–Oil Solutions via Solvent Extraction
The subject of our study is a metalloamphiphile solution consisting of organic malonamide ligand DMDOHEMA (i.e., N,N′-dimethyl-N,N′-dioctylhexylethoxymalonamide, Scheme S1) solvating Eu(III) nitrate in n-heptane oil. The amphiphile DMDOHEMA is an effective solvating ligand for all lanthanide(III) nitrate salts, and Eu(III) was used in this case due to its fluorescent properties that allow for convenient measurement of concentration inside the lab. The system is of technological relevance to f-block metal ion separations and refining via solvent extraction,25 and was selected because it is amenable to SWAXS studies interpreted using the Baxter model26 as well as atomistic MD simulations.24 The malonamide–Eu(III) system is thus employed to investigate how intermolecular interactions around the europium(III) coordination complex drive the attractive ICIs predicted by the Baxter model. Coordinating interactions between the metal ion and solvating ligands (malonamide, water, nitrate, etc.) in similar systems have been previously explored using a variety of techniques,27,28 but there have been none that link intermolecular interactions to the wider self-assembling solution structure. Vibrational spectroscopic measurements were also taken from the solutions in the present study, providing qualitative agreement with previous studies on similar systems. These data, along with additional supporting light scattering and tensiometry measurements, are presented in the Supporting Information.
Previous studies using small-angle scattering interpreted with the Baxter model have shown that the strength of ICIs between aggregates formed in amphiphile–oil solutions depends on the concentration of metal ion as well as pH value.21,29 Samples are therefore prepared at different acidity, with and without Eu(III), by mixing 0.5 M DMDOHEMA in n-heptane with aqueous nitrate phases (Table 1). Eu(III) nitrate transfers into the organic DMDOHEMA–heptane solution to form the metalloamphiphile complex.30 The goal in this present study is to investigate the impact of the coordination complex (europium nitrate) and acid (nitric acid) on the aggregate structures and ICIs between aggregates that leads to mesoscale ordering in the metalloamphiphile–oil solutions. Toward this goal, a two-pronged approach is adopted involving experimental SWAXS and atomistic MD simulations to probe solutions (a)–(d) (Table 1).
Table 1. Concentrations (M) of Solutes in Heptane Solutions.
| soln | DMDOHEMA | H2O | HNO3 | Eu(NO3)3 |
|---|---|---|---|---|
| a | 0.5 | 0.15 | ||
| b | 0.5 | 0.15 | 0.06 | |
| c | 0.5 | 0.33 | 0.31 | |
| d | 0.5 | 0.33 | 0.31 | 0.06 |
Qualitative Comparison of SWAXS between Experiment and Simulation
X-ray scattering techniques are among the few appropriate methods for probing ICIs and the resulting mesoscopic ordering in amphiphile solutions.31 SWAXS is particularly sensitive to metalloamphiphile–oil solutions because X-rays are scattered by regions of electron density inhomogeneity caused by the aggregation of polar molecular groups (amphiphile headgroups, water, nitric acid, europium nitrate) that have more electrons than the surrounding aliphatic media. Interactions between these electron-dense nanoscale aggregate cores cause fluctuations in electron density inhomogeneity that extend into the mesoscale. The SAXS (low-q) region yields information on nano-to-mesoscale structures and interactions, whereas the WAXS (high-q) region is sensitive to the shorter-range molecular structure. As illustrated in Figure 2i, experimental SWAXS data were collected from solutions (a)–(d), as well as from a control system of pure n-heptane. All data converge at high q values, which are dominated by a peak at around 1.4 Å–1 that corresponds to the correlations from the aliphatic background. At low q (0.05–0.6 Å–1), the scattering profile of pure n-heptane is low and flat, indicating the absence of electron density inhomogeneity in the >10 Å size range. In contrast, the scattering intensity increases in this region for solutions (a)–(d), indicating inhomogeneous electron density profiles that manifest from structures in solution with >10 Å in cross section. Such increases in scattering intensity are ascribed to particle scattering, as would be expected from a dispersion of nanoscale aggregates of amphiphile in the heptane solvent medium and is typical for solvent extraction organic phases.32 The intensity of scattering in the low-q range, as well as the slope, changes drastically across the four samples (a)–(d). This indicates that the nanoscale aggregate structures and/or the mesoscale ordering driven by ICIs is sensitive to the presence of acid and the coordinating Eu(III) complex, as expected from previous studies.21,29
Figure 2.

Experimental (i) and simulated (ii) SWAXS for solutions (a)–(d) and pure n-heptane solution. In the experimental X-ray data, the background scattering from air and the sample holder has been subtracted. The simulated results are normalized with respect to the experimental scattering intensity of the pure n-heptane system at 1.4 Å–1. The low-q limit is restricted by the size of the simulation box by q > 2π/(D/2) based on Bragg’s law. Note that D/2, instead of D, is used due to the minimum-image convention in computer simulations under the periodic boundary conditions, where each atom is interacting only with the closest image of the remaining atoms in the system. See Supporting Information for details on the calculation of the SWAXS from atomistic simulations.
Of the four samples, solution (a) shows the lowest scattering intensity in the low-q region and reaches a plateau at q < 0.2 Å–1. Such a low and flat scattering profile suggests that the aggregates are small, with minimal ICIs to drive mesoscale ordering. Under acidic conditions without Eu (solution (c)), the scattering intensity increases and is more sloped, which might suggest an increase in both number and size of aggregates, as well as mesoscopic ordering driven by attractive ICIs between aggregates.15 Incorporation of the coordinating europium nitrate complex to form metalloamphiphile species in solutions (b) and (d) produces even more intense scattering and steeper slopes in the low-q region, especially for the acidic system (d). This suggests more extended structure and dynamics driven by attractive ICIs between the aggregates in the presence of coordinating metal salt, particularly under acidic conditions.
Complementary to the SWAXS experiments, atomistic MD simulations were conducted on solutions (a)–(d) using the component concentrations experimentally obtained (Table 1), as well as a system of pure n-heptane (see Supporting Information for simulation details). Generating SWAXS data from the simulation trajectory offers a method for directly corroborating the simulation with experiment. This approach was first demonstrated on amphiphile–oil systems by Ferru and co-workers in 2014,22,33 although poor agreement was observed in the presence of metal ions.33 Therefore, we adopted an alternative method for generating SWAXS from the simulation that involved a more accurate description of electron density obtained from direct calculation of atomic form factors using quantum methods (see Supporting Information for detailed discussion). In all of the resulting simulated SWAXS data in Figure 2ii, a high-q peak at around 1.4 Å–1 is observed, in good agreement with the experimental data. Also in line with the experimental data, the heptane scattering at 0.1 < q < 0.6 Å–1 is low and flat, indicating the absence of electron density inhomogeneity in the >10 Å size range. For solutions (a)–(d), the shapes of the simulated data functions in the 0.1 < q < 0.6 Å–1 region are similar to the experimental SAXS, with profiles that are typical for particle scattering. Both experimental and simulated SWAXS data for samples (a)–(d) show a minimum at q ≈ 0.6 Å–1 with an intensity I(q) of 0.015–0.025 au (relative to the heptane solvent peak). Most encouraging is that the simulated and experimental SWAXS converge at similar I(q) at the low-q limit of 0.1 Å–1. The intensity at the low-q limit is a product of all the structural correlations in the system—including correlations between atoms in the same molecule as well as correlations between nanoscale aggregates—and so is highly sensitive to solution mesostructure. These agreements help justify the use of atomistic MD simulation for investigating our metalloamphiphile solutions.
A qualitative understanding of structure within the simulations may be gained from the inspection of snapshots after reaching equilibrium from atomistic simulations. The dispersion of aggregated species is visualized more clearly by hiding the heptane and amphiphile molecules to reveal only the hydrophilic solute that resides at the center of the aggregate cores (i.e., water, HNO3, Eu(III), NO3–), and these are shown in Figure 3a–d. The simulation for solution (a), which was formed under neutral conditions without complexing Eu(III) ions, is shown in Figure 3a. Here, the water molecules in the 0.5 M DMDOHEMA–heptane are quite uniformly dispersed into small globular clusters containing no more than 4 water molecules. When the Eu(III) nitrate complex is incorporated within the 0.5 M DMDOHEMA–heptane under neutral conditions (Figure 3b), aggregates containing up to 3 Eu(III) nuclei are assembled with attendant nitrate anions to satisfy neutrality in the low dielectric constant solvent. The water molecules are drawn to these clusters so that large, swollen aggregates pertain. In the simulation of solution (c) (Figure 3c), which was formed under acidic conditions without Eu(III), the water and nitric acid are dispersed in extended aggregates containing many molecules that are linked through H-bonding interactions. When the europium nitrate complex is incorporated into the phase under acidic conditions (Figure 3d), aggregates containing up to 3 Eu(III) nuclei are formed and the water and nitric acid molecules are drawn to the metalloamphiphile complex. It is noteworthy that all of these systems are single phase, with the hydrophilic molecules dispersed in aggregate clusters of varying sizes, which is qualitatively consistent with the particle scattering profiles observed with SWAXS (Figure 2).
Figure 3.
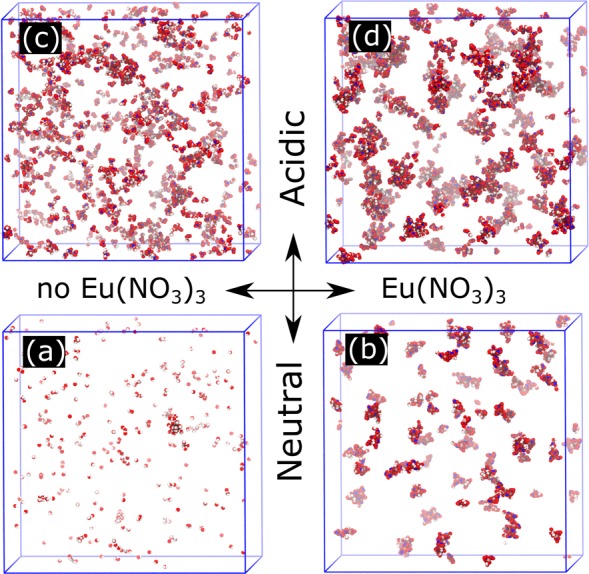
Snapshots of the last simulation frames for solutions (a)–(d). Only H2O, HNO3, Eu3+, and NO3– are shown (heptane and DMDOHEMA omitted for the display). See Figure S9 for the complete figures.
Defining Aggregate Morphology
Our experimental SWAXS data describe the overall shape of aggregates, which is averaged over time and all the existing aggregates in the systems. To determine the size of the average aggregate structures in real space quantitatively, the generalized indirect Fourier transform (GIFT) method34,35 was employed to interpret the normalized background-subtracted SAXS data (see Figure S4 and previous publications36,37 for extended discussion of GIFT method). Using GIFT, we simultaneously approximated the structure factor scattering contribution that arises from all concentrated aggregate/particle systems using the Percus–Yevick (PY) structure factor formula for hard spheres so as to generate the pair distance distribution functions (PDDF). The PDDFs in Figure 4 correspond to probability distribution functions p(r) at given scattering distances r between points in the polar assemblies (i.e., the aggregate cores).
Figure 4.

PDDFs generated from the background subtracted SAXS data for the solutions (a)–(d) formed under conditions described in Table 1. Insets show morphologies of the aggregate polar cores that are suggested by the PDDF functions.
The PDDFs for solutions (a) and (b) in Figure 4 (upper) are asymmetrical bell-shaped curves with positive displacement into high r, which is suggestive of globular aggregates that can be described as ellipsoids with a long and a short axis.38 For solutions (c) and (d), the positive displacement at high r is much more defined, producing ski-slope-shaped functions that suggest more elongated rod-shaped aggregates38 that can also be described with a long and a short axis. The average short axis diameter can be estimated from the second inflection point on these functions, whereas the maximum linear extent of the aggregate long axis is taken from where the PDDF decays to 0. The morphologies of the aggregates that are suggested by the PDDF functions are depicted by the shapes in the insets. Dynamic light scattering (DLS) measurements were also performed to support the PDDF analysis. The DLS measurements are generally supportive of the SWAXS data analysis in that larger particles form when acid and/or Eu(III) is incorporated into the clusters. They also suggest that the attractive interaggregate interactions (see results of Baxter modeling below) are leading to larger overall assemblies. Full discussion of DLS results can be found in the Supporting Information.
In the MD simulations, the positions of all the atoms are saved over the simulation time so that data may be interpreted to understand the average morphology of the aggregate cores and compare with the PDDFs. In the simulation snapshots in Figure 5, aggregate polar cores are formed from the headgroups of DMDOHEMA molecules that encapsulate molecules of water, HNO3, and europium nitrate complex. The aggregates are polydisperse and dynamic in all four simulations (as expected for an aggregating solution system) so that aggregates are merging, dissipating, and emerging from solution over time. This makes it difficult to quantitatively define time- and weight-averaged aggregate morphologies that can be directly compared to the PDDFs. However, through inspection of multiple simulation snapshots while tracking the positions of the polar atoms that make up the aggregate core over time, representative morphologies may be selected. A time-averaged visualization of the aggregate core is extracted by plotting the accumulated distributions of the polar core atoms over time in a given aggregate, shown by the orange regimes in the bottom row of Figure 5. In calculating the accumulated distributions, the translation and rotation of the polar core atoms are first removed to exclude the influence of diffusion behaviors, and then the distributions of the polar core atoms are plotted in a single figure.39 Consequently, the orange region illustrates the shape of the hydrophilic core (i.e., the region sensitive to SAXS) of a given aggregate that was selected as representative in the system.
Figure 5.

Morphologies of representative aggregates in the simulations of solutions (a)–(d). In the upper row, the structures of single aggregates are plotted. In the lower row, the accumulated distributions of the aggregate core (DMDOHEMA headgroup, H2O, HNO3, Eu3+, and NO3–) are included to highlight the morphology of the polar core of such aggregates that produces the background-subtracted experimental SAXS signal from which the PDDFs in Figure 4 were generated. Each of the accumulated distributions is based on a simulation duration of 5 ns in a given system, except 0.5 ns for (c).
The average morphologies of aggregate polar cores suggested from the experimentally derived PDDFs (Figure 4) are generally comparable to the time-averaged structures of the representative aggregates in the simulation (Figure 5). Both simulation and experiment suggest approximately spherical aggregate morphologies in the neutral solutions (a) and (b), with substantial swelling of the aggregate core from 10 to 15 Å initiated by the incorporation of the europium nitrate complex. In the acidic solutions (c) and (d), the aggregates have more elongated morphology. Therefore, in terms of size and shape of the aggregates, the selected structures isolated from the simulation are reflective of experiment.
Quantitative Measurement of Metalloamphiphile Aggregates
When Eu(III) is incorporated into the amphiphile–oil solution, the metal complex seeds aggregation around it with water and amphiphile molecules interacting in the outer coordination sphere. Therefore, the Eu(III) ions in solutions (b) and (d) sit at the center of aggregates that do not dissipate over nanosecond time scales, even though the structures are dynamic with molecules exchanging between the aggregates. Because the majority of aggregates in solutions (b) and (d) have a Eu(III) ion in the core, Eu-centered radial distribution functions (RDFs) may be used to quantitatively measure the time-averaged aggregate structure over the course of the simulations.
In solutions (b) and (d), the simulations show aggregates that contain 1, 2, and 3 Eu(III) ions in varying proportions (Figure 6i). In the neutral system (b), the mononuclear aggregate predominates, whereas under acidic conditions more dinuclear and trinuclear species occur. Assuming that the Eu(III) ions assemble linearly in the aggregate cores, we can describe elliptical species with a short axis and a long axis diameter in the same manner as in the PDDFs in Figure 4. This assumption allows us to measure the simulated structures using a 2D (short axis + long axis) model that is directly comparable to the interpretation of the experimental SWAXS-generated real-space functions (Figure 4). Although a triangularly arranged trinuclear complex is observed (Figure S12), this species is in the minority compared to the linear trinuclear complex. The lengths can be approximated based on the Eu–Eu and Eu–DMDOHEMA headgroup RDFs (Figure S11). The short axis diameter is double the average distance between the central Eu ion and the headgroup of the associated DMDOHEMA. The long axis is calculated by including the distance between neighbor Eu3+ ions along the length of the core. The estimated long and short axis diameters for the 1, 2, and 3 Eu-containing aggregates are shown in Figure 6ii for solutions (b) and (d). According to these measurements, the metalloamphiphile aggregates in the simulation for solution (b) have an average short axis diameter of 10.4 Å and a maximum long axis length of 21.6 Å. In the simulation for solution (d) the aggregates are more swollen and longer, with a short axis of 15.6 Å and long axis of up to 27.2 Å. These dimensions are generally supportive of the dimensions derived from the PDDFs generated from experimental SAXS in Figure 4, especially considering the completely dissimilar approaches taken in measuring and defining aggregate morphology in these dynamic systems. However, the discrepancies between the aggregate morphologies measured from simulation and experimental SAXS must be addressed.
Figure 6.

(i) A histogram showing the relative number of mono-, di-, and trinuclear metalloamphiphile aggregates in solutions (b) (blue) and (d) (red). (ii) Shapes and lengths of the long axis and short axis of linear Eu-centered aggregates containing 1, 2, and 3 Eu3+ per aggregate for solutions (b) and (d), and their respective structures (iii).
The long axis diameter of solution (b) measured using the Eu-centered RDF (Figure 6) is concurrent with the maximum extent of the PDDF function in Figure 4, whereas the short axis diameter is underestimated by about 30%. This may stem from the assumption that Eu ions arrange linearly, which is violated in the simulation of solution (b) by a small number of aggregates with trigonally arranged Eu ions (Figure S12). These have a longer short axis diameter (15 Å) and would increase the apparent average aggregate short axis of the aggregates in the simulation if taken into account. In contrast, in solution (d) the short axis diameter measured from the Eu-centered RDF (Figure 6) is concurrent with the short axis diameter from the PDDF (Figure 4), whereas the maximum long axis diameter is significantly underestimated relative to the PDDF. This may be due to the assumption that the aggregate morphology is completely described by Eu–DMDOHEMA correlations, which is not true in all cases. For example, the simulation snapshots suggest that elongated aggregates form as a result of H-bonding interactions between water and nitric acid molecules in solution (d) and do not necessarily involve Eu centers (Figure S13). The underestimation of the long axis length obtained from the Eu-centered RDF from the simulation of solution (d) may stem from ignoring these aggregates in our measurements.
Modeling SAXS Data as Adhesive Spheres
The strength of the attractive ICIs between aggregates that leads to mesoscopic ordering in the amphiphile–oil systems can be estimated by fitting the normalized background subtracted experimental SWAXS data (Figure S5) with the Baxter sticky hard sphere model.13−15 This model hinges on the assumption that the aggregates can be described as spherical particles with surface adhesion approximated by a narrow attractive potential well, as conceptualized in Figure 1. In solutions (a)–(d), the spherical particles are represented by the aggregate cores that are made up from N, O, and Eu atoms from the amphiphile headgroups and hydrophilic solute (i.e., water, acid, Eu nitrate). The size and number of spherical particles approximated by the Baxter model depends partly on the concentration of hydrophilic solutes (Table 1) that are assumed to reside completely within the core but also depends heavily on the aggregating behavior of the amphiphile. The amphiphile participates in the aggregates as well as existing as unassociated monomers in solution so that the average aggregation number (N) of DMDOHEMA molecules in each aggregate and the monomer concentration ([mono]) are vital parameters that control the size and number of spherical particles approximated by the Baxter model. Monomer concentration was determined via tensiometric measurement of the aqueous–organic interface with variable concentration of amphiphile/metalloamphiphile in heptane (Supporting Information) and is comparable with those reported in the literature for similar systems.26,40,41 Subsequently, only two parameters were optimized during the fitting of the SAXS data using the Baxter sticky hard sphere model: the average aggregation number (N) of amphiphiles and the strength of the attractive sticky interaction U/kBT. The resulting fits are shown in Figure S5, and selected metrics from the model are shown in Table 2.
Table 2. Selected Metrics Used in the Baxter Model Fitting of Experimental SAXS Data (First Six Columns) and Comparable Parameters Derived from Simulation (Last Two Columns).
| atomistic simulation results |
|||||||
|---|---|---|---|---|---|---|---|
| soln | [mono]a (M) | Nb | no. of Eu/agg | Dcore (Å) | U (kBT) | [mono]a,c (M) | Nb,c |
| a | 0.159 | 4 | 11 | –0.73 | 0.040 ± 0.002 | 7.2 ± 0.5 | |
| b | 0.08 | 13 | 2.2 | 19 | –1.42 | 0.032 ± 0.002 | 9.8 ± 0.5 |
| c | 0.08 | 9 | 17 | –1.28 | 0.030 ± 0.001 | 9.2 ± 0.5 | |
| d | 0.05 | 14 | 1.7 | 20 | –1.47 | 0.032 ± 0.001 | 11.1 ± 0.6 |
DMDOHEMA monomer concentration.
DMDOHEMA aggregation number.
The standard deviations are provided as the error bars.
In general, the results from the Baxter model—showing an increase of N and decrease of [mono] in the solutions containing the coordination complex ((b) and (d))—confirm that the metal complex enhances molecular aggregation. The metal complex also drives aggregation on the mesoscale, where the interaggregate interaction potential (U/kBT) is significantly enhanced in solutions (b) and (d). The values of [mono] and N in Table 2 can be used to calculate the average number of Eu ions in each aggregate (no. of Eu/agg). We found that in both metalloamphiphile solutions there are on average ca. 2 Eu3+ per aggregate, independently corroborating the phenomenon of multinuclear metalloaggregates predicted by the MD simulations. The diameters of the spherical cores (Dcore) in the Baxter model are between 10 and 25% larger than the short axis diameter of the ellipsoids suggested by the PDDFs (Figure 4). The origin of this discrepancy is probably due to the contribution of the long axis into the spherical description that is especially pronounced in the more elongated aggregates formed under acidic conditions.
To bridge the sticky sphere model with the MD simulations, the simulation data were analyzed to derive values for N and [mono]. The MD simulations show that the aggregates are made up of DMDOHEMA molecules that may be associated through inner-sphere coordination into the europium nitrate complex (i.e., dative interactions between malonamide O and Eu(III) ion), aggregated in the outer-sphere of the complex, associated with a cluster of 1 or more water/HNO3 molecules, or self-associated. Therefore, RDF between DMDOHEMA headgroups may be used to estimate N and [mono] in the simulations for solutions (a)–(d). As shown in Figure 7i, the calculated RDFs show two major correlation peaks between the DMDOHEMA headgroups in all of the simulations, with the first peak in the shaded gray area located at around 6 Å and the second in the shaded pink at around 8 Å. While the height (i.e., correlation strength) of the first peak changes only slightly from system to system, the intensity of the second peak becomes greatly elevated in the presence of acid and/or Eu3+ ions. To understand the origin of the two major peaks in the RDF, the relative orientations between the headgroups of neighboring DMDOHEMA molecules were explored. As shown in Figure 7ii, parallel or partially parallel orientations of neighboring DMDOHEMA molecules arise when they are within the first correlation shell of around 6 Å (structures shaded in gray). When in the second correlation shell of around 8 Å (structures shaded in pink), the arrangement of DMDOHEMA is more disordered. It is thus suggested that, in the absence of water, acid, and Eu nitrate, the DMDOHEMA headgroups prefer to stack on top of each other due to their roughly planar nature, with the hydrocarbon tail extending outward. The presence of water, acid, or Eu nitrate molecules stimulates the formation of polar pools where DMDOHEMA molecules arrange their headgroups to increase the interaction with the hydrophilic solute in the core. Figure 7iii depicts a metalloamphiphile aggregate in the simulation, where the 6 and 8 Å correlations between DMDOHEMA headgroups are shown by the black and red lines and the time-averaged position of the hydrophilic solute (europium, nitrate, water) is shown in orange. This shows that both the first and second correlation peaks contribute to the aggregation behavior and must be taken into account to estimate N.
Figure 7.

(i) RDFs between the central carbon atoms on the DMDOHEMA headgroups. (ii) The orientations between neighbor DMDOHEMA headgroups contributing to the first (6 Å) and the second (8 Å) RDF peaks are illustrated in black-shaded and red-shaded snapshots, respectively. (iii) Structure of one aggregate highlighting the first (black line) and the second (red line) correlations between DMDOHEMA headgroups. These link the DMDOHEMA molecules that surround the polar core of the aggregate where the accumulative positions of water, HNO3, Eu, and nitrate molecules are shown in orange.
Values for N and [mono] calculated using the DMDOHEMA RDFs are shown in Table 2, where they are compared with the parameters used in the Baxter model. In agreement with the Baxter model, the simulation shows that the formation of metalloamphiphile complex as well as the presence of acid increases N and decreases [mono]. Solution (c) shows the best agreement in N between the MD simulation and Baxter model (<10% difference) and solution (a) the worst (about 42% difference). The value for [mono] in solution (a) is significantly lower in the simulation than for experiment, which may stem from the self-association of DMDOHEMA molecules into chains (Figure S14). These chains, although large in length, have very small cross-sectional areas, which would not scatter X-rays significantly in the SAXS region. This means that the self-associated DMDOHEMA chains contribute to N and [mono] calculated from the simulation (particularly for solution (a)), but do not contribute significantly to the experimental SAXS and are therefore ignored by the Baxter model.
Molecular Origins of Intercluster Interactions
The Baxter model shows that the attractive interaction (U/kBT) between aggregates increases drastically upon incorporation of europium nitrate into the amphiphile–oil solution. By bridging our atomistic MD simulation with the sticky sphere model we are presented with an opportunity to investigate how molecular-level structural dynamics influence the ICIs that drive mesoscale ordering. Our large-scale atomistic MD simulations were performed for tens of nanoseconds for solutions of about (160 Å)3 in space in order to encapsulate the necessary spatial and chronological scales at which mesoscale phenomena manifest.
The Eu(III) ion serves as a probe to explore interactions that lead to aggregation by analyzing the Eu-centered RDF. For the mononuclear species that are dominant in solution (b) (Figure 6i), the Eu(III) ion sits at the center of approximately spherical aggregates, providing a convenient reference point at the center of a particle that best reflects the Baxter model. The Baxter model describes the system as hard spherical particles with surface adhesion, i.e., the aggregates undergo inelastic collisions. The concept of a hard sphere is difficult to define in aggregating amphiphile solutions, which is a major drawback in applying particle models to solution systems in general. However, we may relate the hard sphere diameter to the diameter of the aggregate polar core, which would be roughly fixed through strong H-bonding and coordination interactions as opposed to the flexible aliphatic chains that comprise the aggregate corona. The central concept of the attractive ICIs is surface adhesion, which means that the particles attract only when they collide. In the case of the mononuclear Eu-containing aggregates in the simulation of solution (b), the attractive interactions should therefore occur at a Eu–Eu distance of approximately 10.4 Å (i.e., 2 times the radius of the polar core, Figure 8i), assuming that Eu resides in the center.
Figure 8.

(i) Two Eu-centered aggregates are touching, where the red shadow areas represent their polar core regions. (ii) RDF between Eu3+ ions in the solution (b). The typical correlation peaks are labeled with (1–6), with corresponding structures provided in (iii). (1–3) stand for dinuclear Eu3+ within the same aggregates. (4, 5) denote H-bonds (red dotted lines) bridging aggregates. Green dotted lines stand for Eu–oxygen dative bonds. (6) originates from the dipolar correlation (highlighted sticks) between NO3– coordinated to the left Eu3+ and the DMDOHEMA headgroup coordinating the right Eu3+ ion. In (6′) all the coordinated ligands are plotted, where the red shadow areas highlight the inner core region of the two aggregates.
The calculated RDFs represent interactions controlling molecular scale structural dynamics (e.g., dynamics within aggregate cores) disproportionately as these occur on the picosecond time scale, whereas interactions involved in the dynamics of nanoscale species (i.e., the diffusion and collisions of aggregates) happen at much slower nanosecond time scales. Therefore, molecular interactions that mediate the inelastic collision of aggregates will happen much less frequently than molecular interactions that mediate between associated species (e.g., the bridging interactions between dinuclear Eu complexes). The Eu–Eu RDF calculated from the simulation of system (b) is presented in Figure 8ii. In the short distance range of 4.9–5.7 Å, intense peaks (1–3) correspond to the Eu–Eu correlations in the dinuclear and trinuclear complexes. Although these interactions are dynamic on the molecular picosecond scale, they are fixed relative to the mesoscale nanosecond time frame as the bridged Eu centers diffuse together within the core of the same aggregate, leading to their intense and sharp character.
The most significant feature at the intercluster length scale is a broad and shallow peak at 10–12 Å. Both the position and character of this peak are consistent with an interaggregate interaction. The broad character would arise, in part, from the rotational averaging of the not-quite-spherical aggregates, and the weak intensity is expected from an interaction that occurs from collisions between diffusing aggregates on the nanosecond time scale. The strength of such intercluster interactions was estimated from the peak height to be −0.7 kBT (ΔG = −kBT ln(g(r))), which is in the right energy range to correspond to the “sticky” interaction that is featured in the Baxter model (i.e., −2 to 0 kBT). Although this value is about half of the strength of the attractive potential derived from modeling the SAXS data (Table 2), the peak is unique to the mononuclear aggregate. The total strength of the attractive potential in the simulation would also include the correlations from the di- and trinuclear aggregates, but these are difficult to deconvolute unambiguously from the Eu–Eu RDF.
By inspection of the simulation structures, the Eu–Eu RDF peak at 10–12 Å, as well as the intercluster interactions, is ascribed to a Eu–Eu correlation between two adjacent aggregates bridged through intermolecular dipolar interactions between coordinated nitrate and DMDOHEMA molecules (structure 6 in Figure 8iii). Such correlations could be dynamically stable up to the simulation durations of tens of nanoseconds.
To quantitatively calibrate the contribution of the dipolar interactions, we calculated the values of the dipolar interaction energies between Eu-centered mononuclear aggregates in solution (b) within the distance range of 10–12 Å. See Figure S10 for the distribution of the obtained results. The average value of −0.6 ± 1.5 kBT of the dipolar interactions is in good agreement with the value of ΔG = −0.7 kBT of the intercluster interactions. This strongly supports the significant contribution of the intermolecular dipolar interactions to the overall intercluster interaction energy predicted by the sticky particle model. It is noteworthy that such dipolar attractive interactions are expected to compete with the hydrophobic interactions between the DMDOHEMA tailgroups and the solvent molecules (n-heptane). The former favors the formation of aggregates, whereas the latter dissolves them in the organic environment. The calculated effective interactions ΔG < 0 indicate that the dipolar attraction dominates the intercluster interactions.
Aside from the peak at 10–12 Å, weak oscillations occur from 7 to 10.4 Å. Such weak oscillations correspond to Eu–Eu correlations that are facilitated by H-bonds with the assistance of water molecules after the aggregates merge following an inelastic collision. Just like in classical surfactant-based microemulsions (e.g., water–AOT–oil),42 the collisions are sometimes accompanied by an exchange of core material (e.g., water, acid). The transient dimeric aggregate that forms as a result of the inelastic collision has a very short lifetime, which is why the peaks that correspond to these species are weak relative to the short-range correlations between Eu atoms in the same complex.
The MD simulations suggest that the attractive interaggregate interaction is facilitated through specific dipolar intermolecular bonds between molecules. These interactions, although influencing mesoscale order, are actually short-ranged and only activate when the molecules are brought into close proximity as the aggregates collide. This is entirely consistent with the narrow delta function used to approximate the surface adhesion in the Baxter model for sticky spheres, which has been demonstrated in numerous publications to provide a close approximation for SAXS and small-angle neutron scattering (SANS) data collected from ion–amphiphile–oil systems.11,12 This perspective, hinging on specific intermolecular dipolar interactions, contrasts with the current adhesive particle-based hypothesis for the origin of attractive interaction that infers van der Waals forces between groups of atoms in the aggregate cores according to Hamaker theory.14 Unlike intermolecular dipolar interactions, van der Waals interaction between particles has the form of an extended Lennard-Jones potential: not a narrow delta function for surface adhesion as predicted by Baxter. Therefore, the specific intermolecular interactions that are shown to facilitate interaggregate attraction in our study are more consistent with the fundamental physics of the Baxter model for hard sticky spheres.
Conclusions
In a general sense, our study bridges the molecular scale with the mesoscale by addressing a fundamental question: how do molecular solutions manifest mesoscale properties? We converged theoretical and experimental approaches to investigate the molecular scale structural dynamics that give rise to longer-ranged behavior in a metalloamphiphile solution. On finding good agreements between experiment and simulation in terms of scattering data and aggregate morphology, we drew upon the simulations to target the molecular origins of the attractive ICIs known to drive mesoscale behaviors in solution. Our results suggest that the attractive forces between aggregates predicted by modeling the SAXS data are produced from dipolar interactions between molecules embedded in adjacent aggregates. This interpretation, hinging on specific interactions between pairs of molecules, contrasts with collective interpretations of van der Waals forces between group atoms within the aggregate cores. Our results highlight the importance of molecular phenomena in driving mesoscale ordering within materials.
Acknowledgments
This work and the use of the Advanced Photon Source are supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences and Geosciences, under Contract DE-AC02-06CH11357. B.Q. gratefully acknowledges the discussion with Joshua Dempster and the computing resources provided on Blues, a high-performance computing cluster operated by the Laboratory Computing Resource Center at Argonne National Laboratory. M.O.d.l.C acknowledges the support from U.S. Department of Energy Award DE-FG02-08ER46539.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00306.
Detailed experiment and simulation methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cölfen H.; Mann S. Higher-Order Organization by Mesoscale Self-Assembly and Transformation of Hybrid Nanostructures. Angew. Chem., Int. Ed. 2003, 42 (21), 2350–2365. 10.1002/anie.200200562. [DOI] [PubMed] [Google Scholar]
- Mann S. Life as a Nanoscale Phenomenon. Angew. Chem., Int. Ed. 2008, 47 (29), 5306–5320. 10.1002/anie.200705538. [DOI] [PubMed] [Google Scholar]
- Liu T.; Diemann E.; Li H.; Dress A. W. M.; Muller A. Self-assembly in aqueous solution of wheel-shaped Mo154 oxide clusters into vesicles. Nature 2003, 426 (6962), 59–62. 10.1038/nature02036. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Song Y.-F.; Cronin L.; Liu T. Self-Assembly of Organic–Inorganic Hybrid Amphiphilic Surfactants with Large Polyoxometalates as Polar Head Groups. J. Am. Chem. Soc. 2008, 130 (44), 14408–14409. 10.1021/ja805644a. [DOI] [PubMed] [Google Scholar]
- Bakker H. J. Structural Dynamics of Aqueous Salt Solutions. Chem. Rev. 2008, 108, 1456–1473. 10.1021/cr0206622. [DOI] [PubMed] [Google Scholar]
- Griffiths P. C.; Fallis I. A.; Tatchell T.; Bushby L.; Beeby A. Aqueous solutions of transition metal containing micelles. Adv. Colloid Interface Sci. 2008, 144, 13–23. 10.1016/j.cis.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Griffiths P. C.; Fallis I. A.; Chuenpratoom T.; Watanesk R. Metallosurfactants: Interfaces and micelles. Adv. Colloid Interface Sci. 2006, 122, 107–117. 10.1016/j.cis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Mancin F.; Scrimin P.; Tecilla P.; Tonellato U. Amphiphilic metalloaggregates: Catalysis, transport, and sensing. Coord. Chem. Rev. 2009, 253, 2150–2165. 10.1016/j.ccr.2009.03.015. [DOI] [Google Scholar]
- Binnemans K.; Görller-Walrand C. Lanthanide-containing liquid crystals and surfactants. Chem. Rev. 2002, 102, 2303–2345. 10.1021/cr010287y. [DOI] [PubMed] [Google Scholar]
- Tasker P. A.; Plieger P. G.; West L. C.. Metal complexes for hydrometallurgy and extraction. In Comprehensive Coordination Chemistry II; Ward M. D., Ed.; Elsevier Ltd.: New York, 2004; Vol. 9, pp 759–808. [Google Scholar]
- Testard F.; Zemb T.; Bauduin P.; Berthon L.. Third-phase formation in liquid/liquid extraction: A colloidal approach. In Ion Exchange and Solvent Extraction: A Series of Advances; Moyer B. A., Ed.; CRC Press: Boca Raton, 2010; Vol. 19, pp 381–428. [Google Scholar]
- Zemb T.; Bauer C.; Bauduin P.; Belloni L.; Dejugnat C.; Diat O.; Dubois V.; Dufreche J.-F.; Dourdain S.; Duvail M.; Larpent C.; Testard F.; Pellet-Rostaing S. Recycling metals by controlled transfer of ionic species between complex fluids. En route to ″ienaics″. Colloid Polym. Sci. 2015, 293, 1–22. 10.1007/s00396-014-3447-x. [DOI] [Google Scholar]
- Baxter R. J. Percus-Yevick equation of hard spheres with surface adhesion. J. Chem. Phys. 1968, 49, 2770–2774. 10.1063/1.1670482. [DOI] [Google Scholar]
- Nave S.; Mandin C.; Martinet L.; Berthon L.; Testard F.; Madic C.; Zemb T. Supramolecular organisation of tri-n-butyl phosphate in organic diluent on approaching third phase transition. Phys. Chem. Chem. Phys. 2004, 6 (4), 799–808. 10.1039/b311702b. [DOI] [Google Scholar]
- Erlinger C.; Belloni L.; Zemb T.; Madic C. Attractive interactions between reverse aggregates and phase separation in concentrated malonamide extractant solutions. Langmuir 1999, 15 (7), 2290–2300. 10.1021/la980313w. [DOI] [Google Scholar]
- Demars T. J.; Bera M. K.; Seifert S.; Antonio M. R.; Ellis R. J. Revisiting the Solution Structure of Ceric Ammonium Nitrate. Angew. Chem., Int. Ed. 2015, 54, 7534–7538. 10.1002/anie.201502336. [DOI] [PubMed] [Google Scholar]
- Lee O.-S.; Stupp S. I.; Schatz G. C. Atomistic Molecular Dynamics Simulations of Peptide Amphiphile Self-Assembly into Cylindrical Nanofibers. J. Am. Chem. Soc. 2011, 133 (10), 3677–3683. 10.1021/ja110966y. [DOI] [PubMed] [Google Scholar]
- Leung C.-Y.; Palmer L. C.; Kewalramani S.; Qiao B.; Stupp S. I.; Olvera de la Cruz M.; Bedzyk M. J. Crystalline polymorphism induced by charge regulation in ionic membranes. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (41), 16309–16314. 10.1073/pnas.1316150110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C.-Y.; Palmer L. C.; Qiao B. F.; Kewalramani S.; Sknepnek R.; Newcomb C. J.; Greenfield M. A.; Vernizzi G.; Stupp S. I.; Bedzyk M. J.; Olvera de la Cruz M. Molecular Crystallization Controlled by pH Regulates Mesoscopic Membrane Morphology. ACS Nano 2012, 6 (12), 10901–10909. 10.1021/nn304321w. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Demars T.; Olvera de la Cruz M.; Ellis R. J. How Hydrogen Bonds Affect the Growth of Reverse Micelles around Coordinating Metal Ions. J. Phys. Chem. Lett. 2014, 5, 1440–1444. 10.1021/jz500495p. [DOI] [PubMed] [Google Scholar]
- Ellis R. J.; Meridiano Y.; Muller J.; Berthon L.; Guilbaud P.; Zorz N.; Antonio M. R.; Demars T.; Zemb T. Complexation-Induced Supramolecular Assembly Drives Metal-Ion Extraction. Chem. - Eur. J. 2014, 20, 12796–12807. 10.1002/chem.201403859. [DOI] [PubMed] [Google Scholar]
- Ferru G.; Gomes Rodrigues D.; Berthon L.; Diat O.; Bauduin P.; Guilbaud P. Elucidation of the Structure of Organic Solutions in Solvent Extraction by Combining Molecular Dynamics and X-ray Scattering. Angew. Chem., Int. Ed. 2014, 53, 5346–5350. 10.1002/anie.201402677. [DOI] [PubMed] [Google Scholar]
- Rodrigues F.; Ferru G.; Berthon L.; Boubals N.; Guilbaud P.; Sorel C.; Diat O.; Bauduin P.; Simonin J. P.; Morel J. P.; Morel-Desrosiers N.; Charbonnel M. C. New insights into the extraction of uranium(VI) by an N,N-dialkylamide. Mol. Phys. 2014, 112, 1362–1374. 10.1080/00268976.2014.902139. [DOI] [Google Scholar]
- Guilbaud P.; Zemb T. Solute-induced microstructural transition from weak aggregates towards a curved film of surface-active extractants. ChemPhysChem 2012, 13, 687–691. 10.1002/cphc.201100721. [DOI] [PubMed] [Google Scholar]
- Bisel I.; Nicol C.; Charbonnel M. C.; Blanc P.; Baron P.. Inactive DIAMEX test with the optimized extraction agent DMDOHEMA. In Proceedings of the International Conference on Future Nuclear Systems, GLOBAL’99: Challenge Towards Second Nuclear Era with Advanced Fuel Cycles, Yokohama, Japan, 1997; pp 153–159. [Google Scholar]
- Meridiano Y.; Berthon L.; Crozes X.; Sorel C.; Dannus P.; Antonio M. R.; Chiarizia R.; Zemb T. Aggregation in organic solutions of malonamides: Consequences for water extraction. Solvent Extr. Ion Exch. 2009, 27, 607–637. 10.1080/07366290903270148. [DOI] [Google Scholar]
- Iveson P. B.; Drew M. G. B.; Hudson M. J.; Madic C. Structural studies of lanthanide complexes with new hydrophobic malonamide solvent extraction agents. J. Chem. Soc., Dalton Trans. 1999, 3605–3610. 10.1039/a905579g. [DOI] [Google Scholar]
- Gannaz B.; Antonio M. R.; Chiarizia R.; Hill C.; Cote G. Structural study of trivalent lanthanide and actinide complexes formed upon solvent extraction. Dalton Trans. 2006, 38, 4553–4562. 10.1039/b609492a. [DOI] [PubMed] [Google Scholar]
- Dejugnat C.; Dourdain S.; Dubois V.; Berthon L.; Pellet-Rostaing S.; Dufreche J.-F.; Zemb T. Reverse aggregate nucleation induced by acids in liquid-liquid extraction processes. Phys. Chem. Chem. Phys. 2014, 16, 7339–7349. 10.1039/c4cp00073k. [DOI] [PubMed] [Google Scholar]
- Charbonnel M. C.; Flandin J. L.; Giroux S.; Presson M. T.; Madic C.; Morel J. P.. Extraction of Ln(III) and Am(III) from nitrate media by malonamides and polydentate N-bearing ligands. In Proceedings of the International Solvent Extraction Conference, ISEC 2002; Sole K. C., Cole P. M., Preston J. S., Robinson D. J., Eds.; South African Institute of Mining and Metallurgy: Johannesburg, 2002; Vol. 2, pp 1154–1160. [Google Scholar]
- Gelbart W. M.; Ben-Shaul A. The ″New″ Science of ″Complex Fluids″. J. Phys. Chem. 1996, 100, 13169–13189. 10.1021/jp9606570. [DOI] [Google Scholar]
- Berthon L.; Meridiano Y.; Lagrave S.; Crozes X.; Sorel C.; Zorz N.; Testard F.; Zemb T. In Aggregation of organic extractant phases: consequences on extraction properties; Canadian Institute of Mining, Metallurgy and Petroleum: 2008; pp 1017–1022. [Google Scholar]
- Rodrigues F.; Ferru G.; Berthon L.; Boubals N.; Guilbaud P.; Sorel C.; Diat O.; Bauduin P.; Simonin J. P.; Morel J. P.; Morel-Desrosiers N.; Charbonnel M. C. New insights into the extraction of uranium(VI) by an N,N-dialkylamide. Mol. Phys. 2014, 112 (9–10), 1362–1374. 10.1080/00268976.2014.902139. [DOI] [Google Scholar]
- Fritz G.; Glatter O. Structure and interaction in dense colloidal systems: evaluation of scattering data by the generalized indirect Fourier transformation method. J. Phys.: Condens. Matter 2006, 18, S2403–S2419. 10.1088/0953-8984/18/36/S14. [DOI] [Google Scholar]
- Fritz G.; Bergmann A.; Glatter O. Evaluation of small-angle scattering data of charged particles using the generalized indirect Fourier transformation technique. J. Chem. Phys. 2000, 113, 9733–9740. 10.1063/1.1321770. [DOI] [Google Scholar]
- Ellis R. J.; Anderson T. L.; Antonio M. R.; Braatz A.; Nilsson M. A SAXS study of aggregation in the synergistic TBP-HDBP solvent extraction system. J. Phys. Chem. B 2013, 117, 5916–5924. 10.1021/jp401025e. [DOI] [PubMed] [Google Scholar]
- Ellis R. J.; Audras M.; Antonio M. R. Mesoscopic Aspects of Phase Transitions in a Solvent Extraction System. Langmuir 2012, 28, 15498–15504. 10.1021/la3034879. [DOI] [PubMed] [Google Scholar]
- Glatter O. The interpretation of real-space information from small-angle scattering experiments. J. Appl. Crystallogr. 1979, 12, 166–175. 10.1107/S0021889879012139. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Dejugnat C.; Berthon L.; Dubois V.; Meridiano Y.; Dourdain S.; Guillaumont D.; Pellet-Rostaing S.; Zemb T. Liquid-Liquid Extraction of Acids and Water by a Malonamide: I-Anion Specific Effects on the Polar Core Microstructure of the Aggregated Malonamide. Solvent Extr. Ion Exch. 2014, 32, 601–619. 10.1080/07366299.2014.940229. [DOI] [Google Scholar]
- Meridiano Y.These de Doctorat Thesise; Paris XI: Orsay, 2009. [Google Scholar]
- Rees G. D.; Robinson B. H. Microemulsions and organogels: properties and novel applications. Adv. Mater. 1993, 5, 608–619. 10.1002/adma.19930050903. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.