

Abstract
Inspiratory function is essential for alveolar ventilation and expulsive behaviors that promote airway clearance (e.g., coughing and sneezing). Current evidence demonstrates that inspiratory dysfunction occurs during healthy aging and is accentuated by chronic heart failure (CHF). This inspiratory dysfunction contributes to key aspects of CHF and aging cardiovascular and pulmonary pathophysiology including: i) impaired airway clearance and predisposition to pneumonia; ii) inability to sustain ventilation during physical activity; iii) shallow breathing pattern that limits alveolar ventilation and gas exchange; and iv) sympathetic activation that causes cardiac arrhythmias and tissue vasoconstriction. The diaphragm is the primary inspiratory muscle, hence, its neuromuscular integrity is a main determinant of the adequacy of inspiratory function. Mechanistic work within animal and cellular models has revealed specific factors that may be responsible for diaphragm neuromuscular abnormalities in CHF and aging. These include phrenic nerve and neuromuscular junction alterations as well as intrinsic myocyte abnormalities, such as changes in the quantity and quality of contractile proteins, accelerated fiber atrophy, and shifts in fiber type distribution. CHF, aging, or CHF in the presence of aging disturbs the dynamics of circulating factors (e.g., cytokines and angiotensin II) and cell signaling involving sphingolipids, reactive oxygen species, and proteolytic pathways, thus leading to the previously listed abnormalities. Exercise-based rehabilitation combined with pharmacological therapies targeting the pathways reviewed herein hold promise to treat diaphragm abnormalities and inspiratory muscle dysfunction in CHF and aging.
Keywords: respiratory muscle, fiber atrophy, weakness, dyspnea, exercise tolerance, fatigue
1. Introduction
Inspiratory muscles are essential for ventilatory and non-ventilatory activities. Beyond being active during breathing, inspiratory muscles are recruited near-maximally during expulsive behaviors, e.g., sneezing or coughing [1–3]. Therefore, loss of inspiratory muscle function can compromise gas exchange and the health of the pulmonary system. Heart failure and aging cause skeletal myopathy that affects both limb and inspiratory muscles. Although inspiratory muscles are also ‘skeletal muscles’, their structural, functional, and metabolic properties, and response to stressors or inactivity are strikingly different from limb muscles [4–8]. The diaphragm is the primary inspiratory muscle and, in CHF, abnormalities of the diaphragm occur earlier or to a greater extent than in limb muscles [9–13]. Similarly, aging causes diaphragm abnormalities [14–16,1]. The combined effects of aging and CHF are unclear because experimental models typically address each condition separately. We can deduce, from clinical measures of inspiratory function, that CHF accentuates aging-induced diaphragm abnormalities. Diaphragm dysfunction will contribute to decrease quality of life as well as enhance morbidity and mortality associated with CHF or aging. Therefore, it is imperative to understand the causes of inspiratory (or diaphragm) abnormalities to develop rational pharmacological and rehabilitation strategies that improve quality of life, reduce cardiovascular and pulmonary complications, and increase longevity in CHF patients and the elderly.
2. Evidence of inspiratory dysfunction – Heart Failure and Aging
In the clinical setting, inspiratory dysfunction is documented via ‘respiratory muscle tests’ such as maximal inspiratory pressure [17]. Considering that the diaphragm is the primary inspiratory muscle, investigators assume that abnormal outcomes of respiratory muscle tests reflect diaphragm muscle dysfunction. Because clinical tests are generally voluntary and measurements are performed mainly at the mouth or nostrils, we consider that clinical tests reflect abnormalities of the ‘inspiratory system’ (phrenic motor neurons, neuromuscular junction, and muscles). Thus, we refer to clinical measures as indicators of inspiratory (dys)function in the current review.
Inspiratory dysfunction has received greater attention in CHF than healthy aging, but evidence of inspiratory dysfunction in aging predates studies in CHF [18]. Specifically, results from the initial studies that optimized the technique to measure maximal inspiratory pressure and defined reference values were some of the first indications that aging impairs inspiratory function [19]. In a cross-sectional analysis, Black & Hyatt reported that age was inversely correlated with maximal inspiratory pressure (MIP). Ever since, age has been considered an important determinant of MIP and has been used in prediction equations to define normal values in healthy subjects [17]. Although the relationship between age and MIP has been considered linear, targeted analyses of older age groups have revealed a steeper decline in MIP for subjects older than 65 years of age. Individuals in age groups averaging 25 to 65 years of age demonstrate an approximate 30% decline in MIP with age [20], whereas MIP decreases by 60 to 70% in subjects 80 years of age and older [21,22]. These observations (illustrated in Fig. 1) and measurements of transdiaphragmatic pressure [23,24] are consistent with age-induced inspiratory dysfunction. Inspiratory dysfunction in aging may reflect overall neuromuscular abnormalities due to sarcopenia. Future studies will have to define whether aging affects diaphragm and limb muscles in a similar manner. Differences are likely to exist because of the lifelong activity of the diaphragm and aging-specific changes in respiratory system mechanics.
Figure 1. Decline in maximal inspiratory pressure with aging.

Data are combined mean values from Neder et al. [20] and Enright et. al. [21]. Dotted line indicates mean value for subjects 20 to 29 years old.
Chronic heart failure exacerbates the impairment in inspiratory function due to age. An early study by Hammond et al. [10] demonstrated that patients with severe biventricular CHF had a ~50% decrease in MIP. Several studies followed, which focused on more homogenous groups, and showed that CHF patients have decreased MIP [25–31]. Results from volitional as well as non-volitional tests using phrenic nerve stimulation have confirmed the decrease in inspiratory (mouth or transdiaphragmatic) pressure in CHF [31–33]. The inability to generate normal inspiratory pressure is independent of the etiology of disease [34,31,10,35]. Notably, the prevalence of this inspiratory dysfunction in CHF, defined arbitrarily as MIP < 70% predicted [35,36], does appear to depend on age. In a general outpatient population of CHF patients (age 50–60 yrs and NYHA classes I to III), the prevalence of inspiratory dysfunction is 30–50% [36,35,37]. However, conservative estimates suggest that approximately 60% of CHF patients (Class II and III) with 67 ± 9 years of age have inspiratory dysfunction [38], whereas the prevalence was 70–75% for a group of older patients (75 ± 11 years of age) with acute exacerbation of heart failure [39].
An important aspect often overlooked is that the level and prevalence of inspiratory dysfunction in CHF depends on the stage of the disease. Patients with severe CHF (Class III or IV) are weaker than patients with mild CHF (Class I) [40,32,31]. This concept is illustrated in Fig 2A, showing a cross-sectional observation of progressively lower MIP in patients ranging from New York Heart Association Class I to IV. Measurements of trans-diaphragmatic pressure with magnetic phrenic nerve stimulation have also added support to this notion of concurrent worsening of diaphragm weakness and disease severity [33]. In addition to lower maximal inspiratory pressure, the diminished ability to sustain submaximal efforts also characterizes respiratory dysfunction in CHF [35,41,42]. Specifically, the time to task-failure is substantially shorter when patients perform inspiratory efforts against a submaximal pressure-threshold load (Fig. 2B, and [35]) or isocapnic hyperpnoea [43]. In summary, inspiratory dysfunction is highly prevalent in older CHF patients or those in advanced stages of the disease.
Figure 2. Inspiratory dysfunction in heart failure patients.

A) Progressive decline in maximal inspiratory pressure (%predicted) in heart failure patients going from New York Heart Association (NYHA) class I to IV. Data are replotted from Filusch et al [32]. B) Heart failure patients have diminished endurance during a submaximal inspiratory load endurance test (protocol as in ref. [35]). Data shown in panel B were kindly provided by Dr. Gaspar Chiappa (Universidade Federal do Rio Grande do Sul, Brazil) and are from 18 CHF patients (age 64 ± 4 yrs) and 8 controls (age 62 ± 2 yrs) [131].
3. Relevance of inspiratory muscle dysfunction to CHF and aging (Fig. 3)
Figure 3. Relevance of diaphragm abnormalities to cardiovascular and respiratory pathophysiology in aging and heart failure.

Loss of diaphragm force is caused by contractile apparatus dysfunction and fiber atrophy, whereas slower shortening velocity is determined by contractile apparatus dysfunction and fiber type shifts. These diaphragm alterations trigger cardiovascular and pulmonary pathophysiological responses. SNA, sympathetic nervous activity. Solid arrows and lines are relevant for CHF and aging, while the dotted line is relevant mainly for CHF. The model illustrated here was expanded from concepts originally developed by others [53,3,1,45].
A low percentage of the maximal inspiratory pressure is utilized during quiet breathing [15,2,44]. However, decreases in maximal inspiratory muscle function, such as those seen in aging and CHF, mandate that ventilatory behaviors occur at a higher percentage of the maximal value. Loss of submaximal diaphragm function must be compensated for by increases in motor unit firing frequency and recruitment [45], which implicates diaphragm work being performed at a higher percentage of maximal capacity and a mismatch between input (phrenic nerve activity) and output (diaphragm force). The net result of diminished maximal and submaximal inspiratory function is sensation of dyspnea, compromised ability to sustain elevated ventilation during physical activity, and exercise intolerance in CHF [46,42,47].
Compensatory adaptation to loss of inspiratory muscle function also includes changes in breathing pattern. The inability to generate force, coupled with diminished lung compliance, leads to minute ventilation being achieved with low tidal volume and high breathing rate. This ‘shallow breathing’ is a common characteristic of moderate and severe CHF [48–50]. A consequence of a shallow breathing pattern is an increase in the ratio of dead space-to-tidal volume (VD/VT). Elevated VD/VT, which in CHF reflects an inefficient breathing pattern [48], compromises alveolar ventilation and gas exchange within the lungs. Importantly, markers of impaired gas exchange during exercise have greater prognostic value than VO2max in patients with CHF [51].
A shallow breathing pattern also elevates sympathetic activity, through the interaction of central respiratory and sympathetic neural circuits [52]. Additionally, sympathetic activity increases further because inspiratory muscle dysfunction promotes accumulation of metabolites which stimulate group IV phrenic afferent nerve fibers [53]. Stimulation of these fibers triggers reflex sympathetic activation [53,54]. Altogether, this enhanced sympathetic activity predisposes CHF patients to cardiac arrhythmias and a high risk of death [55] or vasoconstriction in limb muscles that limits whole-body exercise tolerance (relevant for CHF and aging) [53,56]. Accordingly, inspiratory muscle unloading reduces sympathetic nerve activity and increases exercise tolerance in CHF patients [57–59].
In the presence of diaphragm weakness, none of the aforementioned compensatory responses preserves cough, another expulsive behavior requiring near-maximal recruitment of inspiratory muscles [45,44]. Therefore, the inability to generate normal inspiratory pressures can impair airway clearance and predisposes individuals to pulmonary infections. Pneumonia is a common pulmonary complication with aging [60–62], and CHF patients have increased risk of hospitalization due to pneumonia [63,64]. While multiple factors will determine the higher incidence of pneumonia with aging and CHF, there is a likely contribution from the loss of inspiratory function.
The integrative observations presented in this section highlight the significance and impact of inspiratory (muscle) dysfunction in the health status and prognosis of elderly subjects and CHF patients. However, these observations are mainly of indirect nature. To establish causality, it is necessary to test the impact of therapies that specifically improve diaphragm function on clinically-relevant outcomes. Altogether, the aspects discussed above emphasize the importance of understanding the pathophysiological processes and the need for the development of new therapeutic strategies for inspiratory dysfunction. Inspiratory dysfunction in CHF does not correlate with markers of left ventricular function [65,39], is unaffected by acute decompensation of heart failure [39], and is not reversed by heart transplant [42]. These observations indicate that mechanisms beyond cardiac abnormalities per se are responsible for inspiratory dysfunction in CHF [12]. The findings of dysfunction using phrenic nerve stimulation are consistent with abnormalities in respiratory system mechanics, diaphragm neuromuscular transmission, excitation-contraction coupling, muscle fiber size, and the contractile apparatus.
4. Respiratory system mechanics
Chest wall compliance and lung elastic recoil decrease with aging (reviewed in [18]). The overall impact of these changes is diminished respiratory system compliance, and increased residual volume and functional residual capacity with age. The latter causes flattening of the diaphragm that diminishes its force-generating capacity. The effects of CHF on lung volumes and mechanics are the opposite of aging. CHF decreases residual volume and functional residual capacity, e.g. [66]. The disease also increases lung stiffness [67,68]. These alterations will minimize diaphragm flattening and its impact on force generation, but will exacerbate the contribution of diminished respiratory system compliance to inspiratory dysfunction in older CHF patients. The net outcome of changes in respiratory system mechanics in CHF and aging is increased work of breathing [18,67], which is accentuated by physical activity in CHF [67].
5. Neuromuscular abnormalities
The impact of aging on diaphragm phrenic motor neuron and neuromuscular junction has been reviewed in detail recently [1]. Briefly, aging causes remodeling (enlargement and fragmentation) and loss of synaptic contact in individual neuromuscular junctions [69,70]. The associated decrease in neurotrophic factors due to denervation and neuromuscular junction abnormalities plays an important role on diaphragm dysfunction in aging [1]. The effect of CHF on diaphragm neuromuscular physiology is less clear. One study has shown that CHF causes expansion of the neuromuscular junction and enhanced expression of an embryonic-type subunit of nicotinic acetylcholine receptors [71]. Overall, the pattern of changes elicited by CHF in the diaphragm neuromuscular junction is consistent with neurodegeneration and denervation [1,72,73]. Therefore, the development of CHF in the elderly might accelerate the loss of innervation and associated neurotrophic factors and contribute to inspiratory dysfunction. Currently, it is unclear whether alterations in the neuromuscular junction precede (and cause) or are a consequence of intrinsic diaphragm muscle abnormalities in CHF.
6. Intrinsic diaphragm muscle abnormalities
6.1 - Isometric and isotonic contractile properties
Measurements of inspiratory muscle function in humans such as MIP and twitch transdiaphragmatic pressure reflect mostly isometric properties. Direct measurements of diaphragm muscle function in vitro and in situ show that isometric force normalized for cross-sectional area (‘specific force’) is depressed by 15–30% in heart failure [74–76,13,77–79] as well as aging [16,15,14,80,81]. The decrease in isometric force is seen in both twitch and maximal tetanic contractions (e.g., Fig. 4) and is independent of the etiology of disease, being evident in models of dilated and ischemic CHF [74–76,13,77–79,82,83].
Figure 4. Heart failure causes diaphragm isometric and isotonic contractile dysfunction.

Data are from intact diaphragm bundles from adult control (open circles) and CHF mice (closed circles). Specific force, force (in Newton) normalized to cross-sectional area (cm2). Replotted from Ahn et al. [83]. The effects of aging on contractile properties are similar to those shown herein.
Respiratory muscle tests that represent isotonic properties in humans are available [84,85], but to our knowledge these have not been applied to determine aging or CHF effects. However, animal studies have shown that isotonic contractile properties are impaired by CHF or aging. CHF decreases maximal shortening velocity by 20–30% in rodents [86,78,74,83]. Hence, peak power output, which is the product of shortening velocity and specific force, displays the most pronounced degree of diaphragm contractile dysfunction: 35–50% decrease in diaphragm peak power [83,74]. The effects of CHF on isotonic contractile properties are illustrated in Fig. 4B–C.
Aging studies have produced equivocal results for isotonic properties, but species-differences may explain this variance. Old rats and mice (24 mo old, ~75% survival) show increases or no change in maximal shortening velocity or power measured in intact diaphragm bundles [87,88]. Conversely, in hamsters, aging decreased diaphragm maximal shortening velocity and peak power [89]. Ongoing studies by our group suggest that diaphragm bundles from mice in advanced stages of aging (30 mo old, <50% survival rate) also show decreases in maximal shortening velocity and peak power [90], which are consistent with those seen in limb muscles [91,92]. Thus, impairments in diaphragm isotonic contractile properties may have a delayed onset and be more relevant in very old age. We cannot exclude, however, that discrepant results reported among species reflect differences in protocols and analytical approaches to examine isotonic properties. Ideally, the effects of age on diaphragm isotonic contractile function would have to be resolved using skinned single fibers from human samples. This approach would allow the determination of changes in isotonic contractile function specific to each MHC isoform. Yet, testing of skinned fibers from diaphragm of healthy subjects is not a trivial task because collection of biopsies has to be performed during a medically prescribed surgery in the thoracic or abdominal compartment.
Decreases in shortening velocity and power are highly relevant because diaphragm activities require muscle shortening. For instance, inspiratory time diminishes during physical activity due to higher breathing frequency, while inspiratory pressure developed during each breath increases (i.e., becomes more negative) to achieve an elevated tidal volume. Additionally, reflex responses such as coughing and sneezing elicit a very rapid and deep inspiration. Hence, declines in isotonic function will compromises breathing during moderate-to-high intensity physical activity and expulsive behaviors that demand fast and powerful diaphragm contractions. Overall, the impairments in both isometric and isotonic contractile properties in electrically-stimulated bundles in vitro are the first line of evidence that aging and CHF disrupt diaphragm excitation-contraction coupling, the contractile apparatus, or both. A switch in fiber type distribution may also account for the functional changes seen in intact bundles. Fiber atrophy is another important component of the inability to generate normal absolute force and power. These aspects are discussed in detail below.
6.2 - Excitation-contraction coupling
Technical challenges in isolating intact single fibers of the diaphragm have prevented extensive analysis of E-C coupling. Based on findings reported in limb muscles of old [93] and CHF animals [94,95] and patients [96], it is reasonable to speculate that aging or CHF impairs diaphragm calcium release. In diaphragm preparations, CHF slows calcium reuptake, which appears to be caused by decreases in sarcoplasmic reticulum calcium-ATPase expression [97–99]. Thus, abnormalities in E-C coupling may contribute to diaphragm dysfunction in CHF and aging.
6.3 - Contractile apparatus
Isometric and isotonic contraction dysfunction may also be explained at the level of the sarcomere. In permeabilized diaphragm single fibers, where calcium concentration is controlled externally, CHF decreases maximal specific force and Ca2+ sensitivity in all fiber types [100,75]. Maximal specific force is determined by the total number of cross-bridges, the fraction of cross-bridges in the strongly bound force-generating state, and the force generated per cross-bridge [101,102]. The loss of maximal force in CHF is due to a decrease in the number of cross-bridges and force per cross bridge [103,78,79].
Modifications in myosin or thin-filament proteins may account for these effects of CHF on diaphragm single fiber contractile properties. CHF causes a proportional loss of diaphragm titin and myosin heavy chain (MHC) [75,104]. Loss of titin leads to wider myofilament lattice spacing and destabilization of the sarcomere that, respectively, lowers calcium sensitivity and maximal force [105,104,106]. The decrease in MHC content lessens the total number of available cross-bridges and contributes to diaphragm weakness [79,78,75]. Similarly, aging-induced decreases in myofibrillar protein content are associated with the specific force deficit in intact diaphragm bundles [80,14]. However, loss of MHC does not fully explain impairments in contractile function in CHF. The remaining myosin in diaphragm of CHF animals is abnormal as determined by in vitro motility assay showing ~20% slower sliding velocity, which occurs without a clear shift in MHC or myosin light chain isoforms [86]. CHF also slows cross-bridge kinetics in all fiber types [75]. Slowed sliding (shortening) velocity might be caused by decreases in myosin ATPase activity [107,101], which have been shown in all fiber types from limb muscle of CHF patients [108]. The sluggish cross-bridge kinetics is most likely related to a diminished rate of transition from weak to strong-binding state. Thus, post-translational myofibrillar abnormalities appear to be a major contributor to decreases in specific force, shortening velocity, and power determined in intact diaphragm bundles.
6.4 - Fiber type distribution and myofibrillar protein isoforms
Aging and CHF cause modest shifts in diaphragm fiber type composition. The diaphragm of old rats have 5% more type I fibers [109,88] and 10–15% more type IIb fibers [81,88] than young animals, which occurs due to proportional decreases in type IIa [109,88] and IIx/d fibers [81,88]. In old mice, Greising et al. [14] found no change in the percentage of type I fibers, with a shift to increased type IIa and decreased type IIx/d fibers. The effects of CHF (in animal studies) are generally the opposite of those elicited by aging, but findings are inconsistent among studies. Some groups have reported increases in type I and IIa fibers accompanied by decreases in type IIx/d and IIb fibers [110,13,76,111], while other groups have found no difference in fiber type distribution [82,112,113]. Variable results are also seen in studies with human diaphragm biopsies. Tikunov et al [114] reported higher type I and lower type II fiber percentage in patients with severe CHF undergoing heart transplant or left ventricular assist device placement. In contrast, Lindsay et al. [9] observed no difference in fiber type distribution in a similar population of patients. With limited numbers of subjects (n = 7–12 per group) and wide age ranges, it is difficult to draw conclusions based on studies in patients. However, correlational analysis of age vs. MHC type IIb within CHF patients suggests that older CHF patients have fewer type IIb fibers [114]. Tikunov et al have also found a shift from fast to slow myosin light chain, tropomyosin, and troponin (C, I, and T) isoforms in CHF. Aging causes a decrease in the fast myosin light chain 3 isoform in limb muscle that contributes to slow shortening velocity [115], and this may also occur in the diaphragm. Overall, MHC and thin-filament protein adaptations will compromise diaphragm function during expulsive behaviors as fibers with slow myofibrillar protein isoforms have slower shortening velocity and lower peak power than fibers rich in fast isoforms.
6.5 - Diaphragm fiber Atrophy
Aging (sarcopenia) and CHF (cardiac cachexia) cause loss of muscle mass due to fiber atrophy. The diaphragm is highly susceptible to atrophy [5], and aging causes atrophy of type II fibers [14,69,116]. The effects of CHF on diaphragm fiber atrophy in animals seem dependent on animal model used (pressure- vs. volume overload), duration of CHF, and severity of disease. Pigs with CHF induced by supraventricular tachycardia have 20–40% lower cross-sectional area of type I, IIa, and IIB fibers [13]. In rats, CHF induced by myocardial infarction results in a 15% to 25% decrease in fiber cross-sectional area in some studies [11,117,97], but unchanged fiber cross-sectional area has been reported by our group [112] and others [75,111]. Similarly, there was no diaphragm atrophy in rats during late stages of CHF due to aortic stenosis [86]. The only study testing diaphragm atrophy in humans showed no change in fiber diameter for severe CHF (heart transplant) patients compared to controls, but participants had a wide age range (18 – 70 yrs) and were mostly males. We are currently working to define the effects of disease severity, age, and sex on diaphragm fiber atrophy in CHF. It is possible that diaphragm atrophy occurs in the early or mild-to-moderate stages of CHF that precede increased work of breathing. In the transition to severe CHF, the elevated work of breathing might restore fiber cross-sectional area to normal values. This pattern would mask fiber atrophy and the elevated catabolic state that is typical of severe CHF. Alternatively, the lack of diaphragm fiber hypertrophy with elevated work of breathing in CHF could reflect anabolic resistance [118]. Ultimately, diaphragm atrophy, when present, plays an important role in inspiratory dysfunction.
6.6 - Fatigue characteristics
Diaphragm abnormalities elicited by aging and CHF predispose the muscle to fatigue. Indeed, CHF accelerates isometric diaphragm fatigue in situ [77] and in vitro [82]. Conversely, in diaphragm of old animals, isometric fatigue resistance in vitro is unchanged or even increased [81,15]. It must be acknowledged that outcomes of isometric fatigue tested with standard protocols in vitro, in the presence of muscle weakness as occurs with aging and CHF, may not translate to the condition in vivo. Isometric fatigue is generally tested using matched stimulus frequency, i.e., mimicking a fixed phrenic motor neuron firing frequency and fully recruited motor units. Weakened muscles develop lower initial forces in matched-frequency protocols and, therefore, lower tension-time index that is a primary determinant of metabolic rate and fatigue in vitro [119]. Thus, it is common that weak muscles show attenuated rate of isometric fatigue in vitro [120–122,15,81]. However, the diaphragm force required to sustain breathing is unchanged (or even increased in CHF) in the presence of weakness. Therefore, a protocol using stimulus frequency adjusted to match initial specific force between conditions provides a better representation of the situation in vivo. The use of an ‘initial force-matched’ protocol reveals accelerated fatigue rate in the presence of diaphragm weakness [120]. Tests in patients show that CHF diminishes inspiratory muscle endurance, e.g., Fig 2. and refs [35,43]. Inspiratory endurance tests involve diaphragm shortening, and diaphragm function declines faster during repetitive shortening contractions [123,124]. Thus, isotonic fatigue properties of the diaphragm would be more relevant for in vivo function, and such measurements are still lacking in aging or CHF.
7. Inflammatory and neuroendocrine factors
7.1 - Renin-angiotensin system
The renin-angiotensin system is hyperactive in CHF and aging. Angiotensin II signaling is a hallmark of activation of the renin-angiotensin system. CHF raises systemic levels of angiotensin II, whereas aging promotes local activation of the renin-angiotensin system without necessarily elevating circulating angiotensin II levels [125]. Importantly, angiotensin II infusion in mice causes diaphragm atrophy [126]. A likely mechanism behind this atrophy is that angiotensin II stimulates reactive oxygen species (ROS) production by NAD(P)H oxidase and mitochondria [127,128]. This angiotensin II response is relevant because independent studies have implicated ROS as causative agents in diaphragm atrophy and contractile dysfunction [83,5,129].
While the direct effects of angiotensin II on diaphragm contractile function are unknown, antagonism of angiotensin II type I receptor prevents elevation in diaphragm ROS and loss of specific force with mechanical ventilation [130]. Blockade of the renin-angiotensin system with angiotensin-converting enzyme inhibitors prevents diaphragm weakness in CHF animals and patients [26,79]; however, inspiratory dysfunction is prevalent in CHF patients receiving angiotensin-converting enzyme inhibitors [35,40]. In fact, 89% of CHF patients with inspiratory dysfunction were on angiotensin-converting enzyme inhibitors [131]. General benefits of inhibition of the renin-angiotensin system are also seen in aging [125,132], but the effects on the diaphragm are unknown.
7.2 - Cytokines
Inflammatory cytokines are elevated in CHF and aging. Tumor necrosis factor-alpha (TNFα) and interleukin 6 (IL-6) have been considered putative circulating factors that cause diaphragm abnormalities in CHF. Injection of TNFα in vivo, exposure to TNFα in vitro, or cardiac-specific overexpression of TNFα cause a 15–20% loss of diaphragm specific force [133,134,121]. The effects of this cytokine on diaphragm abnormalities are especially notable because the loss of specific force in animals with cardiac-specific overexpression of TNFα occurs in the absence of atrophy [121]. Administration of IL-6, however, does not reduce specific force, but causes atrophy in diaphragm fiber types I, IIa, IIb [135]. Thus, results from studies in vivo and in vitro suggest TNFα and IL-6 signaling as triggers of loss of specific force and atrophy, respectively, in the diaphragm with CHF. The difficulty in focusing on cytokines for systemic treatment is that, to date, a plethora of clinical trials has yielded neutral or negative results due to the complex innate immune response and modulation of cardiac function in heart failure (reviewed in ref. [136]). Thence, it is important to understand the myocyte-specific mechanisms of dysfunction to facilitate the development of diaphragm-targeted therapies. Intracellular downstream effectors of TNFα and IL-6 include sphingolipid signaling, reactive oxygen species (ROS), and activation of proteolytic pathways that are discussed below.
8. Cellular mediators of diaphragm abnormalities in CHF
8.1 - Sphingolipid signaling
Sphingolipids act as second-messengers in several pathways. The enzyme sphingomyelinase generates ceramide and is a critical component of sphingolipid signaling. Cytokines and angiotensin II activate sphingomyelinase [137–139], and CHF causes a 20% increase in the activity of the neutral isoform of sphingomyelinase [82]. Accordingly, there is an accumulation of ceramide (↑20%) in the diaphragm of CHF animals [82]. Experiments in vitro and in vivo support neutral sphingomyelinase and ceramide as mediators of diaphragm abnormalities. Recombinant sphingomyelinase activates calpain and causes atrophy in C2C12 myotubes [140,141]. In diaphragm bundles, exposure to sphingomyelinase or ceramide in vitro mimics the effects of CHF: decreased specific force, calcium-sensitivity, and fatigue resistance by disrupting contractile apparatus function [120,142]. A recent study has shown that neutral sphingomyelinase activation plays a causative role in diaphragm weakness induced by sepsis [140], a condition that, like CHF, is characterized by heightened cytokine levels. Therefore, inhibition of neutral sphingomyelinase may be protective against diaphragm dysfunction in CHF. Diaphragm weakness stimulated by sphingomyelinase is mediated by reactive oxygen species from NAD(P)H oxidases and mitochondria as well as activation of calpain [143,144,142,120,140].
8.2 - Reactive oxygen species, NAD(P)H oxidase, and mitochondrial abnormalities
The accumulation of ROS causes redox imbalance leading to protein oxidation that triggers diaphragm atrophy and impairs contractile function [145,146,129]. Systemic redox imbalance in CHF worsens as the disease progresses and is highest in severe stages of the disease [147,148]. This pattern parallels the progression of diaphragm weakness [32,40,31]. Importantly, CHF heightens ROS emission in the diaphragm, and despite reports of increased diaphragm antioxidant enzyme activity in CHF [149], markers of oxidation are increased by CHF in the diaphragm [86,150].
The main sources of ROS in diaphragm are NAD(P)H oxidases and mitochondria [151–153]. CHF heightens diaphragm mRNA and protein levels of Nox2 subunits of NAD(P)H oxidase. Phosphorylation of the Nox2 subunit p47phox is a critical step for enzyme activation and ROS production [154,153], and CHF increases phosphorylation of p47phox in the diaphragm [83]. Mice deficient in p47phox, which lack Nox2 activity [155], are protected from CHF-induced increase in diaphragm ROS emission and impairments in isometric and isotonic contractile properties [83]. These observations suggest that p47phox is required for diaphragm abnormalities in CHF. Mitochondrial ROS emission is also elevated in CHF [112,150]. Systemic administration of mitochondria-targeted antioxidant blocks diaphragm mitochondrial ROS and normalizes diaphragm specific force in CHF [112]. Thus, mitochondrial ROS are a crucial component of the signaling pathway that culminates in diaphragm dysfunction in CHF. A plausible mechanism that reconciles findings in CHF is a cross-talk between Nox and mitochondria through ROS-induced ROS release [156,127,153,157].
In aging, mitochondria abnormalities are well-defined, and mitochondrial ROS have been implicated in aging-induced skeletal muscle dysfunction [158]. Aging increases mitochondrial DNA deletions in human diaphragm [159]. The pattern of mitochondrial DNA mutation is consistent with, and possibly a cause of, decreases in the activity of electron transport chain complexes I and IV found in the diaphragm of old rats [160]. Further evidence for a role of mitochondria in aging-induced skeletal muscle weakness comes from studies in transgenic mice. The overexpression of mitochondrial catalase in these mice prevented aging-induced increases in hydrogen peroxide emission and weakness in limb muscles [161]. The protection against contractile dysfunction conferred by mitochondrial catalase overexpression appears to be mediated through effects on the contractile apparatus and excitation-contraction coupling. An interesting factor that has emerged as a trigger of elevated skeletal muscle mitochondrial ROS in aging is denervation [158]. Fiber denervation heightens emission of mitochondrial ROS, which can affect the metabolic and contractile properties of innervated fibers surrounding the denervated one [162].
Proteins of the myofilament and excitation-contraction coupling are sensitive to ROS [163,164,161,165–167] such that exposure to exogenous ROS mimics the effects of CHF on muscle function by decreasing specific force [168,169], calcium sensitivity [167], and fatigue resistance [146,170–173]. The oxidant modification most often linked to contractile dysfunction is protein carbonylation. Protein carbonyls are increased in diaphragm homogenates in CHF [150] but not aging [174], and oxidation of MHC (CHF [86]) and RyR (aging limb muscle [93]) are associated with muscle weakness. Notably, carbonylation is an irreversible modification that, despite being a marker of redox imbalance and enhancing protein susceptibility to degradation [175], plays a lesser role in regulation of protein function. Instead, contractile function is modulated by oxidation of protein thiols and redox regulation of protein phosphorylation [176,177,166,167,102], and these are prominent candidate mechanisms of loss of diaphragm specific force and power with aging and CHF. Our research efforts are currently focused on exploring thiol oxidation as a molecular mechanism of diaphragm dysfunction in CHF and aging.
8.3 - Proteolytic pathways
Diaphragm atrophy and degradation of myofibrillar proteins requires activation of proteolytic pathways. All of the aforementioned pathways (cytokines, angiotensin II, sphingomyelinase, and ROS) activate proteolytic signaling in muscle [126,128,178,179,145]. Protein degradation is largely dependent on the ubiquitin-proteasome pathway [180]. In aging, the specific proteolytic pathways associated with diaphragm abnormalities are unclear. In CHF, markers of ubiquitin-proteasome pathway activation are elevated, and inhibition of the proteasome blunts diaphragm MHC degradation and loss of specific force [100]. These findings suggest that activation of the ubiquitin-proteasome pathway is an important component of diaphragm abnormalities in CHF. However, protein cleavage and dislodging from myofibrils precedes degradation of myofibrillar proteins by the proteasome [180]. These antecedent processes are performed at least partially by calpains [181], which are activated by calcium. CHF dysregulates calcium homeostasis in the diaphragm such that intracellular calcium concentration is elevated and calpain activity is increased two-fold [97]. Calpain activation, which causes diaphragm weakness in sepsis and mechanical ventilation [182,183], is a plausible process that mediates diaphragm weakness in CHF, but a cause-and-effect relationship has not been established.
9. Therapeutic strategies to counteract inspiratory dysfunction
9.1 - Endurance training
Endurance exercise training promotes several cardiovascular and muscular benefits in CHF and aging [184,185], including improvements in inspiratory muscle function in CHF [186]. Endurance training prevents the loss of diaphragm specific force in animals with CHF with preserved ejection fraction or after injection of TNF-α [187,149]. However, endurance training does not prevent morphological abnormalities of the neuromuscular junction in CHF [71]. Thus, it is unclear if the improvements in diaphragm function are a direct effect of endurance training on the diaphragm or a secondary response to modulation of upstream circulating factors that trigger diaphragm abnormalities. One important aspect to consider is that endurance training can cause atrophy in diaphragm fibers in healthy young and old rats [188,189]. These findings suggest that endurance training may be detrimental to inspiratory function during expulsive behaviors, which relies on recruitment of type IIx/b fibers [44]. Therefore, we propose that endurance training needs to be combined with adjuvant therapies targeting the diaphragm and inspiratory function to help patients obtain the greatest long-term benefits of rehabilitation.
9.2 - Inspiratory (‘muscle’) resistance training
Inspiratory muscle resistance training (IMT) has been increasingly recognized as an integral component of the clinical management of CHF patients [190]. This therapy can provide additional benefits for CHF patients beyond those associated with endurance training alone [190–192]. In CHF, IMT increases maximal inspiratory pressure and endurance [35,131,193], reduces sympathetic nerve activity [194,195], heightens limb muscle blood flow [131], and prolongs time to fatigue or performance during whole-body exercise [196,191,197,190]. The technical and clinical aspects of IMT in CHF have been reviewed in detail elsewhere [190]. Importantly, CHF patients who undergo IMT pre-surgery have fewer pulmonary complications post-surgery [199]. Recent studies are emerging that suggest potential benefits of inspiratory resistance training in healthy older adults as well [84,200]. In older subjects, inspiratory muscle training increases peak inspiratory flow [84]. These findings are consistent with the notion that IMT might improve diaphragm function during expulsive behaviors and would be beneficial for airway clearance. To our knowledge, there are no specific studies showing that IMT decreases the incidence of pneumonia in the elderly or CHF patients. These extensive investigations are difficult to perform due to the requirements for a large number of patients and prolonged duration of training. Nonetheless, these are important studies that need to be done.
The cellular and molecular bases of improved inspiratory function with IMT in the human diaphragm are unknown. Inspiratory muscle training increases diaphragm thickness in CHF patients and old subjects [131,200], suggesting fiber hypertrophy similar to that seen in healthy young rats undergoing a protocol that simulates IMT [201–203]. This approach also heightens diaphragm citrate synthase and cytochrome c oxidase activities in animals [204,205], suggesting greater mitochondrial volume density post-training. Neuromuscular adaptations are an important component of strength gains with resistance training in limb muscles. Therefore, the functional benefits of IMT in CHF and aging might result from diaphragm fiber hypertrophy along with metabolic and neuromuscular adaptations that have yet to be defined.
9.3 - Pharmacological agents
The existing knowledge of mechanisms of diaphragm weakness in CHF and aging support the use of drugs targeting proteolytic pathways or the myofilament. Bortezomib is a proteasome inhibitor that prevents protein degradation by the ubiquitin proteasome pathway. Systemic administration of bortezomib prevents loss of diaphragm MHC content and attenuates the decrease in maximal specific force in CHF rats [100]. A potential complication of ‘anti-atrophy’ agents for systemic use in CHF is an exacerbation of pathophysiological left ventricular remodeling and hypertrophy. Off-target effects on the left ventricle illustrate the need for isolating pathways and compounds specific to the diaphragm (or to skeletal muscles in general).
Other pharmaceutical agents combat the loss in diaphragmatic specific force by targeting myofibrillar proteins. In this regard, the calcium sensitizer levosimendan interacts with troponin C to increase calcium sensitivity. Exposure of diaphragm fibers (slow and fast isoforms) to levosimendan in vitro enhances calcium sensitivity in CHF animals [206]. Clinicians have been using levosimendan as a cardiac inotropic agent to treat acute or decompensated heart failure [207]. An off-label use of the drug could be the treatment of diaphragm dysfunction in patients with inspiratory dysfunction. New classes of calcium sensitizers have also been developed to target fast skeletal troponin C [208]. Human and animal diaphragm fibers exposed to fast troponin activators in vitro have increased calcium sensitivity, which translates into higher force generation within the physiological range of calcium concentrations [209,11]. In vitro treatment of intact diaphragm bundles from CHF rats with the fast troponin activator CK-2127107 increased submaximal diaphragm force to values equivalent to bundles from untreated control animals [11]. Diaphragm type II fibers are recruited mainly during expulsive behaviors [44,2,3]. Hence, fast troponin activators likely enhance inspiratory function during sneezing and coughing and might improve the patient’s ability to clear the airways.
Myosin activators are an alternative (or adjuvant) to troponin activators. A recent study showed that the myosin activator omecamtiv mecarbil increases calcium sensitivity of slow diaphragm fibers in healthy animals [210]. The efficacy of omecamtiv mecarbil to enhance diaphragm calcium sensitivity in pre-clinical models of CHF has not been tested. Early findings from clinical trials suggest that omecamtiv mecarbil diminishes dyspnea in CHF patients [211], and improved diaphragm function could contribute to the effects reported. Overall, inhibitors of proteolytic pathways and activators of myofibrillar protein function hold therapeutic potential for diaphragm dysfunction. Continued research on pathways upstream of proteolysis and post-translational modification of myofibrillar proteins will help elucidate new drug targets to treat diaphragm abnormalities in CHF.
10. Summary and conclusions
Inspiratory dysfunction occurs with aging and is accentuated by CHF. Diaphragm neuromuscular and intrinsic myocyte abnormalities play a major role in the inspiratory dysfunction caused by CHF. Thus, diaphragm abnormalities contribute to key aspects of cardiovascular and pulmonary pathophysiology in CHF and aging including: i) impaired airway clearance and predisposition to pneumonia; ii) inability to sustain ventilation during physical activity; iii) shallow breathing pattern that limits alveolar ventilation and gas exchange; and iv) sympathetic activation that causes cardiac arrhythmias and tissue vasoconstriction. Loss of neurotrophic factors and activation of sphingolipid signaling, reactive oxygen species, and proteolytic pathways dictate changes in excitation-contraction coupling as well as the quantity and quality of myofibrillar proteins that lead to isometric and isotonic contractile dysfunction. Endurance and inspiratory resistance training combined with calcium sensitizing agents are current treatment options for inspiratory dysfunction, but these have yet to be optimized. The development of novel therapies will depend on research to further define receptors involved and specific cellular pathways leading to dysfunction.
Figure 5. Circulating factors and intra-myocyte pathways leading to diaphragm abnormalities in heart failure.
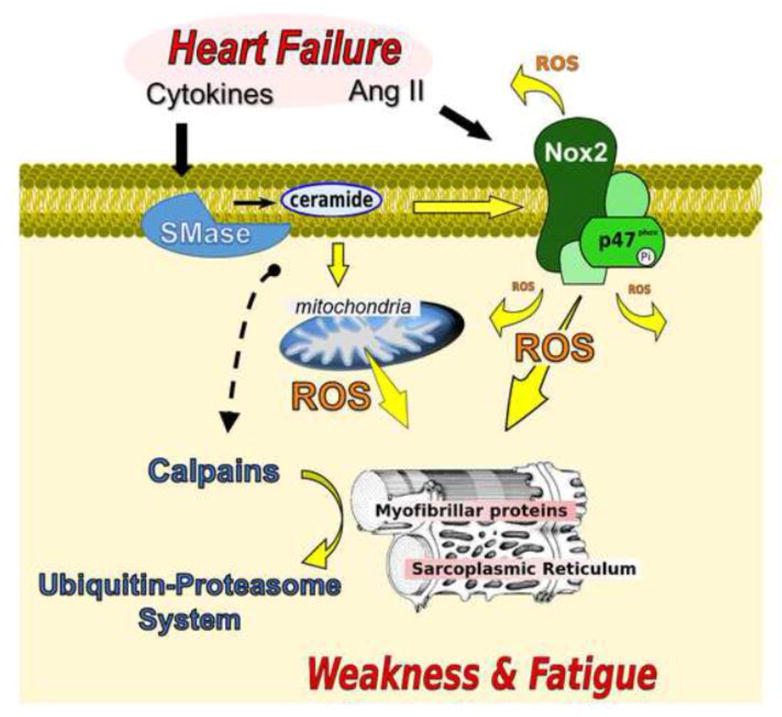
Angiotensin II (Ang II), sphingomyelinase (SMase), NAD(P)H oxidase 2 (Nox2), reactive oxygen species (ROS). Heart failure increases diaphragm neutral SMase activity and ceramide content [82]. SMase and ceramide cause diaphragm contractile dysfunction through ROS from mitochondria and Nox2 [142,144,143], and activation of calpain [140]. ROS play a causative role in diaphragm contractile dysfunction in heart failure [150,83,112]. Heart failure increases diaphragm calpain and proteasome activity [97,100], and proteasome inhibition blunts contractile dysfunction. Notably, ROS stimulates the ubiquitin-proteasome system [178,212].
Acknowledgments
Our research in this area is supported by grants from National Heart, Lung, and Blood Institute (R00-HL098453, R01-HL130318) and the American Heart Association (13GRNT17160000) to L. Ferreira. We would like to thank Jeremey Clark and Christine Coombes (Office of Communications, College of Health and Human Performance, University of Florida) for their assistance in preparation of illustrations for this article.
References
- 1.Elliott JE, Greising SM, Mantilla CB, Sieck GC. Functional impact of sarcopenia in respiratory muscles. Respir Physiol Neurobiol. 2015 doi: 10.1016/j.resp.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantilla CB, Seven YB, Zhan WZ, Sieck GC. Diaphragm motor unit recruitment in rats. Respir Physiol Neurobiol. 2010;173(1):101–106. doi: 10.1016/j.resp.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantilla CB, Sieck GC. Phrenic motor unit recruitment during ventilatory and non-ventilatory behaviors. Respir Physiol Neurobiol. 2011;179(1):57–63. doi: 10.1016/j.resp.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sieck GC, Ferreira LF, Reid MB, Mantilla CB. Mechanical properties of respiratory muscles. Compr Physiol. 2013;3(4):1553–1567. doi: 10.1002/cphy.c130003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powers SK, Wiggs MP, Sollanek KJ, Smuder AJ. Ventilator-induced diaphragm dysfunction: cause and effect. Am J Physiol Regul Integr Comp Physiol. 2013;305(5):R464–477. doi: 10.1152/ajpregu.00231.2013. ajpregu.00231.2013 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Callahan LA, Supinski GS. Sepsis-induced myopathy. Crit Care Med. 2009;37(10 Suppl):S354–367. doi: 10.1097/CCM.0b013e3181b6e439. 00003246-200910001-00010 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358(13):1327–1335. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- 8.Hooijman PE, Beishuizen A, Witt CC, de Waard MC, Girbes AR, Spoelstra-de Man AM, Niessen HW, Manders E, van Hees HW, van den Brom CE, Silderhuis V, Lawlor MW, Labeit S, Stienen GJ, Hartemink KJ, Paul MA, Heunks LM, Ottenheijm CA. Diaphragm muscle fiber weakness and ubiquitin-proteasome activation in critically ill patients. Am J Respir Crit Care Med. 2015;191(10):1126–1138. doi: 10.1164/rccm.201412-2214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindsay DC, Lovegrove CA, Dunn MJ, Bennett JG, Pepper JR, Yacoub MH, Poole-Wilson PA. Histological abnormalities of muscle from limb, thorax and diaphragm in chronic heart failure. Eur Heart J. 1996;17(8):1239–1250. doi: 10.1093/oxfordjournals.eurheartj.a015042. [DOI] [PubMed] [Google Scholar]
- 10.Hammond MD, Bauer KA, Sharp JT, Rocha RD. Respiratory muscle strength in congestive heart failure. Chest. 1990;98(5):1091–1094. doi: 10.1378/chest.98.5.1091. [DOI] [PubMed] [Google Scholar]
- 11.Hwee DT, Kennedy AR, Hartman JJ, Ryans J, Durham N, Malik FI, Jasper JR. The Small-Molecule Fast Skeletal Troponin Activator, CK-2127107, Improves Exercise Tolerance in a Rat Model of Heart Failure. J Pharmacol Exp Ther. 2015;353(1):159–168. doi: 10.1124/jpet.114.222224. jpet.114.222224 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Stassijns G, Lysens R, Decramer M. Peripheral and respiratory muscles in chronic heart failure. Eur Respir J. 1996;9(10):2161–2167. doi: 10.1183/09031936.96.09102161. [DOI] [PubMed] [Google Scholar]
- 13.Howell S, Maarek JM, Fournier M, Sullivan K, Zhan WZ, Sieck GC. Congestive heart failure: differential adaptation of the diaphragm and latissimus dorsi. J Appl Physiol. 1995;79(2):389–397. doi: 10.1152/jappl.1995.79.2.389. [DOI] [PubMed] [Google Scholar]
- 14.Greising SM, Mantilla CB, Gorman BA, Ermilov LG, Sieck GC. Diaphragm muscle sarcopenia in aging mice. Exp Gerontol. 2013;48(9):881–887. doi: 10.1016/j.exger.2013.06.001. S0531-5565(13)00202-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greising SM, Mantilla CB, Medina-Martinez JS, Stowe JM, Sieck GC. Functional impact of diaphragm muscle sarcopenia in both male and female mice. Am J Physiol Lung Cell Mol Physiol. 2015;309(1):L46–52. doi: 10.1152/ajplung.00064.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cacciani N, Ogilvie H, Larsson L. Age related differences in diaphragm muscle fiber response to mid/long term controlled mechanical ventilation. Exp Gerontol. 2014;59:28–33. doi: 10.1016/j.exger.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 17.ATS/ERS Statement on respiratory muscle testing. Am J Respir Crit Care Med. 2002;166(4):518–624. doi: 10.1164/rccm.166.4.518. [DOI] [PubMed] [Google Scholar]
- 18.Janssens JP. Aging of the respiratory system: impact on pulmonary function tests and adaptation to exertion. Clin Chest Med. 2005;26(3):469–484. vi–vii. doi: 10.1016/j.ccm.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Black LF, Hyatt RE. Maximal respiratory pressures: normal values and relationship to age and sex. Am Rev Respir Dis. 1969;99(5):696–702. doi: 10.1164/arrd.1969.99.5.696. [DOI] [PubMed] [Google Scholar]
- 20.Neder JA, Andreoni S, Lerario MC, Nery LE. Reference values for lung function tests. II. Maximal respiratory pressures and voluntary ventilation. Braz J Med Biol Res. 1999;32(6):719–727. doi: 10.1590/s0100-879x1999000600007. [DOI] [PubMed] [Google Scholar]
- 21.Enright PL, Adams AB, Boyle PJ, Sherrill DL. Spirometry and maximal respiratory pressure references from healthy Minnesota 65- to 85-year-old women and men. Chest. 1995;108(3):663–669. doi: 10.1378/chest.108.3.663. [DOI] [PubMed] [Google Scholar]
- 22.Enright PL, Kronmal RA, Manolio TA, Schenker MB, Hyatt RE. Respiratory muscle strength in the elderly. Correlates and reference values. Cardiovascular Health Study Research Group. Am J Respir Crit Care Med. 1994;149(2 Pt 1):430–438. doi: 10.1164/ajrccm.149.2.8306041. [DOI] [PubMed] [Google Scholar]
- 23.Tolep K, Higgins N, Muza S, Criner G, Kelsen SG. Comparison of diaphragm strength between healthy adult elderly and young men. Am J Respir Crit Care Med. 1995;152(2):677–682. doi: 10.1164/ajrccm.152.2.7633725. [DOI] [PubMed] [Google Scholar]
- 24.Polkey MI, Harris ML, Hughes PD, Hamnegard CH, Lyons D, Green M, Moxham J. The contractile properties of the elderly human diaphragm. Am J Respir Crit Care Med. 1997;155(5):1560–1564. doi: 10.1164/ajrccm.155.5.9154857. [DOI] [PubMed] [Google Scholar]
- 25.Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med. 2003;168(1):10–48. doi: 10.1164/rccm.2206020. 168/1/10 [pii] [DOI] [PubMed] [Google Scholar]
- 26.Coirault C, Hagege A, Chemla D, Fratacci MD, Guerot C, Lecarpentier Y. Angiotensin-converting enzyme inhibitor therapy improves respiratory muscle strength in patients with heart failure. Chest. 2001;119(6):1755–1760. doi: 10.1378/chest.119.6.1755. [DOI] [PubMed] [Google Scholar]
- 27.McParland C, Krishnan B, Wang Y, Gallagher CG. Inspiratory muscle weakness and dyspnea in chronic heart failure. Am Rev Respir Dis. 1992;146(2):467–472. doi: 10.1164/ajrccm/146.2.467. [DOI] [PubMed] [Google Scholar]
- 28.Carmo MM, Barbara C, Ferreira T, Branco J, Ferreira S, Rendas AB. Diaphragmatic function in patients with chronic left ventricular failure. Pathophysiology. 2001;8(1):55–60. doi: 10.1016/s0928-4680(01)00065-7. S0928468001000657 [pii] [DOI] [PubMed] [Google Scholar]
- 29.Evans SA, Watson L, Hawkins M, Cowley AJ, Johnston ID, Kinnear WJ. Respiratory muscle strength in chronic heart failure. Thorax. 1995;50(6):625–628. doi: 10.1136/thx.50.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witt C, Borges AC, Haake H, Reindl I, Kleber FX, Baumann G. Respiratory muscle weakness and normal ventilatory drive in dilative cardiomyopathy. Eur Heart J. 1997;18(8):1322–1328. doi: 10.1093/oxfordjournals.eurheartj.a015445. [DOI] [PubMed] [Google Scholar]
- 31.Ambrosino N, Opasich C, Crotti P, Cobelli F, Tavazzi L, Rampulla C. Breathing pattern, ventilatory drive and respiratory muscle strength in patients with chronic heart failure. Eur Respir J. 1994;7(1):17–22. doi: 10.1183/09031936.94.07010017. [DOI] [PubMed] [Google Scholar]
- 32.Filusch A, Ewert R, Altesellmeier M, Zugck C, Hetzer R, Borst MM, Katus HA, Meyer FJ. Respiratory muscle dysfunction in congestive heart failure--the role of pulmonary hypertension. Int J Cardiol. 2011;150(2):182–185. doi: 10.1016/j.ijcard.2010.04.006. S0167-5273(10)00221-4 [pii] [DOI] [PubMed] [Google Scholar]
- 33.Hughes PD, Polkey MI, Harrus ML, Coats AJ, Moxham J, Green M. Diaphragm strength in chronic heart failure. Am J Respir Crit Care Med. 1999;160(2):529–534. doi: 10.1164/ajrccm.160.2.9810081. [DOI] [PubMed] [Google Scholar]
- 34.Daganou M, Dimopoulou I, Alivizatos PA, Tzelepis GE. Pulmonary function and respiratory muscle strength in chronic heart failure: comparison between ischaemic and idiopathic dilated cardiomyopathy. Heart. 1999;81(6):618–620. doi: 10.1136/hrt.81.6.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dall’ago P, Chiappa GR, Guths H, Stein R, Ribeiro JP. Inspiratory muscle training in patients with heart failure and inspiratory muscle weakness: a randomized trial. J Am Coll Cardiol. 2006;47(4):757–763. doi: 10.1016/j.jacc.2005.09.052. [DOI] [PubMed] [Google Scholar]
- 36.Ribeiro JP, Chiappa GR, Neder JA, Frankenstein L. Respiratory muscle function and exercise intolerance in heart failure. Curr Heart Fail Rep. 2009;6(2):95–101. doi: 10.1007/s11897-009-0015-7. [DOI] [PubMed] [Google Scholar]
- 37.Tager T, Schell M, Cebola R, Frohlich H, Dosch A, Franke J, Katus HA, Wians FH, Jr, Frankenstein L. Biological variation, reference change value (RCV) and minimal important difference (MID) of inspiratory muscle strength (PImax) in patients with stable chronic heart failure. Clin Res Cardiol. 2015;104(10):822–830. doi: 10.1007/s00392-015-0850-3. [DOI] [PubMed] [Google Scholar]
- 38.Bosnak-Guclu M, Arikan H, Savci S, Inal-Ince D, Tulumen E, Aytemir K, Tokgozoglu L. Effects of inspiratory muscle training in patients with heart failure. Respir Med. 2011;105(11):1671–1681. doi: 10.1016/j.rmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Verissimo P, Casalaspo TJ, Goncalves LH, Yang AS, Eid RC, Timenetsky KT. High prevalence of respiratory muscle weakness in hospitalized acute heart failure elderly patients. PLoS One. 2015;10(2):e0118218. doi: 10.1371/journal.pone.0118218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kasahara Y, Izawa PK, Watanabe S, Osada N, Omiya K. The Relation of Respiratory Muscle Strength to Disease Severity and Abnormal Ventilation During Exercise in Chronic Heart Failure Patients. Research in Cardiovascular Medicine. 2015;4(4):e228944. doi: 10.5812/cardiovascmed.28944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hart N, Kearney MT, Pride NB, Green M, Lofaso F, Shah AM, Moxham J, Polkey MI. Inspiratory muscle load and capacity in chronic heart failure. Thorax. 2004;59(6):477–482. doi: 10.1136/thx.2003.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mancini DM, LaManca JJ, Donchez LJ, Levine S, Henson DJ. Diminished respiratory muscle endurance persists after cardiac transplantation. Am J Cardiol. 1995;75(5):418–421. doi: 10.1016/s0002-9149(99)80571-2. S0002914999805712 [pii] [DOI] [PubMed] [Google Scholar]
- 43.Mancini DM. Pulmonary factors limiting exercise capacity in patients with heart failure. Prog Cardiovasc Dis. 1995;37(6):347–370. doi: 10.1016/s0033-0620(05)80018-0. [DOI] [PubMed] [Google Scholar]
- 44.Sieck GC, Fournier M. Diaphragm motor unit recruitment during ventilatory and nonventilatory behaviors. J Appl Physiol (1985) 1989;66(6):2539–2545. doi: 10.1152/jappl.1989.66.6.2539. [DOI] [PubMed] [Google Scholar]
- 45.Mantilla CB, Sieck GC. Impact of diaphragm muscle fiber atrophy on neuromotor control. Respir Physiol Neurobiol. 2013;189(2):411–418. doi: 10.1016/j.resp.2013.06.025. S1569-9048(13)00231-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mancini DM, Henson D, LaManca J, Levine S. Respiratory muscle function and dyspnea in patients with chronic congestive heart failure. Circulation. 1992;86(3):909–918. doi: 10.1161/01.cir.86.3.909. [DOI] [PubMed] [Google Scholar]
- 47.Manning HL, Schwartzstein RM. Pathophysiology of dyspnea. N Engl J Med. 1995;333(23):1547–1553. doi: 10.1056/NEJM199512073332307. [DOI] [PubMed] [Google Scholar]
- 48.Woods PR, Olson TP, Frantz RP, Johnson BD. Causes of breathing inefficiency during exercise in heart failure. J Card Fail. 2010;16(10):835–842. doi: 10.1016/j.cardfail.2010.05.003. S1071-9164(10)00209-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark AL, Chua TP, Coats AJ. Anatomical dead space, ventilatory pattern, and exercise capacity in chronic heart failure. Br Heart J. 1995;74(4):377–380. doi: 10.1136/hrt.74.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokoyama H, Sato H, Hori M, Takeda H, Kamada T. A characteristic change in ventilation mode during exertional dyspnea in patients with chronic heart failure. Chest. 1994;106(4):1007–1013. doi: 10.1378/chest.106.4.1007. [DOI] [PubMed] [Google Scholar]
- 51.Ponikowski P, Francis DP, Piepoli MF, Davies LC, Chua TP, Davos CH, Florea V, Banasiak W, Poole-Wilson PA, Coats AJ, Anker SD. Enhanced ventilatory response to exercise in patients with chronic heart failure and preserved exercise tolerance: marker of abnormal cardiorespiratory reflex control and predictor of poor prognosis. Circulation. 2001;103(7):967–972. doi: 10.1161/01.cir.103.7.967. [DOI] [PubMed] [Google Scholar]
- 52.Baekey DM, Molkov YI, Paton JF, Rybak IA, Dick TE. Effect of baroreceptor stimulation on the respiratory pattern: insights into respiratory-sympathetic interactions. Respir Physiol Neurobiol. 2010;174(1–2):135–145. doi: 10.1016/j.resp.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dempsey JA, Romer L, Rodman J, Miller J, Smith C. Consequences of exercise-induced respiratory muscle work. Respir Physiol Neurobiol. 2006;151(2–3):242–250. doi: 10.1016/j.resp.2005.12.015. S1569-9048(06)00109-1 [pii] [DOI] [PubMed] [Google Scholar]
- 54.Hill JM. Discharge of group IV phrenic afferent fibers increases during diaphragmatic fatigue. Brain Res. 2000;856(1–2):240–244. doi: 10.1016/s0006-8993(99)02366-5. [DOI] [PubMed] [Google Scholar]
- 55.Del Rio R, Marcus NJ, Schultz HD. Carotid chemoreceptor ablation improves survival in heart failure: rescuing autonomic control of cardiorespiratory function. J Am Coll Cardiol. 2013;62(25):2422–2430. doi: 10.1016/j.jacc.2013.07.079. S0735-1097(13)04006-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller JD, Smith CA, Hemauer SJ, Dempsey JA. The effects of inspiratory intrathoracic pressure production on the cardiovascular response to submaximal exercise in health and chronic heart failure. Am J Physiol Heart Circ Physiol. 2007;292(1):H580–592. doi: 10.1152/ajpheart.00211.2006. 00211.2006 [pii] [DOI] [PubMed] [Google Scholar]
- 57.Olson TP, Joyner MJ, Dietz NM, Eisenach JH, Curry TB, Johnson BD. Effects of respiratory muscle work on blood flow distribution during exercise in heart failure. J Physiol. 2010;588(Pt 13):2487–2501. doi: 10.1113/jphysiol.2009.186056. jphysiol.2009.186056 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mancini D, Donchez L, Levine S. Acute unloading of the work of breathing extends exercise duration in patients with heart failure. J Am Coll Cardiol. 1997;29(3):590–596. doi: 10.1016/s0735-1097(96)00556-6. S0735109796005566 [pii] [DOI] [PubMed] [Google Scholar]
- 59.O’Donnell DE, D’Arsigny C, Raj S, Abdollah H, Webb KA. Ventilatory assistance improves exercise endurance in stable congestive heart failure. Am J Respir Crit Care Med. 1999;160(6):1804–1811. doi: 10.1164/ajrccm.160.6.9808134. [DOI] [PubMed] [Google Scholar]
- 60.Morimoto K, Suzuki M, Ishifuji T, Yaegashi M, Asoh N, Hamashige N, Abe M, Aoshima M, Ariyoshi K Adult Pneumonia Study G-J. The burden and etiology of community-onset pneumonia in the aging Japanese population: a multicenter prospective study. PLoS One. 2015;10(3):e0122247. doi: 10.1371/journal.pone.0122247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Welte T, Torres A, Nathwani D. Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax. 2012;67(1):71–79. doi: 10.1136/thx.2009.129502. [DOI] [PubMed] [Google Scholar]
- 62.Wroe PC, Finkelstein JA, Ray GT, Linder JA, Johnson KM, Rifas-Shiman S, Moore MR, Huang SS. Aging population and future burden of pneumococcal pneumonia in the United States. J Infect Dis. 2012;205(10):1589–1592. doi: 10.1093/infdis/jis240. [DOI] [PubMed] [Google Scholar]
- 63.Mor A, Thomsen RW, Ulrichsen SP, Sorensen HT. Chronic heart failure and risk of hospitalization with pneumonia: a population-based study. Eur J Intern Med. 2013;24(4):349–353. doi: 10.1016/j.ejim.2013.02.013. S0953-6205(13)00079-4 [pii] [DOI] [PubMed] [Google Scholar]
- 64.Jackson ML, Neuzil KM, Thompson WW, Shay DK, Yu O, Hanson CA, Jackson LA. The burden of community-acquired pneumonia in seniors: results of a population-based study. Clin Infect Dis. 2004;39(11):1642–1650. doi: 10.1086/425615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer FJ, Borst MM, Zugck C, Kirschke A, Schellberg D, Kubler W, Haass M. Respiratory muscle dysfunction in congestive heart failure: clinical correlation and prognostic significance. Circulation. 2001;103(17):2153–2158. doi: 10.1161/01.cir.103.17.2153. [DOI] [PubMed] [Google Scholar]
- 66.Torchio R, Gulotta C, Greco-Lucchina P, Perboni A, Montagna L, Guglielmo M, Milic-Emili J. Closing capacity and gas exchange in chronic heart failure. Chest. 2006;129(5):1330–1336. doi: 10.1378/chest.129.5.1330. [DOI] [PubMed] [Google Scholar]
- 67.Cross TJ, Sabapathy S, Beck KC, Morris NR, Johnson BD. The resistive and elastic work of breathing during exercise in patients with chronic heart failure. Eur Respir J. 2012;39(6):1449–1457. doi: 10.1183/09031936.00125011. 09031936.00125011 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agostoni P, Pellegrino R, Conca C, Rodarte JR, Brusasco V. Exercise hyperpnea in chronic heart failure: relationships to lung stiffness and expiratory flow limitation. J Appl Physiol (1985) 2002;92(4):1409–1416. doi: 10.1152/japplphysiol.00724.2001. [DOI] [PubMed] [Google Scholar]
- 69.Prakash YS, Sieck GC. Age-related remodeling of neuromuscular junctions on type-identified diaphragm fibers. Muscle Nerve. 1998;21(7):887–895. doi: 10.1002/(sici)1097-4598(199807)21:7<887::aid-mus6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 70.Cardasis CA, LaFontaine DM. Aging rat neuromuscular junctions: a morphometric study of cholinesterase-stained whole mounts and ultrastructure. Muscle Nerve. 1987;10(3):200–213. doi: 10.1002/mus.880100303. [DOI] [PubMed] [Google Scholar]
- 71.de Souza PA, de Souza RW, Soares LC, Piedade WP, Campos DH, Carvalho RF, Padovani CR, Okoshi K, Cicogna AC, Matheus SM, Dal-Pai-Silva M. Aerobic training attenuates nicotinic acethylcholine receptor changes in the diaphragm muscle during heart failure. Histol Histopathol. 2015;30(7):801–811. doi: 10.14670/HH-11-581. [DOI] [PubMed] [Google Scholar]
- 72.Wu P, Chawla A, Spinner RJ, Yu C, Yaszemski MJ, Windebank AJ, Wang H. Key changes in denervated muscles and their impact on regeneration and reinnervation. Neural Regen Res. 2014;9(20):1796–1809. doi: 10.4103/1673-5374.143424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adams L, Carlson BM, Henderson L, Goldman D. Adaptation of nicotinic acetylcholine receptor, myogenin, and MRF4 gene expression to long-term muscle denervation. J Cell Biol. 1995;131(5):1341–1349. doi: 10.1083/jcb.131.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Hees HW, van der Heijden HF, Hafmans T, Ennen L, Heunks LM, Verheugt FW, Dekhuijzen PN. Impaired isotonic contractility and structural abnormalities in the diaphragm of congestive heart failure rats. Int J Cardiol. 2008;128(3):326–335. doi: 10.1016/j.ijcard.2007.06.080. S0167-5273(07)01103-5 [pii] [DOI] [PubMed] [Google Scholar]
- 75.van Hees HW, van der Heijden HF, Ottenheijm CA, Heunks LM, Pigmans CJ, Verheugt FW, Brouwer RM, Dekhuijzen PN. Diaphragm single-fiber weakness and loss of myosin in congestive heart failure rats. Am J Physiol Heart Circ Physiol. 2007;293(1):H819–828. doi: 10.1152/ajpheart.00085.2007. 00085.2007 [pii] [DOI] [PubMed] [Google Scholar]
- 76.Stassijns G, Gayan-Ramirez G, De Leyn P, de Bock V, Dom R, Lysens R, Decramer M. Effects of dilated cardiomyopathy on the diaphragm in the Syrian hamster. Eur Respir J. 1999;13(2):391–397. doi: 10.1183/09031936.99.13239199. [DOI] [PubMed] [Google Scholar]
- 77.Supinski G, DiMarco A, Dibner-Dunlap M. Alterations in diaphragm strength and fatiguability in congestive heart failure. J Appl Physiol. 1994;76(6):2707–2713. doi: 10.1152/jappl.1994.76.6.2707. [DOI] [PubMed] [Google Scholar]
- 78.Lecarpentier Y, Chemla D, Blanc FX, Pourny JC, Joseph T, Riou B, Coirault C. Mechanics, energetics, and crossbridge kinetics of rabbit diaphragm during congestive heart failure. FASEB J. 1998;12(11):981–989. doi: 10.1096/fasebj.12.11.981. [DOI] [PubMed] [Google Scholar]
- 79.Coirault C, Langeron O, Lambert F, Blanc FX, Lerebours G, Claude N, Riou B, Chemla D, Lecarpentier Y. Impaired skeletal muscle performance in the early stage of cardiac pressure overload in rabbits: beneficial effects of angiotensin-converting enzyme inhibition. J Pharmacol Exp Ther. 1999;291(1):70–75. [PubMed] [Google Scholar]
- 80.Criswell DS, Powers SK, Herb RA, Dodd SL. Mechanism of specific force deficit in the senescent rat diaphragm. Respir Physiol. 1997;107(2):149–155. doi: 10.1016/s0034-5687(96)02509-1. [DOI] [PubMed] [Google Scholar]
- 81.Gosselin LE, Johnson BD, Sieck GC. Age-related changes in diaphragm muscle contractile properties and myosin heavy chain isoforms. Am J Respir Crit Care Med. 1994;150(1):174–178. doi: 10.1164/ajrccm.150.1.8025746. [DOI] [PubMed] [Google Scholar]
- 82.Empinado HM, Deevska GM, Nikolova-Karakashian M, Yoo JK, Christou DD, Ferreira LF. Diaphragm dysfunction in heart failure is accompanied by increases in neutral sphingomyelinase activity and ceramide content. Eur J Heart Fail. 2014;16(5):519–525. doi: 10.1002/ejhf.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ahn B, Beharry AW, Frye GS, Judge AR, Ferreira LF. NAD(P)H oxidase subunit p47phox is elevated, and p47phox knockout prevents diaphragm contractile dysfunction in heart failure. Am J Physiol Lung Cell Mol Physiol. 2015;309(5):L497–505. doi: 10.1152/ajplung.00176.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mills DE, Johnson MA, Barnett YA, Smith WH, Sharpe GR. The effects of inspiratory muscle training in older adults. Med Sci Sports Exerc. 2015;47(4):691–697. doi: 10.1249/MSS.0000000000000474. [DOI] [PubMed] [Google Scholar]
- 85.Romer LM, McConnell AK. Specificity and reversibility of inspiratory muscle training. Med Sci Sports Exerc. 2003;35(2):237–244. doi: 10.1249/01.MSS.0000048642.58419.1E. [DOI] [PubMed] [Google Scholar]
- 86.Coirault C, Guellich A, Barbry T, Samuel JL, Riou B, Lecarpentier Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am J Physiol Heart CircPhysiol. 2007;292(2):H1009–H1017. doi: 10.1152/ajpheart.00438.2006. [DOI] [PubMed] [Google Scholar]
- 87.Lynch GS, Rafael JA, Hinkle RT, Cole NM, Chamberlain JS, Faulkner JA. Contractile properties of diaphragm muscle segments from old mdx and old transgenic mdx mice. Am J Physiol. 1997;272(6 Pt 1):C2063–2068. doi: 10.1152/ajpcell.1997.272.6.C2063. [DOI] [PubMed] [Google Scholar]
- 88.Powers SK, Criswell D, Herb RA, Demirel H, Dodd S. Age-related increases in diaphragmatic maximal shortening velocity. J Appl Physiol (1985) 1996;80(2):445–451. doi: 10.1152/jappl.1996.80.2.445. [DOI] [PubMed] [Google Scholar]
- 89.Zhang YL, Kelsen SG. Effects of aging on diaphragm contractile function in golden hamsters. Am Rev Respir Dis. 1990;142(6 Pt 1):1396–1401. doi: 10.1164/ajrccm/142.6_Pt_1.1396. [DOI] [PubMed] [Google Scholar]
- 90.Ferreira LF, McDonagh B, Kelley RC, Coblentz PD, Patel N. Aging-induced impairments in diaphragm isotonic contractile properties and modifications of proteomic and sphingolipid profile. FASEB J. 2016;30(4):A111. [Google Scholar]
- 91.Graber TG, Kim JH, Grange RW, McLoon LK, Thompson LV. C57BL/6 life span study: age-related declines in muscle power production and contractile velocity. Age (Dordr) 2015;37(3):9773. doi: 10.1007/s11357-015-9773-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14(2):196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci U S A. 2005;102(27):9607–9612. doi: 10.1073/pnas.0500353102. 0500353102 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol. 2003;160(6):919–928. doi: 10.1083/jcb.200211012. jcb.200211012 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rullman E, Andersson DC, Melin M, Reiken S, Mancini DM, Marks AR, Lund LH, Gustafsson T. Modifications of skeletal muscle ryanodine receptor type 1 and exercise intolerance in heart failure. J Heart Lung Transplant. 2013;32(9):925–929. doi: 10.1016/j.healun.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dominguez JF, Howell S. Compartmental analysis of steady-state diaphragm Ca2+ kinetics in chronic congestive heart failure. Cell Calcium. 2003;33(3):163–174. doi: 10.1016/s0143-4160(02)00208-7. S0143416002002087 [pii] [DOI] [PubMed] [Google Scholar]
- 98.MacFarlane NG, Darnley GM, Smith GL. Cellular basis for contractile dysfunction in the diaphragm from a rabbit infarct model of heart failure. Am J Physiol Cell Physiol. 2000;278(4):C739–746. doi: 10.1152/ajpcell.2000.278.4.C739. [DOI] [PubMed] [Google Scholar]
- 99.Peters DG, Mitchell HL, McCune SA, Park S, Williams JH, Kandarian SC. Skeletal muscle sarcoplasmic reticulum Ca(2+)-ATPase gene expression in congestive heart failure. Circ Res. 1997;81(5):703–710. doi: 10.1161/01.res.81.5.703. [DOI] [PubMed] [Google Scholar]
- 100.van Hees HW, Li YP, Ottenheijm CA, Jin B, Pigmans CJ, Linkels M, Dekhuijzen PN, Heunks LM. Proteasome inhibition improves diaphragm function in congestive heart failure rats. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1260–1268. doi: 10.1152/ajplung.00035.2008. 00035.2008 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80(2):853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 102.Perkins WJ, Han YS, Sieck GC. Skeletal muscle force and actomyosin ATPase activity reduced by nitric oxide donor. J Appl Physiol. 1997;83(4):1326–1332. doi: 10.1152/jappl.1997.83.4.1326. [DOI] [PubMed] [Google Scholar]
- 103.Coirault C, Chemla D, Pourny JC, Lambert F, Lecarpentier Y. Instantaneous force-velocity-length relationship in diaphragmatic sarcomere. J Appl Physiol. 1997;82(2):404–412. doi: 10.1152/jappl.1997.82.2.404. [DOI] [PubMed] [Google Scholar]
- 104.van Hees HW, Ottenheijm CA, Granzier HL, Dekhuijzen PN, Heunks LM. Heart failure decreases passive tension generation of rat diaphragm fibers. Int J Cardiol. 2010;141(3):275–283. doi: 10.1016/j.ijcard.2008.12.042. S0167-5273(08)01421-6 [pii] [DOI] [PubMed] [Google Scholar]
- 105.Irving T, Wu Y, Bekyarova T, Farman GP, Fukuda N, Granzier H. Thick-filament strain and interfilament spacing in passive muscle: effect of titin-based passive tension. Biophys J. 2011;100(6):1499–1508. doi: 10.1016/j.bpj.2011.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Udaka J, Ohmori S, Terui T, Ohtsuki I, Ishiwata S, Kurihara S, Fukuda N. Disuse-induced preferential loss of the giant protein titin depresses muscle performance via abnormal sarcomeric organization. J Gen Physiol. 2008;131(1):33–41. doi: 10.1085/jgp.200709888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thompson LV. Age-related decline in actomyosin structure and function. In: Lynch GS, editor. Sarcopenia - Age Related Muscle Wasting and Weakness. Springer; New York, NY: 2011. pp. 75–111. [DOI] [Google Scholar]
- 108.Szentesi P, Bekedam MA, van Beek-Harmsen BJ, van der Laarse WJ, Zaremba R, Boonstra A, Visser FC, Stienen GJ. Depression of force production and ATPase activity in different types of human skeletal muscle fibers from patients with chronic heart failure. J Appl Physiol. 2005;99(6):2189–2195. doi: 10.1152/japplphysiol.00542.2005. [DOI] [PubMed] [Google Scholar]
- 109.Eddinger TJ, Moss RL, Cassens RG. Fiber number and type composition in extensor digitorum longus, soleus, and diaphragm muscles with aging in Fisher 344 rats. J Histochem Cytochem. 1985;33(10):1033–1041. doi: 10.1177/33.10.2931475. [DOI] [PubMed] [Google Scholar]
- 110.De Sousa E, Veksler V, Bigard X, Mateo P, Serrurier B, Ventura-Clapier R. Dual influence of disease and increased load on diaphragm muscle in heart failure. J Mol Cell Cardiol. 2001;33(4):699–710. doi: 10.1006/jmcc.2000.1336. S0022-2828(00)91336-1 [pii] [DOI] [PubMed] [Google Scholar]
- 111.Lima AR, Martinez PF, Damatto RL, Cezar MD, Guizoni DM, Bonomo C, Oliveira SA, Jr, Dal-Pai Silva M, Zornoff LA, Okoshi K, Okoshi MP. Heart failure-induced diaphragm myopathy. Cell Physiol Biochem. 2014;34(2):333–345. doi: 10.1159/000363003. [DOI] [PubMed] [Google Scholar]
- 112.Ferreira LF, Coblentz P, BANP, Yoo JK, Christou DD. Mitochondria-targeted antioxidant treatment prevents elevation in diaphragm mitochondrial reactive oxygen species and weakness in chronic heart failure. FASEB J. 2015;29:812. [Google Scholar]
- 113.Toth MJ, Palmer BM, LeWinter MM. Effect of heart failure on skeletal muscle myofibrillar protein content, isoform expression and calcium sensitivity. Int J Cardiol. 2006;107(2):211–219. doi: 10.1016/j.ijcard.2005.03.024. S0167-5273(05)00568-1 [pii] [DOI] [PubMed] [Google Scholar]
- 114.Tikunov B, Levine S, Mancini D. Chronic congestive heart failure elicits adaptations of endurance exercise in diaphragmatic muscle. Circulation. 1997;95(4):910–916. doi: 10.1161/01.cir.95.4.910. [DOI] [PubMed] [Google Scholar]
- 115.Kim JH, Torgerud WS, Mosser KH, Hirai H, Watanabe S, Asakura A, Thompson LV. Myosin light chain 3f attenuates age-induced decline in contractile velocity in MHC type II single muscle fibers. Aging Cell. 2012;11(2):203–212. doi: 10.1111/j.1474-9726.2011.00774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gosselin LE, Betlach M, Vailas AC, Thomas DP. Training-induced alterations in young and senescent rat diaphragm muscle. J Appl Physiol (1985) 1992;72(4):1506–1511. doi: 10.1152/jappl.1992.72.4.1506. [DOI] [PubMed] [Google Scholar]
- 117.Stassijns G, Gayan-Ramirez G, De Leyn P, Verhoeven G, Herijgers P, de Bock V, Dom R, Lysens R, Decramer M. Systolic ventricular dysfunction causes selective diaphragm atrophy in rats. Am J Respir Crit Care Med. 1998;158(6):1963–1967. doi: 10.1164/ajrccm.158.6.9710028. [DOI] [PubMed] [Google Scholar]
- 118.Jankowska EA, Biel B, Majda J, Szklarska A, Lopuszanska M, Medras M, Anker SD, Banasiak W, Poole-Wilson PA, Ponikowski P. Anabolic deficiency in men with chronic heart failure: prevalence and detrimental impact on survival. Circulation. 2006;114(17):1829–1837. doi: 10.1161/CIRCULATIONAHA.106.649426. [DOI] [PubMed] [Google Scholar]
- 119.Klawitter PF, Clanton TL. Tension-time index, fatigue, and energetics in isolated rat diaphragm: a new experimental model. J Appl Physiol. 2004;96(1):89–95. doi: 10.1152/japplphysiol.00237.2003. 00237.2003 [pii] [DOI] [PubMed] [Google Scholar]
- 120.Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova-Karakashian M, Reid MB. Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol. 2010;299(3):C552–560. doi: 10.1152/ajpcell.00065.2010. ajpcell.00065.2010 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li X, Moody MR, Engel D, Walker S, Clubb FJ, Jr, Sivasubramanian N, Mann DL, Reid MB. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102(14):1690–1696. doi: 10.1161/01.cir.102.14.1690. [DOI] [PubMed] [Google Scholar]
- 122.Greising SM, Ermilov LG, Sieck GC, Mantilla CB. Ageing and neurotrophic signalling effects on diaphragm neuromuscular function. J Physiol. 2015;593(2):431–440. doi: 10.1113/jphysiol.2014.282244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seow CY, Stephens NL. Fatigue of mouse diaphragm muscle in isometric and isotonic contractions. J Appl Physiol (1985) 1988;64(6):2388–2393. doi: 10.1152/jappl.1988.64.6.2388. [DOI] [PubMed] [Google Scholar]
- 124.Zhan WZ, Watchko JF, Prakash YS, Sieck GC. Isotonic contractile and fatigue properties of developing rat diaphragm muscle. J Appl Physiol (1985) 1998;84(4):1260–1268. doi: 10.1152/jappl.1998.84.4.1260. [DOI] [PubMed] [Google Scholar]
- 125.Conti S, Cassis P, Benigni A. Aging and the renin-angiotensin system. Hypertension. 2012;60(4):878–883. doi: 10.1161/HYPERTENSIONAHA.110.155895. [DOI] [PubMed] [Google Scholar]
- 126.Rezk BM, Yoshida T, Semprun-Prieto L, Higashi Y, Sukhanov S, Delafontaine P. Angiotensin II infusion induces marked diaphragmatic skeletal muscle atrophy. PLoS One. 2012;7(1):e30276. doi: 10.1371/journal.pone.0030276. PONE-D-11-19752 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. S0891-5849(11)00427-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Semprun-Prieto LC, Sukhanov S, Yoshida T, Rezk BM, Gonzalez-Villalobos RA, Vaughn C, Michael Tabony A, Delafontaine P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun. 2011;409(2):217–221. doi: 10.1016/j.bbrc.2011.04.122. S0006-291X(11)00729-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Reid MB, Moylan JS. Beyond atrophy: redox mechanisms of muscle dysfunction in chronic inflammatory disease. J Physiol. 2011;589(Pt 9):2171–2179. doi: 10.1113/jphysiol.2010.203356. jphysiol.2010.203356 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kwon OS, Smuder AJ, Wiggs MP, Hall SE, Sollanek KJ, Morton AB, Talbert EE, Toklu HZ, Tumer N, Powers SK. AT1 receptor blocker losartan protects against mechanical ventilation-induced diaphragmatic dysfunction. J Appl Physiol (1985) 2015 doi: 10.1152/japplphysiol.00237.2015. jap 00237 02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chiappa GR, Roseguini BT, Vieira PJ, Alves CN, Tavares A, Winkelmann ER, Ferlin EL, Stein R, Ribeiro JP. Inspiratory muscle training improves blood flow to resting and exercising limbs in patients with chronic heart failure. J Am Coll Cardiol. 2008;51(17):1663–1671. doi: 10.1016/j.jacc.2007.12.045. S0735-1097(08)00567-6 [pii] [DOI] [PubMed] [Google Scholar]
- 132.de Cavanagh EM, Inserra F, Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011;89(1):31–40. doi: 10.1093/cvr/cvq285. [DOI] [PubMed] [Google Scholar]
- 133.Hardin BJ, Campbell KS, Smith JD, Arbogast S, Smith J, Moylan JS, Reid MB. TNF-alpha acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J Appl Physiol. 2008;104(3):694–699. doi: 10.1152/japplphysiol.00898.2007. 00898.2007 [pii] [DOI] [PubMed] [Google Scholar]
- 134.Stasko SA, Hardin BJ, Smith JD, Moylan JS, Reid MB. TNF signals via neuronal-type nitric oxide synthase and reactive oxygen species to depress specific force of skeletal muscle. J Appl Physiol (1985) 2013;114(11):1629–1636. doi: 10.1152/japplphysiol.00871.2012. japplphysiol.00871.2012 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Janssen SP, Gayan-Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, Decramer M. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation. 2005;111(8):996–1005. doi: 10.1161/01.CIR.0000156469.96135.0D. [DOI] [PubMed] [Google Scholar]
- 136.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–1268. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–150. doi: 10.1038/nrm2329. nrm2329 [pii] [DOI] [PubMed] [Google Scholar]
- 138.Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82(1):27–44. doi: 10.1139/o03-091. o03-091 [pii] [DOI] [PubMed] [Google Scholar]
- 139.Berry C, Touyz R, Dominiczak AF, Webb RC, Johns DG. Angiotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide. Am J Physiol Heart Circ Physiol. 2001;281(6):H2337–2365. doi: 10.1152/ajpheart.2001.281.6.H2337. [DOI] [PubMed] [Google Scholar]
- 140.Supinski GS, Alimov AP, Wang L, Song XH, Callahan LA. Neutral sphingomyelinase 2 is required for cytokine-induced skeletal muscle calpain activation. Am J Physiol Lung Cell Mol Physiol. 2015;309(6):L614–624. doi: 10.1152/ajplung.00141.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schwagerl PJ, Talbert EE, Nguyen LM, Powers SK, Ferreira LF. Sphingomyelinase promotes atrophy in C2C12 myotubes. FASEB Journal. 2011;25:LB602. [Google Scholar]
- 142.Ferreira LF, Moylan JS, Stasko S, Smith JD, Campbell KS, Reid MB. Sphingomyelinase depresses force and calcium sensitivity of the contractile apparatus in mouse diaphragm muscle fibers. J Appl Physiol. 2012;112(9):1538–1545. doi: 10.1152/japplphysiol.01269.2011. japplphysiol.01269.2011 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Loehr JA, Abo-Zahrah R, Pal R, Rodney GG. Sphingomyelinase promotes oxidant production and skeletal muscle contractile dysfunction through activation of NADPH oxidase. Front Physiol. 2014;5:530. doi: 10.3389/fphys.2014.00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bost ER, Frye GS, Ahn B, Ferreira LF. Diaphragm dysfunction caused by sphingomyelinase requires the p47(phox) subunit of NADPH oxidase. Respir Physiol Neurobiol. 2015;205:47–52. doi: 10.1016/j.resp.2014.10.011. S1569-9048(14)00283-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Powers SK, Ji LL, Kavazis AN, Jackson MJ. Reactive oxygen species: impact on skeletal muscle. Compr Physiol. 2011;1(2):941–969. doi: 10.1002/cphy.c100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ferreira LF, Reid MB. Muscle-derived ROS and thiol regulation in muscle fatigue. J Appl Physiol. 2008;104(3):853–860. doi: 10.1152/japplphysiol.00953.2007. [DOI] [PubMed] [Google Scholar]
- 147.Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65(5):245–248. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nishiyama Y, Ikeda H, Haramaki N, Yoshida N, Imaizumi T. Oxidative stress is related to exercise intolerance in patients with heart failure. Am Heart J. 1998;135(1):115–120. doi: 10.1016/s0002-8703(98)70351-5. [DOI] [PubMed] [Google Scholar]
- 149.Mangner N, Linke A, Oberbach A, Kullnick Y, Gielen S, Sandri M, Hoellriegel R, Matsumoto Y, Schuler G, Adams V. Exercise training prevents TNF-alpha induced loss of force in the diaphragm of mice. PLoS One. 2013;8(1):e52274. doi: 10.1371/journal.pone.0052274. PONE-D-12-31375 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Supinski GS, Callahan LA. Diaphragmatic free radical generation increases in an animal model of heart failure. J Appl Physiol. 2005;99(3):1078–1084. doi: 10.1152/japplphysiol.01145.2004. [DOI] [PubMed] [Google Scholar]
- 151.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88(4):1243–1276. doi: 10.1152/physrev.00031.2007. 88/4/1243 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sakellariou GK, Jackson MJ, Vasilaki A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic Res. 2014;48(1):12–29. doi: 10.3109/10715762.2013.830718. [DOI] [PubMed] [Google Scholar]
- 153.Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.05.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110(10):1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. 110/10/1364 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182(3):751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797(6–7):897–906. doi: 10.1016/j.bbabio.2010.01.032. S0005-2728(10)00043-5 [pii] [DOI] [PubMed] [Google Scholar]
- 157.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hepple RT. Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci. 2014;6:211. doi: 10.3389/fnagi.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Torii K, Sugiyama S, Tanaka M, Takagi K, Hanaki Y, Iida K, Matsuyama M, Hirabayashi N, Uno Y, Ozawa T. Aging-associated deletions of human diaphragmatic mitochondrial DNA. Am J Respir Cell Mol Biol. 1992;6(5):543–549. doi: 10.1165/ajrcmb/6.5.543. [DOI] [PubMed] [Google Scholar]
- 160.Torii K, Sugiyama S, Takagi K, Satake T, Ozawa T. Age-related decrease in respiratory muscle mitochondrial function in rats. Am J Respir Cell Mol Biol. 1992;6(1):88–92. doi: 10.1165/ajrcmb/6.1.88. [DOI] [PubMed] [Google Scholar]
- 161.Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci U S A. 2014;111(42):15250–15255. doi: 10.1073/pnas.1412754111. 1412754111 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jackson MJ. Redox regulation of muscle adaptations to contractile activity and aging. J Appl Physiol (1985) 2015;119(3):163–171. doi: 10.1152/japplphysiol.00760.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu KY, Zweier JL, Becker LC. Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ Res. 1997;80(1):76–81. doi: 10.1161/01.res.80.1.76. [DOI] [PubMed] [Google Scholar]
- 164.Hamilton SL, Reid MB. RyR1 modulation by oxidation and calmodulin. Antiox Redox Signal. 2000;2(1):41–45. doi: 10.1089/ars.2000.2.1-41. [DOI] [PubMed] [Google Scholar]
- 165.Fedorova M, Kuleva N, Hoffmann R. Reversible and irreversible modifications of skeletal muscle proteins in a rat model of acute oxidative stress. Biochim Biophys Acta. 2009;1792(12):1185–1193. doi: 10.1016/j.bbadis.2009.09.011. S0925-4439(09)00222-1 [pii] [DOI] [PubMed] [Google Scholar]
- 166.Prochniewicz E, Lowe DA, Spakowicz DJ, Higgins L, O’Conor K, Thompson LV, Ferrington DA, Thomas DD. Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am J Physiol Cell Physiol. 2008;294(2):C613–C626. doi: 10.1152/ajpcell.00232.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol. 1998;509( Pt 2):565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nethery D, Stofan D, Callahan L, DiMarco A, Supinski G. Formation of reactive oxygen species by the contracting diaphragm is PLA(2) dependent. J Appl Physiol. 1999;87(2):792–800. doi: 10.1152/jappl.1999.87.2.792. [DOI] [PubMed] [Google Scholar]
- 169.Callahan LA, She ZW, Nosek TM. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J Appl Physiol. 2001;90(1):45–54. doi: 10.1152/jappl.2001.90.1.45. [DOI] [PubMed] [Google Scholar]
- 170.Ferreira LF, Gilliam LA, Reid MB. L-2-oxothiazolidine-4-carboxylate reverses glutathione oxidation and delays fatigue of skeletal muscle in vitro. J Appl Physiol. 2009;107:211–216. doi: 10.1152/japplphysiol.00001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moopanar TR, Allen DG. The activity-induced reduction of myofibrillar Ca2+ sensitivity in mouse skeletal muscle is reversed by dithiothreitol. J Physiol. 2006;571(Pt 1):191–200. doi: 10.1113/jphysiol.2005.101105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Reid MB, Haack KE, Franchek KM, Valberg PA, Kobzik L, West MS. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J Appl Physiol. 1992;73(5):1797–1804. doi: 10.1152/jappl.1992.73.5.1797. [DOI] [PubMed] [Google Scholar]
- 173.Supinski G, Nethery D, Stofan D, DiMarco A. Effect of free radical scavengers on diaphragmatic fatigue. Am J Respir Crit Care Med. 1997;155(2):622–629. doi: 10.1164/ajrccm.155.2.9032204. [DOI] [PubMed] [Google Scholar]
- 174.Criswell DS, Shanely RA, Betters JJ, McKenzie MJ, Sellman JE, Van Gammeren DL, Powers SK. Cumulative effects of aging and mechanical ventilation on in vitro diaphragm function. Chest. 2003;124(6):2302–2308. doi: 10.1378/chest.124.6.2302. [DOI] [PubMed] [Google Scholar]
- 175.Smuder AJ, Kavazis AN, Hudson MB, Nelson WB, Powers SK. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic Biol Med. 2010;49(7):1152–1160. doi: 10.1016/j.freeradbiomed.2010.06.025. S0891-5849(10)00390-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res. 2013;112(2):382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Williams DL, Jr, Swenson CA. Disulfide bridges in tropomyosin. Effect on ATPase activity of actomyosin. Eur J Biochem. 1982;127(3):495–499. [PubMed] [Google Scholar]
- 178.Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve. 2007;35(4):411–429. doi: 10.1002/mus.20743. [DOI] [PubMed] [Google Scholar]
- 179.Nikolova-Karakashian MN, Reid MB. Sphingolipid Metabolism, Oxidant Signaling, and Contractile Function of Skeletal Muscle. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2011.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 2006;33(2):155–165. doi: 10.1002/mus.20442. [DOI] [PubMed] [Google Scholar]
- 181.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83(3):731–801. doi: 10.1152/physrev.00029.2002. 83/3/731 [pii] [DOI] [PubMed] [Google Scholar]
- 182.Supinski GS, Wang W, Callahan LA. Caspase and calpain activation both contribute to sepsis-induced diaphragmatic weakness. J Appl Physiol. 2009;107(5):1389–1396. doi: 10.1152/japplphysiol.00341.2009. 00341.2009 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nelson WB, Smuder AJ, Hudson MB, Talbert EE, Powers SK. Cross-talk between the calpain and caspase-3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit Care Med. 2012;40(6):1857–1863. doi: 10.1097/CCM.0b013e318246bb5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hirai DM, Musch TI, Poole DC. Exercise training in chronic heart failure: improving skeletal muscle O2 transport and utilization. Am J Physiol Heart Circ Physiol. 2015;309(9):H1419–1439. doi: 10.1152/ajpheart.00469.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gielen S, Laughlin MH, O’Conner C, Duncker DJ. Exercise training in patients with heart disease: review of beneficial effects and clinical recommendations. Prog Cardiovasc Dis. 2015;57(4):347–355. doi: 10.1016/j.pcad.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 186.Adamopoulos S, Schmid JP, Dendale P, Poerschke D, Hansen D, Dritsas A, Kouloubinis A, Alders T, Gkouziouta A, Reyckers I, Vartela V, Plessas N, Doulaptsis C, Saner H, Laoutaris ID. Combined aerobic/inspiratory muscle training vs. aerobic training in patients with chronic heart failure: The Vent-HeFT trial: a European prospective multicentre randomized trial. Eur J Heart Fail. 2014;16(5):574–582. doi: 10.1002/ejhf.70. [DOI] [PubMed] [Google Scholar]
- 187.Bowen TS, Rolim NP, Fischer T, Baekkerud FH, Medeiros A, Werner S, Bronstad E, Rognmo O, Mangner N, Linke A, Schuler G, Silva GJ, Wisloff U, Adams V. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail. 2015;17(3):263–272. doi: 10.1002/ejhf.239. [DOI] [PubMed] [Google Scholar]
- 188.Powers SK, Criswell D, Lieu FK, Dodd S, Silverman H. Exercise-induced cellular alterations in the diaphragm. Am J Physiol. 1992;263(5 Pt 2):R1093–1098. doi: 10.1152/ajpregu.1992.263.5.R1093. [DOI] [PubMed] [Google Scholar]
- 189.Powers SK, Criswell D, Lieu FK, Dodd S, Silverman H. Diaphragmatic fiber type specific adaptation to endurance exercise. Respir Physiol. 1992;89(2):195–207. doi: 10.1016/0034-5687(92)90050-7. [DOI] [PubMed] [Google Scholar]
- 190.Cahalin LP, Arena R, Guazzi M, Myers J, Cipriano G, Chiappa G, Lavie CJ, Forman DE. Inspiratory muscle training in heart disease and heart failure: a review of the literature with a focus on method of training and outcomes. Expert Rev Cardiovasc Ther. 2013;11(2):161–177. doi: 10.1586/erc.12.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Laoutaris ID, Adamopoulos S, Manginas A, Panagiotakos DB, Kallistratos MS, Doulaptsis C, Kouloubinis A, Voudris V, Pavlides G, Cokkinos DV, Dritsas A. Benefits of combined aerobic/resistance/inspiratory training in patients with chronic heart failure. A complete exercise model? A prospective randomised study. Int J Cardiol. 2013;167(5):1967–1972. doi: 10.1016/j.ijcard.2012.05.019. S0167-5273(12)00630-4 [pii] [DOI] [PubMed] [Google Scholar]
- 192.Winkelmann ER, Chiappa GR, Lima CO, Viecili PR, Stein R, Ribeiro JP. Addition of inspiratory muscle training to aerobic training improves cardiorespiratory responses to exercise in patients with heart failure and inspiratory muscle weakness. American heart journal. 2009;158(5):768 e761–767. doi: 10.1016/j.ahj.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 193.Marco E, Ramirez-Sarmiento AL, Coloma A, Sartor M, Comin-Colet J, Vila J, Enjuanes C, Bruguera J, Escalada F, Gea J, Orozco-Levi M. High-intensity vs. sham inspiratory muscle training in patients with chronic heart failure: a prospective randomized trial. Eur J Heart Fail. 2013;15(8):892–901. doi: 10.1093/eurjhf/hft035. hft035 [pii] [DOI] [PubMed] [Google Scholar]
- 194.Di Lisa F, De Tullio R, Salamino F, Barbato R, Melloni E, Siliprandi N, Schiaffino S, Pontremoli S. Specific degradation of troponin T and I by mu-calpain and its modulation by substrate phosphorylation. Biochem J. 1995;308( Pt 1):57–61. doi: 10.1042/bj3080057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jaenisch RB, Hentschke VS, Quagliotto E, Cavinato PR, Schmeing LA, Xavier LL, Dal Lago P. Respiratory muscle training improves hemodynamics, autonomic function, baroreceptor sensitivity, and respiratory mechanics in rats with heart failure. J Appl Physiol (1985) 2011;111(6):1664–1670. doi: 10.1152/japplphysiol.01245.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Montemezzo D, Fregonezi GA, Pereira DA, Britto RR, Reid WD. Influence of inspiratory muscle weakness on inspiratory muscle training responses in chronic heart failure patients: a systematic review and meta-analysis. Arch Phys Med Rehabil. 2014;95(7):1398–1407. doi: 10.1016/j.apmr.2014.02.022. S0003-9993(14)00184-1 [pii] [DOI] [PubMed] [Google Scholar]
- 197.Palau P, Dominguez E, Nunez E, Schmid JP, Vergara P, Ramon JM, Mascarell B, Sanchis J, Chorro FJ, Nunez J. Effects of inspiratory muscle training in patients with heart failure with preserved ejection fraction. European journal of preventive cardiology. 2014;21(12):1465–1473. doi: 10.1177/2047487313498832. [DOI] [PubMed] [Google Scholar]
- 198.Darnley GM, Gray AC, McClure SJ, Neary P, Petrie M, McMurray JJ, MacFarlane NG. Effects of resistive breathing on exercise capacity and diaphragm function in patients with ischaemic heart disease. Eur J Heart Fail. 1999;1(3):297–300. doi: 10.1016/s1388-9842(99)00027-6. S1388-9842(99)00027-6 [pii] [DOI] [PubMed] [Google Scholar]
- 199.Hulzebos EH, Helders PJ, Favie NJ, De Bie RA, Brutel de la Riviere A, Van Meeteren NL. Preoperative intensive inspiratory muscle training to prevent postoperative pulmonary complications in high-risk patients undergoing CABG surgery: a randomized clinical trial. JAMA. 2006;296(15):1851–1857. doi: 10.1001/jama.296.15.1851. 296/15/1851 [pii] [DOI] [PubMed] [Google Scholar]
- 200.Souza H, Rocha T, Pessoa M, Rattes C, Brandao D, Fregonezi G, Campos S, Aliverti A, Dornelas A. Effects of inspiratory muscle training in elderly women on respiratory muscle strength, diaphragm thickness and mobility. J Gerontol A Biol Sci Med Sci. 2014;69(12):1545–1553. doi: 10.1093/gerona/glu182. [DOI] [PubMed] [Google Scholar]
- 201.Smith BK, Martin AD, Vandenborne K, Darragh BD, Davenport PW. Chronic intrinsic transient tracheal occlusion elicits diaphragmatic muscle fiber remodeling in conscious rodents. PLoS One. 2012;7(11):e49264. doi: 10.1371/journal.pone.0049264. PONE-D-12-18557 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rollier H, Bisschop A, Gayan-Ramirez G, Gosselink R, Decramer M. Low load inspiratory muscle training increases diaphragmatic fiber dimensions in rats. Am J Respir Crit Care Med. 1998;157(3 Pt 1):833–839. doi: 10.1164/ajrccm.157.3.9512103. [DOI] [PubMed] [Google Scholar]
- 203.Bisschop A, Gayan-Ramirez G, Rollier H, Gosselink R, Dom R, de Bock V, Decramer M. Intermittent inspiratory muscle training induces fiber hypertrophy in rat diaphragm. Am J Respir Crit Care Med. 1997;155(5):1583–1589. doi: 10.1164/ajrccm.155.5.9154861. [DOI] [PubMed] [Google Scholar]
- 204.Keens TG, Chen V, Patel P, O’Brien P, Levison H, Ianuzzo CD. Cellular adaptations of the ventilatory muscles to a chronic increased respiratory load. J Appl Physiol Respir Environ Exerc Physiol. 1978;44(6):905–908. doi: 10.1152/jappl.1978.44.6.905. [DOI] [PubMed] [Google Scholar]
- 205.Akabas SR, Bazzy AR, DiMauro S, Haddad GG. Metabolic and functional adaptation of the diaphragm to training with resistive loads. J Appl Physiol (1985) 1989;66(2):529–535. doi: 10.1152/jappl.1989.66.2.529. [DOI] [PubMed] [Google Scholar]
- 206.van Hees HW, Andrade Acuna G, Linkels M, Dekhuijzen PN, Heunks LM. Levosimendan improves calcium sensitivity of diaphragm muscle fibres from a rat model of heart failure. Br J Pharmacol. 2011;162(3):566–573. doi: 10.1111/j.1476-5381.2010.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Silvetti S, Nieminen MS. Repeated or intermittent levosimendan treatment in advanced heart failure: An updated meta-analysis. Int J Cardiol. 2015;202:138–143. doi: 10.1016/j.ijcard.2015.08.188. [DOI] [PubMed] [Google Scholar]
- 208.Russell AJ, Hartman JJ, Hinken AC, Muci AR, Kawas R, Driscoll L, Godinez G, Lee KH, Marquez D, Browne WFt, Chen MM, Clarke D, Collibee SE, Garard M, Hansen R, Jia Z, Lu PP, Rodriguez H, Saikali KG, Schaletzky J, Vijayakumar V, Albertus DL, Claflin DR, Morgans DJ, Morgan BP, Malik FI. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat Med. 2012;18(3):452–455. doi: 10.1038/nm.2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hooijman PE, Beishuizen A, de Waard MC, de Man FS, Vermeijden JW, Steenvoorde P, Bouwman RA, Lommen W, van Hees HW, Heunks LM, Dickhoff C, van der Peet DL, Girbes AR, Jasper JR, Malik FI, Stienen GJ, Hartemink KJ, Paul MA, Ottenheijm CA. Diaphragm fiber strength is reduced in critically ill patients and restored by a troponin activator. Am J Respir Crit Care Med. 2014;189(7):863–865. doi: 10.1164/rccm.201312-2260LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nagy L, Kovacs A, Bodi B, Pasztor ET, Fulop GA, Toth A, Edes I, Papp Z. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br J Pharmacol. 2015 doi: 10.1111/bph.13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Valentova M, von Haehling S. An overview of recent developments in the treatment of heart failure: update from the ESC Congress 2013. Expert Opin Investig Drugs. 2014;23(4):573–578. doi: 10.1517/13543784.2014.881799. [DOI] [PubMed] [Google Scholar]
- 212.Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol. 2003;285(4):C806–812. doi: 10.1152/ajpcell.00129.2003. 00129.2003 [pii] [DOI] [PubMed] [Google Scholar]