

Abstract
Next-generation sequencing (NGS) technologies that have advanced rapidly in the past few years possess the potential to classify diseases, decipher the molecular code of related cell processes, identify targets for decision-making on targeted therapy or prevention strategies, and predict clinical treatment response. Thus, NGS is on its way to revolutionize oncology. With the help of NGS, we can draw a finer map for the genetic basis of diseases and can improve our understanding of diagnostic and prognostic applications and therapeutic methods. Despite these advantages and its potential, NGS is facing several critical challenges, including reduction of sequencing cost, enhancement of sequencing quality, improvement of technical simplicity and reliability, and development of semiautomated and integrated analysis workflow. In order to address these challenges, we conducted a literature research and summarized a four-stage NGS workflow for providing a systematic review on NGS-based analysis, explaining the strength and weakness of diverse NGS-based software tools, and elucidating its potential connection to individualized medicine. By presenting this four-stage NGS workflow, we try to provide a minimal structural layout required for NGS data storage and reproducibility.
Keywords: sequence alignment, single-nucleotide polymorphism, mutation annotation, pathway analysis, template preparation
Introduction
Over the past few years, rapid advances in next-generation sequencing (NGS) technologies have enabled researchers to generate enormous numbers of sequence reads at markedly reduced prices; this has not only led to unprecedented extension of the scope of genome-based research projects but also made NGS to revolutionize biological and biomedical research, including human disease studies.1,2 Moreover, NGS technologies are becoming more affordable and are replacing the microarray-based genotyping methods limited to interrogating regions of known sequence variation.2 To date, diverse large-scale projects have been performed incorporating NGS technologies to characterize numerous cancers, including renal cancer,3 melanoma,4 hepatocellular carcinoma,5 acute monocytic leukemia,6 and head and neck squamous cell carcinoma.7 Despite these successes, there is still a growing next-generation gap between the generation of massively parallel sequencing data and the ability to analyze and interpret the resulting information. If this gap cannot be closed, the coveted $1,000 genome could come with a $20,000 analysis price tag.8 Although numerous computational tools dedicated to specific aspects of NGS data analysis have been developed in the past few years, most have project-specific features and their functionality and parameterization are complicated. This is especially challenging for bench scientists and investigators, who are redirected to this new field and in the early stages of acquiring its technical knowledge. Although a recent study summarized the potentials and challenges of NGS-based cancer genome analysis, it did not provide a conceptual strategy or a detailed example of how to cope with the complexity of this type of analysis.9 In this review, we introduce an NGS-based workflow consisting of four major stages for the utilization of NGS data for the purpose of individualized medicine. The introduction of a conceptual NGS workflow might also forge the basis for collaboration between computational biologists, who develop the analytical methods; bioinformaticians, who utilize diverse data resources and implement software and tools; and clinicians, who act as the system end user and are responsible for shaping a new clinical practice.
Primary Stage: Template Preparation, Sequencing, and Imaging
The primary stage of the proposed NGS workflow includes three interconnected parts: template preparation, sequencing, and imaging. Each NGS platform utilizes a unique combination of specific protocols to interconnect these three parts, and this combination determines the type, coverage, and quality of the NGS data. However, all NGS platforms monitor the sequential addition of nucleotides into immobilized templates containing spatially arrayed deoxyribonucleic acid (DNA) molecules (Fig. 1). The main differences among NGS platforms are in template generation and the methods of recording and identifying sequences.10 The following sections explain each part of this stage in detail.
Figure 1.

Workflow of the primary stage. This primary stage consists of four parts that include preparation of tissue materials, creation of sequencing template, perform imaging, and sequencing. Their unique combination determines how the genetic sequence information is generated from tissues of different organisms. Although the focus of this stage of workflow is on the latter three parts, the importance of selection and preparation of tissue materials for NGS analysis cannot be neglected. The biased preparation of tissue materials could lead to fatal result of NGS workflow.
Template preparation
The first part of the primary stage of the NGS workflow is to randomly break genomic DNA for generating sequence templates, which should serve as a representative material source of targeted genomic nucleic acids (Fig. 1). There are three well-established approaches for template creation as follows: clonally amplified template, single-molecule template, and circle template. The first one, as the name suggests, focuses on sequence amplification, which is based on polymerase chain reaction (PCR). The raw sample concentration of amplification processes should be less than 20 ng/μL, generally depending on the NGS platform (eg, Illumina: ∼15 ng/μL; Roche/454: 5–10 ng/μL). For this approach, a library of fragment templates with adaptors of priming sequence sites is created. Amplification of template can be performed via either the emulsion PCR (ePCR)11 or bridge PCR (bPCR).12 For example, the sequencing by oligonucleotide ligation and detection (SOLiD), Roche/454, and Polonator platforms mainly rely on ePCR, while in Illumina platform, bPCR is mainly applied. After amplification, millions of DNA molecules can be separately captured by the targeted adaptor primers. Then, the NGS sequencing can be performed through a platform such as Roche/454, which provides the PicoTiterPlate wells for this process.13
Compared to the clonally amplified template, the approach of the single-molecule template is more straightforward and requires less preparation materials (<1 μg). For this approach, single-molecule templates are prepared and usually immobilized on a solid surface, where single DNA polymerase molecules can bind to the immobilized primer template for the subsequent NGS process.14,15 Another advantage of this approach is its independence from the PCR, which reduces the sequencing error rate and avoids amplification bias of the AT-rich and GC-rich parts of the target sequences. Furthermore, larger DNA molecules (up to tens of thousands of base pairs) can be applied for this approach to prolong the read length16 and to facilitate the read-time sequencing (RTS) methods.17
The circle template is a recently developed library preparation method that is able to reduce error rate dramatically and increase efficiency for the sequencing process.18 For this method, double-stranded (ds) DNA is denatured and then single-stranded DNA is circularized. Afterward, random primers and the Phi29 polymerase are applied to perform rolling circle replication, during which multiple tandem-copy dsDNA products are generated and sequenced simultaneously by any high-throughput sequencing technology. Each read is computationally grouped into a read family according to the original location of the circle.18 It is noteworthy that DNA damage that occurred during the process of template preparation will prevent accuracy and efficacy in the circle template-related approach.
Because it generates a representative material source, the template preparation step determines the quality of the NGS data, and therefore, it is ultimately crucial for all stages of the NGS workflow. The choice of a library preparation approach will be dependent on the task at hand. The chosen approach for this part should be highly robust and sensitive in order to reduce error rates. For an investigator who wants to conduct a quantitative NGS analysis, such as transcriptome or gene expression profile analysis, the single-molecule template is recommended in order to avoid the risk of sequence amplification bias. Investigators who intend to conduct a qualitative NGS analysis, including methylation analysis or mutational analysis, are recommended to apply the amplified template to capture complete genomic sequences without arbitrary sequence loss. However, the substitution and biased presentation of AT-rich and GC-rich regions will be the most common error type when the amplification template is used. The circle template-related approach is especially suitable to deal with cancer profiling, including diploid and rare-variant calling, microbial diversity, immunogenetics, and environmental sampling. The potential of this approach is now being increasingly recognized.
Sequencing and imaging
The next two parts of the primary stage of the NGS workflow are sequencing and imaging. The central strategy for these steps is to utilize the spatially separated immobilization of templates generated from the previous part and to record large numbers of simultaneous sequencing reactions. Four major technologies currently exist for this purpose as follows: (1) the complementary metal-oxide semiconductor (CMOS) used by Ion Torrent Personal Genome Machine (PGM)19; (2) single-molecular real-time (SMRT) sequencing used by Pacific Biosciences (PacBio)15; (3) incorporation of a fluorescently labeled reversible terminator (FLRT) in the synthesis process used by Illumina Genome Analyzer (IGA)20; and (4) a combination of emPCR and pyrosequencing used by Roche/454.21 The CMOS is a nonoptical sequencing method, which has been designed with the facility of the ion-sensitive field-effect transistor (ISFET) to detect the hydrogen ions released by DNA polymerase during the synthesis process. The DNA fragments are ligated to adapters and amplified via ePCR onto beads that are loaded into proton-sensing wells on a silicon wafer. As sequencing proceeds, the incorporation of each base releases a hydrogen ion associated with a signal that can be detected by ISFET.19
SMRT and FLRT are the main optical sequencing methods in the current market. Briefly, these methods specifically incorporate dye-labeled modified nucleotides into the DNA sequence synthesis process. Using fluorescent imaging, these dye-labeled nucleotides can be efficiently cleaved, and the corresponding signals can be emitted and recorded. Because there are essential differences in sequencing the clonally amplified template and single-molecule template, there are four well-established approaches for sequencing and imaging with regard to template selection as follows: cyclic reversible termination (CRT), sequencing by ligation (SBL), single-nucleotide addition via pyrosequencing (SAPY), and RTS (Table 1). The study by Metzker22 explains the functional principles of these four approaches in detail.
Table 1.
Sequencing methods of NGS.
| METHOD | READ LENGTH (BP) | ACCURACY (%) | SPEED (READS/HOUR) | COST PER 1 MEGABASE |
|---|---|---|---|---|
| CRT | 50–300 | 98 | 45,000,000 | $0.1 |
| SBL | 85–100 | 99.9 | 7,000,000 | $0.13 |
| SAPY | 700 | 99.9 | 40,000 | $10 |
| RTS | 14,000 | 99.9 | 500,000,000 | $0.13–$0.60 |
CRT is a cyclic sequencing approach including nucleotide incorporation, fluorescence imaging, and signal emission and recording22 and is suitable for both aforementioned templates. For instance, the Genome Analyzer (GA) developed by Illumina/Solexa applies the clonally amplified template coupled with a four-color CRT cycle,20 whereas the HeliScope, a single-molecule sequencer developed by Helicos BioSciences, uses the single-molecule template combined with a one-color CRT cycle.23 Despite these successful applications, care must be taken with the compound that will function as a reversible terminator to stop each cycle within CRT. Inappropriate use of the terminator compound will reduce the quality of CRT and dramatically increase sequencing error. Currently, several compounds including 3′-O-allyl-2′-deoxyribonucleoside triphosphates (dNTPs)24 and 3′-O-azidomethyl-dNTPs25 have been shown to be efficient reversible terminators. SBL, also a cyclic sequencing approach, has only been applied with the clonally amplified template.26,27 In 2008, Valouev et al developed SOLiD, a commercial form of SBL. Their study successfully demonstrated the efficiency of SOLiD sequencing for the creation of a high-resolution, nucleosome position map of Caenorhabditis elegans.27 Furthermore, they found that, in general, SOLiD data have the tendency to underrepresent AT-rich and GC-rich regions. It has also been shown that SOLiD lacks the ability to sequence the palindromic region efficiently.28 Moreover, substitution is another common error type in SOLiD data. SAPY is a nonelectrophoretic and bioluminescence method that has been improved by Roche/454 in order to increase the read length and data quality. However, insertion and deletion are still the common error types due to the light signal absorption. This approach has only been applied with the clonally amplified template.29
Finally, RTS is a parallelized DNA sequencing based on the single molecular template (Table 1). This method was invented by the PacBio.15 The current read accuracy is approximately 83% (131/158). By repeated sequencing of the same template more than 15 times, the read accuracy can be improved to >99%.15 The strength of this approach is to use DNA polymerase as the engine to achieve a base pair solution with a natural high catalytic rate and high processivity. The sequencing and imaging process is followed by base calling for identifying the nucleotides in accordance with four fluorescence dyes (Fig. 1). Currently, the most commonly applied base calling method is Phred base-calling, which provides high sensitivity and a low error rate when compared to other methods.30 Both CMOS and SMRT have the potential to become essential parts of the third-generation sequencing technology, which includes nanopore sequencing,31 molecular force spectrometry,32 single-molecule motion sequencing,33 electron microscopy,34 sequencing by tip-enhanced Raman scattering,35 and others.
The primary stage is an instrument-specific stage and is confined to the NGS machinery process to produce digital data from a biochemical process. Thus, choosing an NGS sequencing platform is the first factor to influence downstream data analysis. For example, the Illumina platform is suitable for a wide range of applications, especially whole-genome sequencing, although it can relatively have a high substitution error rate and the sequence quality of the IGA can decrease toward the end of the read. The SOLiD platform has one of the lowest error rates due to the advantage of two base pair color encoding. However, it is limited by its short-read lengths (<100 bp). Therefore, this platform is more suitable for targeted resequencing and exome sequencing. Nevertheless, the limitation of the current state of NGS is not the cost, although $1,000 per genome might be expensive for a routine setting in a standard hospital, but the issue of high throughput, because the latest sequencing methods, such as HiSeq X Ten from Illumina, can only produce in a day about seven genomes with 30× coverage.36 It has been anticipated that third-generation sequencers might have the potential to overcome these limitations.37
Secondary Stage: Alignments, De Novo Assembly, Single-Nucleotide Variant, and Structural Variant Detection
After NGS reads have been generated in the primary stage, the secondary stage of the NGS workflow has goals that can be divided into four main categories: chromatin immunoprecipitation (ChIP)-seq, ribonucleic acid (RNA)-seq, bisulfite (BS) sequencing, and whole-genome/whole-exome sequencing (WGS/WES; Fig. 2A). The goal of ChIP-seq data analysis is to investigate the genome-wide protein–DNA interactions. This analysis approach has facilitated unprecedented extension of the ability to discover and identify protein-binding sites.38 An excellent review by Park summarizes the advantages and challenges for current research and technology using ChIP-seq.39 Figure 2A depicts the ChIP-seq data analysis protocol. RNA-seq has now become a reliable and popular alternative for transcriptomic studies, as it enables a large number of novel applications. Recently, two precise and informative reviews have elucidated that RNA-seq data analysis provides far more precise measurements of the level of transcripts and their isoforms than other methods and that RNA-seq is an unbiased method for investigating complex traits and the pathogenesis of common disorders.40,41 Figure 2A depicts the protocol of RNA-seq data analysis. BS sequencing data analysis has been recently developed with reduced representation BS sequencing technology. This technology is able to profile genome-scale DNA methylation from mammalian genomes at significantly lower cost and higher efficiency than other methylation-related methods.42 Jeltsch and Zhang provided a comprehensive review about BS sequencing data analysis.43 Figure 2A depicts the protocol of the BS method. WGS/WES analysis aims to determine and discover genetic variations based on sequence data, which is also the main focus of our review for this stage. Different aspects have been investigated, including sequence-read alignment, sequence assembly, single-nucleotide variant (SNV) detection, structural variant (SV) detection, and others (Fig. 2B). Therefore, the secondary stage of NGS workflow is a critical part of NGS-based projects. The application of diverse methods within this stage is strongly dependent on the project aim and other factors such as cost, effort, and time.
Figure 2.

Data analysis protocols of the secondary stage. (A) The secondary stage is critical for NGS-based projects. Four different types of data analysis protocols are summarized here. Each of them serves their corresponding investigation purposes and goals. However, they share common procedures that are the read quality control and read mapping: Cufflinks,188 EdgeR,189 DEGseq,190 MACS,191 cisGenome,192 PeakSeq,193 SISSr,194 bismark,195 BS-seeker,196 BSMAP,197 methyKit,198 MAQ,74 bam2 mpg,76 GATK,78 and Samtools.77 (B) Sequence alignment, SNV detection, and SV detection are major parts for the secondary stage of workflow, whose results determine the quality of the downstream analysis in the NGS workflow.
Sequence alignment
The central process of this stage of the NGS workflow is sequence read alignment, which provides the sequence precondition for RNA-seq, ChIP-seq, WGS/WES, and BS sequencing (Fig. 2A). A growing number of sequence-read alignment tools, including Bowtie,44 BWA,45,46 CUSHAW,47 Genome Multitool (GEM),48 Genomic Short-read Nucleotide Alignment Program (GSNAP),49 and TopHat,50 have been developed to handle alignment depending on diverse parameters of mapped reads, error rate, search speed, memory, sensitivity, and alignment accuracy. Bowie is an ultrafast, memory-efficient program for aligning short reads to genomes. Its functional strategy is to use a scheme based on the Burrows–Wheeler Transform (BWT)51 and the FM Index52 for construction of genome indices, which allows Bowtie to align more than 25 million reads per CPU hour to the human genome in a small memory footprint of approximately 1.3 GB.44 In 2012, with the release of the second version, Bowtie improved with fast and memory efficiency, strength of full-text minute index, and speed of hardware-accelerated dynamic programming algorithm to achieve a combination of high speed, sensitivity, and accuracy.53
BWA is an alignment software package for mapping low-divergent sequences against a large reference genome. Its algorithm is based on backward search with BWT,54 which dramatically increases the memory footprint and alignment accuracy independent of the genome size.45 It needs to construct an FM index for the reference genomes to be used. The BWA consists of three algorithms as follows: BWA-backtrack, BWA-SW, and BWA-MEM. The first is designed for short reads up to 100 bp, while the other two couple with long reads from 70 bp to 1 Mbp.46 BWA is slower than Bowtie, but its alignment accuracy outperforms Bowtie slightly. At present, these two tools are the most applied in the field of sequence alignment.
CUSHAW is the first known sequence-read alignment software, whose algorithm is based on a compute unified device architecture parallel programming model. Although CUSHAW applies the same BWT and FM indices as Bowtie does, it is much faster and can provide comparable or even better alignment quality for paired-end alignment than Bowtie and BWA.47 However, CUSHAW is designed to deal with short-read alignment, supporting a maximum read length of 256 bp. Currently, three different versions of this software have been released.47,55,56
The filtration-based GEM is the fastest alignment software on CPU devices. Its functional strategy is to prune the search space without missing sequence-read matches, primed with careful optimizations by application of pigeonhole-like rules and refined by dynamic programming in bit-compressed representations.48 GEM is faster than all currently applied alignment tools on CPU devices and is well suited for aligning long reads due to its filtering-based pruning scale.
The Short Oligonucleotide Analysis Package (SOAP) is another well-established short-read alignment tool, which applies different fast and effective algorithms for indexing the reference genome. There have been four releases to date, which have improved memory reduction, increase of alignment speed, and utility on the GPU.57–60
The GSNAP is a fast and memory-efficient method for aligning both single- and paired-end reads as short as 14 nt and as long as desired. It works by considering complex variants involving multiple mismatches and long indels, and different splicings in individual reads. Moreover, GSNAP permits single-nucleotide polymorphism (SNP)-tolerant alignment by using probabilistic models or a reference database such as dbSNP to increase the precision of sequence alignment. Another additional function of GSNAP is to map reads from DNA treated with sodium BS to investigate the methylation state of genomic sequences. Currently, the major application of GSNAP is RNA-seq analysis.
TopHat is one of the most applicable mapping tools for RNA-seq analysis. It aligns all sequence sites relying on an efficient 2-bit-per-base encoding and a data structure for efficiently using the cache on modern computer processors. It is noteworthy that TopHat considers RNA-seq reads spanning an exon boundary, which would be a major reason for alignment failure in previous mapping strategies.61,62 Furthermore, TopHat utilizes Bowtie for mapping non-junction reads. TopHat2 is the second generation tool, with many significant functional enhancements, including aligning reads from fusion breaks and considering the presence of pseudogenes.63 Currently, TopHat/TopHat2 are often used to localize RNA-seq reads generated from the Illumina and SOLiD platforms.
In general, choosing an aligner is highly dependent on the read length, alignment speed, hardware condition, and time of investigation. In the case of BWA, an MEM algorithm is usually preferred for read length of 70 bp or longer, such as those generated by Illumina, 454, ion torrent, and Sanger sequencing. The MEM algorithm has better accuracy than BWA-SW. Comparisons of the computational performance of these tools have been conducted by two recent studies.56,60 The comparisons were performed on specific data conditions and focused on alignment speed and sensitivity (Table 2). During these comparisons, multiple simulated data were generated for testing. However, it is unknown whether the change of comparison conditions would lead to the change of tool rank. Mappers such as BWA and bowtie that are unaware of splicing are widely used for DNA-seq datasets, while TopHat and SOAP-splice are used for RNA-seq, since these aligners can handle spliced alignments such as mRNA transcripts without introns.
Table 2.
Reads alignment softwares.
| NAME | AVERAGE ALIGNMENTS SPEED (MILLION READS/CPU HOUR) | MAXIMUM SENSITIVITY (%) | ALLOWED GAPPED | REFERENCES |
|---|---|---|---|---|
| Bowtie | ∼2.3 | 96.52 | No | 45,56,60 |
| BWA | ~3 | 94.40 | Yes | 45,56,60 |
| CUSHAW | >30 (GPU) | 96.73 | No | 56,60 |
| GEM | >9 | 95.26 | Yes | 56,60 |
| Soap | ∼3 | 98.12 | Yes | 45,56,60 |
| GSNAP | 1.8∼2.8 | 94 | Yes | 49,199 |
| Tophat/Tophat2 | 5∼10 | 96.1 | Yes | 50,199 |
Notes: In general, the sensitivity and speed are in inverse correlation. The benchmark tests of the following tools have been conducted by different studies under specific conditions; therefore, caution is needed when comparing them with each other.
De novo assembly
Another cornerstone within the second stage is the global assembly of sequence reads into a complete genome (de novo assembly). This process facilitates more cost-effective and accurate genome analysis and removes all possible biases introduced by sequence alignment to a reference genome. Since NGS technologies pose tremendous challenges to de novo assemblers for assembling millions and billions of reads from different organisms, to the present, most de novo assemblers perform well on bacteria and small eukaryotes. For instance, Velvet, a de novo assembler based on the de Bruijn graph approach, has generated several genomes from bacteria to fungi with the ability to leverage short reads in combination with read pairs.64 Edena, another de novo assembler, has been developed based on the classical assembly approach where all overlaps are structured in a graph for exactly assembling accurate contigs from data sets encompassing short reads of the same length. This software has been applied to generate several bacterial genomes with high-quality results.65 Table 3 summarizes the properties of several well-established de novo assemblers.
Table 3.
De novo assemblers.
| NAME | SUPPORTED TECHNOLOGY | ASSEMBLY COVERAGE | ERROR RATE | REFERENCES |
|---|---|---|---|---|
| ABySS | Solexa, SOLiD | 95.6% | 1 per 8 kbp | 66 |
| Celera | Solexa, Sanger, 454 | 95.23% | 1 per 17 kbp | 200 |
| Edena | Illumina | 95.11% | 1 per 4 kbp | 201 |
| Euler | Sanger, 454, Solexa | 92.78% | 1 per 2 kbp | 202 |
| Forge | 454, Solexa, SOLID, Sanger | 93.67% | 1 per 6 kbp | 203 |
| MIRA | Sanger, Illumina, 454 | 94.48% | 1 per 8kbp | 200 |
| PASHA | Illumina | 93.17% | 1 per 7 kbp | 204 |
| SGA | Illumina, Sanger, 454, Ion Torrent | 95.9% | 1 per 83 kbp | 66 |
| SOAPdenovo | Solexa | 94.8% | 1 per 81 kbp | 66 |
| Velvet | Sanger, 454, Solexa, SOLiD | 94.5% | 1 per 18 kbp | 66 |
Notes: The coverage and error rate were measured by different studies under different conditions; therefore, the comparison might not be considered at quantitative level.
Only two recent studies have achieved breakthrough success by assembling human genomes. One study reported the development of a parallel short-read de novo assembler (assembly by short sequences [ABySS]) and its application for assembling a whole-genome sequence of a Yoruba man with 42-fold read redundancy.66 The other study developed an advanced de Bruijn graph-based approach for more efficient and cost-effective de novo assembly and reported the successful generation of complete genomes of an Asian man and an African man by achieving a 71× sequencing depth of the NCBI human reference genome.67 Despite these successful applications, de novo technologies face common challenges that impede their practical utilization. One conventional disadvantage of de novo assembly approaches is low speed, because the assembly of randomly positioned DNA reads is a computationally intensive process. Another disadvantage is the complication of repetitive sequence reads, which results in high error rate and imprecise assembly.68 However, the de novo assembly methods are irreplaceable and essentially important for characterizing unknown sequences of different organisms and discovering the cellular and biological diversity of our world.69 Thus, due to the rapid advances in sequence assembly technology, we anticipate that de novo assembly will become a practical method for creating disease-specific or individual family-specific reference genomes to help determine and detect the biological and cellular underpinnings of diseases and in other ways expand personalized medicine.
SNV detection
Because the SNVs including small insertions and deletions are the most abundant among the various types of mutations causing diseases, approaches for SNV detection have become an indispensable part of downstream NGS analysis. In general, the SNV detection approaches are performed after mapping reads to a reference genome and are critical in both WGS and WES analyses. Based on empirical experience of several genome sequencing studies, the SNV detection approaches can generate 3–4 million SNVs as an initial set for WGS analysis, while for WES analysis, approximately 20,000 SNVs can be found.70–73 Therefore, the essential functionality that diverse SNV detection approaches need to provide is to minimize false-positive rate and maximize high-quality SNV sets for follow-up analysis and interpretation (Table 4).
Table 4.
SNV/SNP detection tools.
| NAME | FEATURES | COVERAGE (%) | ERROR RATE (%) | REFERENCE |
|---|---|---|---|---|
| bam2mpg | Variant calling | 98.23 | 2.34 | 76 |
| GATK | Variant calling, SNV/SNP filter and quality calibration | 97.78 | 2.90 | 205 |
| MAQ | Variant calling | 97.92 | 0.18 | 74 |
| IMPUTE2 | SNP filter and genotype likelihood | 97.16 | 0.88 | 206 |
| Samtools | Variant calling, SNV/SNP filter and quality calibration | 97.86 | 3.30 | 205 |
| SOAPsnp | Variant calling, SNV/SNP filter | 98.12 | 0.16 | 77 |
| SNP array | Variant calling, SNV/SNP filter | 98.43 | 0.13 | 205 |
| VarScan | Variant calling, SNV/SNP filter | 97.67 | 8.50 | 205 |
| MuTect | Tumor Variant calling, SNV filter | NA | <0.24 | 80 |
Notes: The benchmark tests of the following tools have been conducted by different studies under specific conditions. The comparison is with precaution.
One early study developed the software MAQ for read mapping and SNV detection, which uses a Bayesian statistical model and considers the mapping quality and error probabilities from raw sequence quality scores in order to detect SNVs accurately and efficiently.74 A follow-up study applied MAQ combined with a set of critical rules related to read counts, base quality, and SNP quality scores and detected 2.6 million validated, high-quality SNVs from an acute myeloid leukemia genome.75 Another recent study developed the software bam2 mpg for SNV detection from sequence reads of haploid or diploid DNA aligned to a related reference genome. The bam2 mpg tool uses the most probable genotype (MPG) algorithm based on a Bayesian model and applies heterozygosity-dependent prior probability in order to calculate the likelihood of each possible genotype given the observed sequence data.76 A follow-up study utilized this software and successfully developed a ratio score for evaluating mutation-related SNVs in melanoma.4
Although the two aforementioned tools have achieved certain successes in SNV detection, the most frequently applied SNV detection tools are SAMtools77 and Genome Analysis Toolkit (GATK).78 Both implement various utilities for pre- and postprocessing sequence data from different formats for indexing, variant calls, sequence alignment, and others. Both tools were developed during large-scale genome projects, so methods including variant calling within both tools are robust, efficient, and validated with large sequence data. However, SAMtools and GATK depend on multiple parameters for variant calling, and according to the documentation, it is not clear how different parameters of both tools should be interpreted with regard to whether a variant call is correct or how variants should be prioritized.
The low frequency of many important somatic mutations is pervasive in samples of different types of cancer.79 Therefore, precise identification of SNVs with low frequency from heterogeneous cancer samples is a major task and a great challenge for clinical diagnostic approaches. Currently, two widely used SNV detection methods for this purpose are MuTect80 and Strelka.81 MuTect has been developed by using a Bayesian classifier to detect SNVs with very low allele fractions in cancer samples. This method also applies six internal filters to remove artifacts for increasing read quality. Many studies have shown that MuTect could identify important subclonal drive mutations responsible for tumor progression and treatment resistance with high sensitivity and specificity.82–85 The known disadvantage is that the sensitivity of MuTect will decrease when running it with high confidence configuration to control false positives. Strelka is another popular method for detecting somatic SVNs and indels from sequencing data of matched tumor–normal samples. This method is based on a novel Bayesian approach by considering normal samples as a mixture of germline variation with noise and matched tumor samples as a mixture of the normal sample with somatic variation. The Strelka has also been designed to cope with diverse SNV scenarios of matched normal–tumor samples, including identification of alleles with absence in the host’s germline.81 Under a standard configuration, the performance of both methods (MuTect and Strelka) is highly similar.80 However, it is unknown whether a change of performance would follow when the conditions of the benchmark test are changed.
For SNV analysis, it is not only important to apply a tool with a high coverage and a low error rate but also necessary to consider the systematic bias that a chosen reference genome could cause. Although at present plenty of software tools have been developed for the purpose of SNV detections, and some of them have been applied and even proven to be accurate and efficient as mentioned before, the general challenge of SNV detection has not been fully addressed. What is lacking is a concept or method for assessing the accuracy of each individual variant in order to reduce false discovery rate.86 Fortunately, the first well-characterized complete whole-genome reference material (NA12878) has been released recently.87 Common sequencing biases that can result normally in hundreds of thousands of discrepancies between different sequencing approaches for the same human genome can now be dramatically reduced by the utilization of the NA12878. Furthermore, another optimal option would be that one should apply a de novo assembly approach to generate a project-specific reference genome from a control group, then conduct the SNV analysis and subsequently the filtering process to identify the potential candidate SNVs. Afterward, apply a known SNP database such as dbSNP88 to identify the disease-related SNPs among the SNVs, because a SNP is a special SNV found on a population level.
SV detection
Structural variations in the genome can be defined as any DNA sequence alternation other than a single-nucleotide variation, including insertions, deletions, duplications, inversions, translocations, and copy-number variants.89,90 Detection and characterization of genomic SVs are crucial steps for investigating the relationships between genotype and phenotype and understanding the genetic cause of complex diseases including cancer. Many previous studies have reported discoveries or identifications of a large number of SVs within human genomes and revealed the pathological involvements of different types of SVs.91–95 This has led to invoke an increased interest in the study of genomic structural variations and corresponding software developments for SV detections (Table 5). Current SV detection software can be classified into four categories according to the applied strategies for SV discovery:
paired-end mapping (PEM);
split read mapping (SRM);
depth of coverage (DOC); and
assembly-based approach (ASA).
Table 5.
Structural variation detection tools.
Each category has its advantages and limitations. PEM identifies SVs from mapped paired reads generated in a discordant manner, whose distances differ very significantly from a predefined or a usual average distance of paired reads. Therefore, PEM-based methods such as PEMer92 and BreakDancer96 can efficiently detect many kinds of SVs including insertions, deletions, inversions, and tandem duplication, but are not capable of detecting SVs in low-resolution or low-complexity genomic regions with segmental duplication. Moreover, PEM-based methods have difficulty identifying SVs with larger than average size defined in the genome library.97
SRM detects SVs based on unmapped or partially mapped reads that potentially provide accurate position information of breaking points in a genomic region. Usually, these failed mapped reads are split into different fragments by SRM-based methods such as Pindel98 and AGE.99 Only the first and last fragments are further used for being aligned into the reference genome in order to localize the precise start and stop position of SV events. Therefore, the limitation of this type of approach is its tight dependence on the reference genome and the length of split reads. An interesting SV detection tool, DELLY, has been developed recently, which integrated PEM and SRM to accurately delineate SVs at single-nucleotide resolution.100 This tool is suitable for detecting tandem duplication and copy-number variable deletion events as well as balanced rearrangements. A recent survey shows that only integrative approaches such as DELLY are able to meet high discovery criteria of sensitivity and specificity.101
DOC-based methods such as CNVeM102 and Exome CNV103 apply one important feature of massively parallel sequencing data with which several hundred million short sequence reads are efficiently produced to detect SVs based on the density of reads aligning to the reference genome.104 Therefore, DOC takes the advantage of high-coverage NGS data and strongly varies from the aforementioned PEM and SRM, which focus on the genomic position information. There are two major bias factors of this approach: GC content and the presence of repetitive regions in the reference genome. Fortunately, several strategies have been developed to correct these bias factors.105,106 Furthermore, the utility of this kind of approach for investigating insertion, deletion, duplication, and other SVs needs to be investigated.
Entirely different from the three aforementioned approaches, the ASA tries to first reconstruct DNA fragments by assembling overlapping reads with or without a reference genome. The SVs can then be detected by comparing constructed DNA fragments with a reference genome. Therefore, ASA-based tools such as Cortex107 and Magnolya108 have a minimum requirement of read coverage and can discover novel genetic SVs ranging from a single base pair to a large structure variation. The main weakness of the ASA is its high demand on computational resources. This approach is not suitable for discovering SVs from a genomic sequence with low quality.
In summary, the secondary stage is an algorithm-dependent stage, which is vital for NGS-based projects. Carefully considering and choosing appropriate methods and algorithms can dramatically improve the data quality of downstream analysis and reduce error rate.
Tertiary Stage: Statistics, Clustering, and Disease-Specific Mutations
The second stage of the workflow reveals an abundant list of genetic variants. However, not all of them influence key functional factors that change normal cells into highly malignant derivatives, and not all of them achieve survival and prolife ration. Therefore, the important task within the tertiary stage of the NGS workflow is to detect variants that drive selective advantages.
In general, there are five independent approaches to identify functional and driver variants (Fig. 3):
Figure 3.

Five approaches for the tertiary stage on the investigation of disease-driven mutations: (A) Visualization of random data on multisegment plot, 4 Bartonella genomes by the genoPlotR115; (B) Annotate mutation according to an annotation database (http://creativecommons.org/licenses/by-nc-sa/3.0/legalcode); (C) Predict mutation effect through analysis of the protein 3D structure generated by the tool PyMol (www.pymol.org); (C) Analyze the possible mutation effect with a statistical method; and (E) Detect mutation effect via generation of gene signature.
Note: Figure 3B is reused from http://barrymieny.deviantart.com/ under the conditions of the CC BY-NC 3.0 license.
(a) Visualization of genomic variant distribution and relationship with (disease) phenotypes; (b) Annotations of mutations according to existing knowledge, such as publicly available annotation databases including dbSNP,88 Online Mendelian Inheritance in Man (OMIM),109 HapMap,110 Human Gene Mutation Database (HGMD),111 and catalogue of somatic mutations in cancer (COSMIC),112 so that the functional implications of variants can be identified or determined; (c) Application of computational methods or tools to determine or predict the possible functional impact of mutations; and (d) Using statistical methods to analyze the possible mutation effect based on the frequency or location of mutations with regard to a genetic collection or a cohort of patients; and (e) Generating predictive gene signatures by applying a sophisticated machine learning technique, such as random forest, or stepwise regression selection, such as lasso.113
Visualize the distribution of genomic variants and their relationship with (disease) phenotypes
Efficient visualization approaches have been developed to combine disease phenotypes with the genomic variants in individual samples. They are helpful to create hypotheses and to prepare independent validation studies. Impressive examples for these complex visualizations are presented in the Nature series on The Cancer Genome Atlas (TCGA)-based analysis for specific tumor entities. For example, Figure 2 from the study of TCGA114 presents the individual genome-wide mutational changes (somatic exome versus tumor exome) for 195 colorectal cancer (CRC) samples ordered with regard to several disease phenotypes as follows: tumor site, CpG island methylator phenotype expression phenotype, BRAF V600E mutations, methylation cluster, and RNA expression cluster.
The R-environment for statistical computation (https://www.r-project.org/) provides an elaborated toolbox for complex visualization. A more generic tool in this field is ggplot (http://ggplot2.org/), which is based on the grammar of graphics. The ggplot package takes care of many of the details that make plotting an effort providing tools for complex multilayered graphics. More genome-specific tools are provided by the package genoPlotR.115 This R package allows users to read from files with usual formats such as protein table files and blast results files, as well as home-made tabular files, to generate visualization with different layouts. Furthermore, the circular layout is an efficient way to create a visualization of huge amounts of genomic information. The R package circlize116 provides an implementation of circular layout generation in R as well as an enhancement of visual effect. The package uses low-level graphics functions, and self-defined high-level graphics can be easily adapted by users for specific purposes. Together with the seamless connection between the powerful computational and visual environment in R, circlize gives users convenience and freedom to design figures for better understanding genomic patterns based on multidimensional data. Similar software is provided by Circos (http://circos.ca).117 The Broad Institute offers the Integrative Genomics Viewer (IGV), which is a high-performance visualization tool for interactive exploration of large, integrated genomic datasets. It supports a wide variety of data types, including array-based and next-generation sequence data, and genomic annotations (https://www.broadinstitute.org/software/igv/home).
Database-based mutation annotation
Different large-scale projects, including 1,000 Genome projects, Cancer Genome Atlas Network, and the International Cancer Genome Consortium, provide new insights into cancerous genomic functions related to protein-coding and noncoding transcripts, transcription and epigenetic-regulation elements, and conserved genomic region. Databases such as dbNSFP118 were developed for functional prediction and annotation of all potential nonsynonymous SNVs. dbNSFP compiles prediction scores based on algorithms such as SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/), and MutationTaster (http://www.mutationtaster.org/). Furthermore, the world’s largest and most comprehensive human mutation database, COSMIC,112 was updated recently in order to better emphasize the impact of the latest knowledge about cancer-related mutations and allow systematical identification of the impact of known cancerous genes. Its potential has been waiting to be explored. These databases provide different types of valuable genetic information, made easily accessible by software tools such as SnpEff,119 AnnTools,120 ANNOVAR (http://annovar.openbioinformatics.org/en/latest/misc/credit/), and Oncotator (http://www.broadinstitute.org/cancer/cga/oncotator). More recently, another database-based method, combined annotation-dependent depletion (CADD), has been developed by objectively integrating more than 14 million high-frequency human-derived alleles. The score from CADD can quantitatively differentiate functional, deleterious, and disease causal variants across a wide range of functional categories in both research and clinical settings. Its performance has reached a higher level of efficiency when compared with other methods.121 However, the database-related approaches are confined to the “common disease, common variant” hypothesis and are, therefore, not capable of classifying rare variants and mutations.122,123
Computational prediction approaches
If no assessment information for genetic variants and mutations is available, a computation-based prediction of functional impact is possible in order to identify a cancer-relevant or disease-associated functional impact. Some methods use physicochemical properties of a sequence as well as amino acid position information to predict the functional effect of a genetic variant or mutation.124–126 More recently, it has become feasible to characterize somatic noncoding mutations from genomic regulatory sites. Efforts have been made to apply high-throughput genomic data to characterize regulatory binding motifs that are subsequently used to predict the binding sites for diverse transcription factors in regulatory sequence regions.127,128 However, the challenge is how to interpret the mutational effect at system level. For instance, what is the relationship between the changed function of kinases or transcription factors and the cellular functions such as evading apoptosis, sustained angiogenesis, drug resistance, limitless replicative potential, and others?
Frequency- and location-based statistical analysis
Because of their ability to provide adaptive advantages to cancer cells, driver mutations (compared to somatic DNA) are positively selected during the clonal development and evolution of pathological cells and tissues. Therefore, in general, driver mutations occur at a higher frequency than passenger or nonfunctional mutations, which occurs only at a random frequency. Furthermore, for certain genes that possess key functions for cancer development, such as oncogenes and tumor-suppressor genes, although mutations within these genes may be highly variable, most tend to cluster within functional domains or within evolutionarily conserved regions. In this way, they alter the cellular function of these genes for facilitating cancer development, in contrast to passenger mutations, which occur at random locations throughout the genome.129
Statistical methods have been developed to assess mutation frequency or mutation density by location within a given cohort of patients in order to identify the disease-related mutations. These instruments support frequency- and location-based statistical analysis. For instance, the tool mutational significance in cancer (MuSiC) applies various statistical tests including the convolution test, Fisher’s combined P-value test, and the likelihood ratio test (LRT) to distinguish driver mutations from background (passenger) mutation according to mutation frequency and location.130 Reimand and Bader developed a statistical model, ActiveDriver, that focuses on location-related, phosphosite-specific mutation rates across multiple cancer types.131 The model assumes that a missense mutation of cancer genes followed the Poisson probabilities distribution, and P-value-based scores are created for ranking the top candidates that may be driver mutations. In 2012, Hodis et al described a sequence mutation-based permutation framework.132 By application of their framework, they proposed potential candidate genes for positive selection during melanoma development and attempted to investigate the relationship between genes with high mutation burden and environmental factors including ultraviolet light for a cohort of melanoma patients. A specific aspect of their approach is to leverage intron and UTR sequences in each gene locus for calculating the gene-specific basal mutation rates.132 However, it is noteworthy that the frequency- and location-based statistical approaches are generally not able to assess the functional consequence of mutations. Results of these statistical analyses have to be biologically validated.
It is also of interest to combine gene annotation with observed frequencies of variants. Variants of the gene set enrichment analysis may be helpful to discover over- or under-representation of disease-specific variants in functional complexes represented by a specific set of genes.133
Gene signature-based approach
Another popular approach for identification of mutation effects is the generation of a gene signature, which is a selected set of genes together with an algorithm to calculate a signature derived score value. This approach combines a genomic spectrum of variants with a phenotypic outcome. In this setting, logic regression-based methods may be applied. The logic regression is a generalized regression methodology primarily applied when most of the covariates in the data to be analyzed are binary. The goal of logic regression is to find predictors that are Boolean (logical) combinations of the original predictors.134
Many approaches developed for gene expression-based classification and prediction can be translated to the analysis of the prognostic and predictive relevance of mutation and variant spectra. Penalized regression approaches are particularly helpful for studying the relationship of single variants within a large set of potential genomic markers.113
Of specific interest for individualized medicine is the relevance of predictive genomic markers for treatment decisions. A marker is predictive if it contains information with regard to the response of a specific patient to a specific therapy. For example, specific mutations in the KRAS gene impair the response to cetuximab in patients with advanced CRC.135 More and more clinical trials are scanning whole-genome variant spectra for predictive markers, and providing the statistical instruments for this type of purpose is a very active field of research.136,137 However, the predictive marker-based approaches have several potential disadvantages. Often, these approaches lose discrimination power when the test data sets are entirely different than the training data sets. Furthermore, the generation of one or more predictive markers would be highly dependent on the following four factors: (1) purity degree of the patient sample; (2) NGS or microarray platform; (3) the statistical analysis approach that is chosen to build a gene signature; and (4) strong transcriptional dependency within a gene signature. Slight changes in these four factors might result in the selection of entirely different markers. Therefore, it is essential to verify the robustness and uniqueness of a predictive marker before any application.
Quaternary Stage: Pathway- and Network-Based Analyses
System aspects are seen as key to understanding cancer. The understanding of the diversity and frequency of genetic changes leading to deregulation of signaling pathways in CRC is of high interest. Insights into the systems biology of the cancer cell may help to improve cancer treatment.138 Therefore, it is of interest to explore how the components of the system interact. Most approaches are static, ignoring the dynamic behavior of a system over time because of the complexity of human-based time-series studies.
In general, the network- and pathway-based approaches use different types of molecular data and are rarely restricted to genome-wide information on variants and mutations. Transcriptomics, proteomics, metabolomics, and data on methylation and microRNA regulation are typically assessed simultaneously in an integrative analysis. Since a review of the full spectrum of system-related integrative analysis goes beyond the purpose of this review, we restrict our consideration to a few relevant aspects. Again, the Nature series on TCGA analyses tries to express first insights into systemic aspects by informative graphs. See for example Figure 4 from the study of TCGA,114 which combines mutational and transcriptome information and studies up- and downregulation of specific pathways. Several large-scale genome-wide projects give excellent examples of role of pathways in the progression or etiology of different cancer types and indicate the importance of pathway- and network-based analyses for the understanding of disease development.83,138–142 Therefore, pathway- and network-based analysis has been advocated as an important downstream analysis for genome-wide association study.143–145 This type of analysis addresses at least two major issues as follows: it can identify groups of genes directly associated with disease or pathological phenotypes in a way that is easily comprehended by the investigators, and it can successfully separate noisy genetic bystanders caused by the instability of malignant genomes.146 The computational challenges of cancer genome analysis are summarized by Vazquez et al.9
Figure 4.

Whole workflow of variant calling procedure. Output from mark duplicates is considered as phase 1 output while that of print reads were considered to be phase 2 output.
Network-based analysis
A network is defined by nodes and edges expressing the neighborhood between two nodes. Network analysis is a straightforward first approach to systems biology. Algorithms for constructing networks and defining neighborhoods of specific nodes are under study. Often very simple approaches are used: two genes are neighbors if they are quoted in the same paper. However, neighborhood may also be defined by human protein–protein interaction (PPI) maps,147,148 related chemical reactions as proposed in the molecular signaling map,149 or curated maps of human metabolism and regulatory networks.150 Chuang et al developed a PPI-based network analysis with the potential to integrate genome-wide data including sequencing data and gene expression data.151 This approach showed the advantages and high potential of the subnetwork signature for metastatic breast patients.
Visualization of networks is helpful. Hairball plots are typically used, but the best way to plot networks is under discussion.152 Interpreting hairballs is made difficult by several significant shortcomings as follows: (1) their form is determined by layout algorithms; (2) many layout algorithms are stochastic and can produce many different layouts of the same network; (3) layouts of the same network created by different algorithms cannot be easily compared; (4) the layout can be disproportionately affected by very small changes in a network; and (5) layouts of different networks created by the same algorithm cannot be easily compared.
Pathway-based analysis
A pathway encompasses a set of biochemical events that operate within a cellular process and includes a group of genes defined by some biological commonality for certain phenotypes. Pathway-based analysis begins with biological knowledge and can provide concrete and detailed functional or mechanistic insight into the connection between genotype and phenotype. There are several large-scale, public pathway databases, expert-curated and peer-reviewed to ensure high quality, including Reactome,153 KEGG,154 PANTHER Pathway,155 and others. These public pathway databases form the fertile data basis for conducting pathway-based analysis. Recently, a genome-scale molecular metabolic model containing >90 diverse metabolic pathways has been successfully reconstructed based on the 50-year legacy bibliome data combined with some aforementioned pathway databases. This model possesses strong potential related to predicting the outcomes of adaptive evolution. This model has also been applied for identification of key metabolic functions or components corresponding to specific environmental or genetic perturbations.150 A follow-up study utilized this metabolic model and developed a model building algorithm in order to automatically build case-specific cancer-metabolic models for elucidation of drug target effects and prediction of synthetic lethal effects.156
Another recent study by Bordbar et al applied the aforementioned genome-scale metabolic model to elucidate the functional relationship between the activation of macrophages, immune response, and metabolic reprogramming by integrating multiple omics data including transcriptomics, proteomics, and metabolomics.157 The approach of this study is based on the metabolic flux, which has been widely accepted for analyzing metabolism.158,159 The result of this study provides an important indication to delineate metabolic mechanisms as immunotherapeutic targets, which further evidences the strength of pathway-based analysis.
More recently, in order to better understand the dynamic behavior of cancerous cellular systems, Li and Mansmann conducted literature research to collect molecular information for construction of a large-scale human signaling model.149 This published model includes >20,000 biochemical reactions that can be organized into >50 diverse cancer-relevant signaling pathways. In addition, both authors developed the Flux-Comparative-Analysis (FCA) to incorporate the transcriptome data of individual cell lines into this signaling model with the goal of drug response prediction at an individual level. They applied FCA to predict the drug response of NCI-60 cancer cell lines and achieved a promising result, demonstrating the usefulness of pathway-based analysis for the targeted therapy.
The result of this stage can directly influence the individual patient treatment and outcome. However, care must be taken to verify the quality of an applied biological network. The application of an unverified biological network can substantially increase the false-positive rate. A second error source is the quality of the data generated by the previous three stages. Therefore, these four stages are interconnected parts. The following section gives an application example of this four-stage workflow and highlights the key points of the workflow. We would like to finish this chapter with a warning by Sadeh et al.160 Our current understanding of cellular networks is rather incomplete. We overlook important, but so far unknown, genes and mechanisms in the pathways. Moreover, we often only have a partial account of the molecular interactions and modifications of the known players. When analyzing the cell, we look through narrow windows, leaving potentially important events in blind spots. Network reconstruction is naturally confined to what we have observed. Little is known on how the incompleteness of our observations confounds our interpretation of the available data.
Reproducibility
The reproducibility is becoming an essential part of the NGS landscape. The bioinformatics community has developed different systems including Galaxy (http://usegalaxy.org) to address this issue. Our application example demonstrates that our proposed NGS-based four-stage workflow can be implemented in a Galaxy instance. Galaxy, an open-source, web-based platform for biomedical research, is one of the current leading workflow systems. A local galaxy instance can be built up with necessary tools and computational capacities and can be used as an ideal platform for bioinformatics. Since Galaxy manages the tool versions and tool dependencies, it provides opportunities for reproducing identical results even after tool upgrades. Project-specific workflows can be generated and can be used repeatedly in an orderly fashion. Workflows and even complete analyses can also be shared among different galaxy instances, which provide great scope for knowledge sharing and uniformity among consortiums. Another potential platform for reproducible bioinformatics is the Docker (www.docker.com). It is a very recent technology for facilitating reproducibility by encapsulating a complete environment with system tools, scripts, libraries, tool dependencies, etc., into a Linux operating system. Docker containers can be launched on any operating system, and the necessary tools can be used without any interference from the operation system itself. A well-configured and documented Docker image can be shared among different study groups, and the designed workflows can be executed in a reproducible manner. Although Docker has the potential to be recognized as one of the most fundamental workflow systems developed in recent years, we have not incorporated our workflow into the Docker environment because of several security issues raised recently.161 Currently, we are following the projects relating to common workflow language (CWL) very closely and are in close contact with the main developers. We believe that CWL can define the future direction for annotation and development of NGS-related tools. Although there is no final specification of CWL, Galaxy and Docker continue to interact to push the development of CWL, and our proposed workflow has the potential as a draft version of a standard NGS workflow for reproducibility. In the following section, we demonstrate an application workflow within the Galaxy.
Application Example
The aim of this example is to reproduce the genomic profile of colon patients reported by the TCGA,114 which gives a demonstration of the suggested four-stage workflow. The application starts with processing the digitalized genomic data, the BAM files, containing sequence reads from Illumina platform. These BAM files include raw exome reads, which were mapped to the GRCh37-lite reference genome (https://browser.cghub.ucsc.edu/help/assemblies/). Thus, in our example, the primary stage and the beginning of the secondary stage (mapping) have already been performed by TCGA. Five CRC samples (both tumor and normal) were used for our analyses. All the included datasets were downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/) in order to reproduce their published genomic variants. Subsequently, our variant calls were compared with the variant calling made by TCGA to demonstrate the concordance and discordance among the variants. Moreover, variant callers specific for tumor–normal sample pairs such as Mutect80 and Strelka81 were also used to obtain somatic variants in this example.
Among the 24 significantly mutated genes presented by TCGA, we restricted our region of interests to the following six genes: APC, TP53, SMAD4, PIK3CA, KRAS, and ARID1A. The gene list includes some tumor-suppressor genes, such as TP53 and APC, and an oncogene, KRAS, and others frequently associated with cancer.162–164 The secondary stage proceeds with preprocessing raw mapped reads before the variant calling procedure is carried out to filter out noisy background information. There are no gold standard preprocessing steps established for variant calling. The best practice guides (https://www.broadinstitute.org/gatk/guide/best-practices) and common usages (http://varscan.sourceforge.net/support-faq.html) were applied and were grouped into two phases in order to determine their effects over variant calling. Phase 1 preprocessing involved the removal of read duplicates, reordering and sorting the mapped reads, while phase 2 extended the steps from phase 1 with indel realignment and base recalibration. Two of the most popular variant calling tool boxes, GATK (v2.7.4) and VarScan (v2.3.6), were used to create six variant calling procedures common for variant detection. Unified Genotyper (UG-default), HaplotypeCaller (HC-default), and VarScan with default parameters (VS-default) restricted P values (VS-pvalue) and two conservative parameters (VS-cons1 and VS-cons2). These are shown in Supplementary File 1. The SNPs and indels were called with the workflow pictured in Figure 4. Variant callers such as MuTect (v1.1.7), Strelka (v2.0.5), and VarScan (v2.3.6) were used for the detection of somatic variants. Default values were used in all three somatic variant callers.
The resulting variants were then compared with the TCGA variants, which were considered as reference, to determine concordance and discordance among them (Supplementary File 1). In the case of SNP detection, all six variant calling procedures showed similar performance, with a concordance range of 90%–95% (Fig. 5). But a wide variation in indel calling (45%–90%) was observed. The variant calling VS-default could reach the highest true positive rate independent of the preprocessing phases used. However, it also showed a high false-positive rate (Fig. 6). Although most of the preprocessing steps are included in the best practice guides for GATK, this does not seem to increase its performance with UG and HC. Among the GATK variant callers, HC-defaults seem to have a high concordance rate with comparably low false-positive and false-negative rates. Among the VarScan parameter sets, VS-pvalue shows better performance than others. These variants can then be filtered with quality, frequency, etc., depending on the study. Of the somatic variant callers, MuTect from Broad Institute detected many variants among tumor–normal pairs when compared with Strelka and VarScan. Although somatic variants were detected, almost all of them were rejected by the inbuilt filtering algorithms of the variant callers. Several criteria were used for filtering somatic variants. For example, MuTect rejects the variants with strand artifacts, poor mapping regions, triallelic sites, etc., whereas Strelka considers chromosomal mean depth, fraction of base calls filtered, etc. Apart from the filtering criteria, the region of interest also restricts the somatic variant calls and thus resulted in the detection of very few variants (Fig. 7).
Figure 5.

Concordance among TCGA-derived variants and our workflow-derived variants.
Note: X-axis denotes preprocessing steps and Y-axis specifies the percentage of true positive variants when compared with TCGA variants. The results from variant calls were color coded.
Figure 6.

Discordance among the TCGA-derived and workflow-derived variants.
Notes: X-axis denotes the false-negative values, while Y-axis denotes false-positive values. Both axes were log transformed. The pale blue and yellow boxes indicate phase 1 and phase 2 boxplots of the axes variables, respectively. The results from variant callers were color coded.
Figure 7.

Somatic variants called from the region of interest from TCGA dataset.
Notes: X-axis denotes the different variant callers used, while Y-axis denotes the number of somatic mutations detected. Y-axis was log transformed. Results from filtering were color coded.
The tertiary stage involves the annotation of detected variants. The SNP ids were annotated from the dbSNP database.88 The functional predictions of potential nonsynonymous variants were annotated from dbNSFP database,118 which provides prediction scores from SIFT, Polyphen 2, LRT, etc. SnpSift (http://snpeff.sourceforge.net/SnpSift.html) was the tool platform used for the annotation processes. It was followed by predicting effect, effect impact, codon change, etc., performed using SnpEff.119 An additional way to comprehensively understand the effect of SNPs is to visualize the annotated variants. The figures in Supplementary File 2 represent the relative genomic positions of SNPs and the exon regions of genes of interest.
The quaternary stage is network-based analysis, with the aim of associating the genes with pathological phenotypes. We performed the analysis similar to that described in the study of TCGA, counting the mutational frequency of these genes of interest and applying the human molecular signaling map149 to locate the potential molecular influences of these genes on cancer development (Fig. 8). In this way, it is easy for investigators to comprehend or identify the major therapeutic targets for prevention and for halting tumorigenesis from a global molecular perspective.
Figure 8.
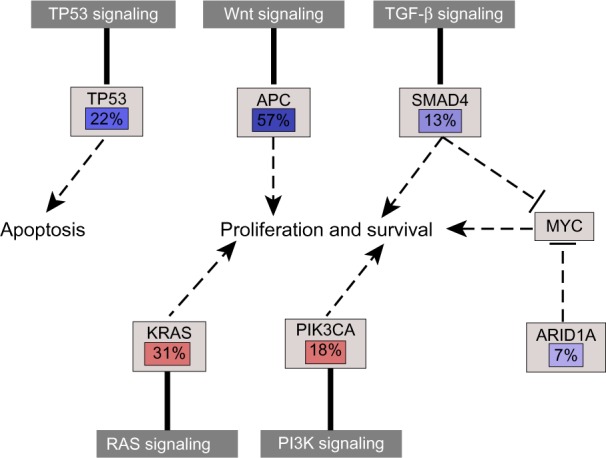
Frequencies of genetic changes leading to deregulation of pathways, which is associated with pathological phenotypes.
Notes: Red denotes activated genes and blue denotes inactivated genes.
Conclusion and Summary
NGS technologies perform massively parallel sequencing, which can facilitate high-throughput genome data sequencing and provide an unprecedented opportunity for genome research. Thus, NGS technologies can become an essential part of individualized precision medicine. Although a large number of NGS-related tools and softwares have been developed for specific purposes with NGS data, there has not been a generalized NGS data analysis protocol that can be interpreted easily and generate results that can be reproduced independently. To face this challenge, we have set out a four-stage NGS workflow that gives an overview for adoption of NGS technology in a clinical context for individualized medicine. The primary stage is an instrument-dependent stage, focused on the elucidation of the effects and influence of different combinations of sequencing protocols on the NGS data. The choice of NGS platform and its related sequencing protocol could be the main factor influencing downstream analysis. The secondary stage is an algorithm-dependent stage, which is a critical part of any NGS-based project. This stage introduces diverse methods and related algorithms for different purposes of processing NGS data and provides an indication of how to process NGS data with optimization of cost, time, and effort. The tertiary stage is an application-dependent stage, which summarizes five kinds of approaches for identification of driver mutations. The quaternary stage is a patient-dependent stage, which is the key stage for combining patient data with molecular modeling approaches. This stage reveals the importance and advantage of pathway- and network-based analyses for the purpose of NGS-related personalized medicine. In summary, this four-stage workflow provides an opportunity to examine the possible benefits of incorporating NGS data into individual patient care and attempts to lay out a workflow structure for optimized storage of NGS data and results.
Different research reports show that clinicians possess critically low knowledge and experience of combining treatments with application of multiplex genomic test results.165,166 This could be the major reason why many clinical centers join molecular tumor boards (MTBs), which include expertise from molecular pathologists, medical oncologists, bioinformaticians, genetic counselors, and others in order to serve as treatment advice organizations for individualized medicine.167,168 Unfortunately, most MTBs still apply gene panels with limited numbers of genes and cannot fully utilize the advantages of NGS sequencing technologies.
We believe that our proposed four-stage workflow could function as guidance for MTBs to resolve the situation of lack of NGS-based experience. For example, the primary stage can provide a clear indication of which sequencing platform should be applied, and how much the related cost for sequencing and downstream data analysis might be. This gives organizers of an MTB a basis for calculation of a treatment budget. The secondary and tertiary stages illustrate how disease-related mutations are identified and classified. The procedures of both stages can be designed and interpreted by MTB researchers in bioinformatics, clinical genetics, and translational science, enabling them to determine the driver mutations or disease-causing mutations. The result of the quaternary stage can be used by pathway analysis specialists, medical and surgical oncologists, and pathologists to predetermine the scope of possible therapeutic interventions that can be discussed with other members of an MTB to determine the optimal treatment option for an individual. Furthermore, regarding the educational efforts, the workflow might foster better cooperation within an MTB and provide basic knowledge for bench scientists and investigators who are redirected to this new field.
Because of rapid advances in NGS technologies, the generation of new data and the corresponding scientific publications are happening at a previously unprecedented rate. Unfortunately, diverse evidence shows that majority of these scientific findings cannot stand the test of time and suffer the problem of irreproducibility.169 Our proposed four-stage workflow can be incorporated into the Galaxy framework170 or other web services to help with constructing a basic framework for scientific reproducibility. Although it has been widely recognized that NGS technologies have the strongest potential for a powerful clinical diagnostic and prognostic application, there still exist multiple challenges for the interpretation of NGS data. These must be overcome in order to make such technologies routine in clinical applications.
A robust clinical protocol is still needed to extract high-quality DNA from different tumor samples in order to create a good sequencing library, especially, because most tumor samples are stored in the form of formalin-fixed, paraffin-embedded (FFPE) samples. FFPE storage may damage DNA or decrease DNA quality.171 Furthermore, many tumor samples are available in limited amounts, such as small core needle biopsies or the small cell blocks generated by separating malignant pleural effusion.172 Fluid biopsies may also offer new potential to harvest DNA of interest.
The criteria for the selection of tumor specimens need to be considered carefully, because of tumor heterogeneity and low quantity of tumor nuclei in some cases.173 Therefore, uniformly high sequence coverage and appropriate analysis approaches are needed.
The nature of NGS data encompasses different technological and biological biases, as well as systematic errors, that may result from different sources including uncertainties in read alignments,174,175 batch effects,175 sequence effects and base calling sequence error,174–177 platform-specific mechanistic problems,178,179 and others.179,180 Therefore, the careful processes within each stage of NGS workflow are critical to reduce the potential error rate for the final result and interpretation.
Many studies provide evidence that genetic intratumor heterogeneity may be the major reason for failure in prognosis, diagnosis, and treatment.181–184 Recently, regarding different cancer types, several studies have shown clear quantitative differences of genetic aberrations between primary tumor sites and metastatic sites or recurrent sites using NGS-based methods.185–187 This trait of tumor heterogeneity might elicit a main reason why a treatment of monotherapy can result in resistance in many cases, and combination therapy might be effective. Given these facts, it is advisable to define and use the tissue material of a tumor carefully, following a clear protocol in order to ensure an appropriate starting point of NGS workflow.
Supplementary Materials
Supplementary File 1. Comparision of variant calling tools for five colon-cancer patients.
Supplementary File 2. Visualization of variants from cancer genes.
Acknowledgments
We thank the Cancer Genome Atlas for the collaboration and professional technical support.
Abbreviations
- ABySS
assembly by short sequences
- ASA
assembly-based approach
- BWA/T
Burrows–Wheeler aligner/transform
- B/SAM
Binary/Sequence alignment/map
- ChIP
chromatin immunoprecipitation
- CIMP
CpG island methylator phenotype
- CMOS
complementary metal-oxide semiconductor
- CNV
copy-number variation
- C/GPU
central/graphic processing unit
- COSMIC
Catalogue of somatic mutations in cancer
- CRC
colorectal cancer
- CRT
cyclic reversible termination
- CT
convolution test
- CUDA
compute unified device architecture
- CWL
common workflow language
- DOC
depth of coverage
- DNA
deoxyribonucleic acid
- dNTP
deoxyribonucleoside triphosphate
- FCPT
Fisher’s combined P-value test
- FDR
false discovery rate
- FFPE
formalin-fixed, paraffin-embedded
- FLRT
fluorescently labeled reversible terminator
- GATK
Genome Analysis Toolkit
- GESEA
gene set enrichment analysis
- GEM
Genome Multitool
- GSNAP
Genomic Short-read Nucleotide Alignment Program
- HC
HaplotypeCaller
- HGMD
Human Gene Mutation Database
- IGA
Illumina Genome Analyzer
- IGV
Integrative Genomics Viewer
- ISFET
ion-sensitive field-effect transistor
- LRT
likelihood ratio test
- MBA
model building algorithm
- MPG
most probable genotype
- MTB
molecular tumor board
- MuSiC
mutational significance in cancer
- NGS
next-generation sequencing
- OMIM
Online Mendelian Inheritance in Man
- PacBio
Pacific Biosciences
- PCR
polymerase chain reaction
- PEM
paired-end mapping
- PGM
Personal Genome Machine
- PPI
protein–protein interaction
- RNA
ribonucleic acid
- RRBS
reduced representation bisulfite sequencing
- RTS
real-time sequencing
- SAPY
single-nucleotide addition via pyrosequencing
- SBL
sequencing by ligation
- SMRT
single-molecular real-time
- SNV/P
single-nucleotide variant/polymorphism
- SOAP
Short Oligonucleotide Analysis Package
- SRM
split read mapping
- SOLiD
sequencing by oligonucleotide ligation and detection
- SV
structural variant
- TCGA
The Cancer Genome Atlas
- UG
Unified Genotyper
- WGS/WES
whole-genome/whole-exome sequencing.
Footnotes
ACADEMIC EDITOR: J. T. Efird, Editor in Chief
FUNDING: This study is funded by the German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
PEER REVIEW: Nine peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2,277 words, excluding any confidential comments to the academic editor.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Contributed to conception and design of this study: JL, URM. Collected the clinical data: JL, AMNB, BG. Performed the analysis: AMNB. Interpreted the data result: JL, AMNB, URM. All the authors drafted the manuscript together. Gave the final approval for publication: URM.
REFERENCES
- 1.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2 A as frequently mutated in melanoma. Nat Genet. 2011;43:442–6. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Totoki Y, Tatsuno K, Yamamoto S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–9. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 6.Yan XJ, Xu J, Gu ZH, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3 A in acute monocytic leukemia. Nat Genet. 2011;43:309–15. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 7.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McPherson JD. Next-generation gap. Nat Methods. 2009;6(11 suppl):S2–5. doi: 10.1038/nmeth.f.268. [DOI] [PubMed] [Google Scholar]
- 9.Vazquez M, de la Torre V, Valencia A. Chapter 14: cancer genome analysis. PLoS Comput Biol. 2012;8(12):e1002824. doi: 10.1371/journal.pcbi.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linnarsson S. Recent advances in DNA sequencing methods – general principles of sample preparation. Exp Cell Res. 2010;316:1339–43. doi: 10.1016/j.yexcr.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 11.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100:8817–22. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedurco M, Romieu A, Williams S, Lawrence I, Turcatti G. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 2006;34:e22. doi: 10.1093/nar/gnj023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leamon JH, Lee WL, Tartaro KR, et al. A massively parallel PicoTiterPlate based platform for discrete picoliter-scale polymerase chain reactions. Electrophoresis. 2003;24:3769–77. doi: 10.1002/elps.200305646. [DOI] [PubMed] [Google Scholar]
- 14.Harris TD, Buzby PR, Babcock H, et al. Single-molecule DNA sequencing of a viral genome. Science. 2008;320:106–9. doi: 10.1126/science.1150427. [DOI] [PubMed] [Google Scholar]
- 15.Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 16.Williams JGK. System and methods for nucleic acid sequencing of single molecules by polymerase synthesis. US Patent. 1998;6:255, 083. [Google Scholar]
- 17.Hardin S, Gao X, Briggs J, Willson R, Tu SC. Methods for real-time single molecule sequence determination. US Patent. 2000;7:329–492. [Google Scholar]
- 18.Lou DI, Hussmann JA, McBee RM, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc Natl Acad Sci U S A. 2013;110(49):19872–7. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–52. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 20.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siqueira JF, Jr, Fouad AF, Rôças IN. Pyrosequencing as a tool for better understanding of human microbiomes. J Oral Microbiol. 2012;4:10743. doi: 10.3402/jom.v4i0.10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzker ML. Sequencing technologies – the next generation. Nat Rev. 2010;11:30. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 23.Braslavsky I, Hebert B, Kartalov E, Quake SR. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci U S A. 2003;100:3960–4. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ju J, Kim DH, Bi L, et al. Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc Natl Acad Sci U S A. 2006;103:19635–40. doi: 10.1073/pnas.0609513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo J, Xu N, Li Z, et al. Four-color DNA sequencing with 3′-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proc Natl Acad Sci U S A. 2008;105:9145–50. doi: 10.1073/pnas.0804023105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shendure J, Porreca GJ, Reppas NB, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–32. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 27.Valouev A, Ichikawa J, Tonthat T, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–63. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang YF, Chen SC, Chiang YS, Chen TH, Chiu KP. Palindromic sequence impedes sequencing-by-ligation mechanism. BMC Syst Biol. 2012;6(suppl 2):S10. doi: 10.1186/1752-0509-6-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8(3):175–85. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 31.Branton D, Deamer DW, Marziali A, et al. The potential and challenges of nanopore sequencing. Nat Biotechnol. 2008;26:1146–53. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng P, Oliver PM, Barrett MJ, Vezenov D. Progress toward the application of molecular force spectroscopy to DNA sequencing. Electrophoresis. 2012;33:3497–505. doi: 10.1002/elps.201200351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding F, Manosas M, Spiering MM, et al. Single-molecule mechanical identification and sequencing. Nat Methods. 2012;9:367–72. doi: 10.1038/nmeth.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bell DC, Thomas WK, Murtagh KM, et al. DNA base identification by electron microscopy. Microsc Microanal. 2012;18:1049–53. doi: 10.1017/S1431927612012615. [DOI] [PubMed] [Google Scholar]
- 35.Treffer R, Lin X, Bailo E, et al. Distinction of nucleobases – a tip-enhanced Raman approach. Beilstein J Nanotechnol. 2011;2:628–37. doi: 10.3762/bjnano.2.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heger M. Illumina Launches Two New NGS Instruments: Desktop Platform and ‘Factory-Scale’ System. Sequence/GenomeWeb; 2014. [Accessed July 26, 2014]. Available at: http://www.genomeweb.com/sequencing/illumina-launches-two-new-ngs-instruments-desktop-platform-and-factory-scale-sys. [Google Scholar]
- 37.Wang Y, Yang Q, Wang Z. The evolution of nanopore sequencing. Front Genet. 2014;5:449. doi: 10.3389/fgene.2014.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Park P. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet. 2013;21:134–42. doi: 10.1038/ejhg.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeltsch A, Zhang YY. The application of next generation sequencing in DNA methylation analysis. Gene. 2010;1:85–101. doi: 10.3390/genes1010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Schmidt B, Maskell DL. CUSHAW: a CUDA compatible short read aligner to large genomes based on the Burrows-Wheeler transform. Bioinformatics. 2012;28(14):1830–7. doi: 10.1093/bioinformatics/bts276. [DOI] [PubMed] [Google Scholar]
- 48.Santiago MS, Sammeth M, Roderic G, Paolo R. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods. 2012;9(12):1185–8. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 49.Wu T, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26(7):873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trapnell C, Pachter L, Salzberg S. Tophat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burrows M, Wheeler DJ. A Block Sorting Lossless Data Compression Algorithm. Palo Alto: Digital Equipment Corporation; 1994. p. 124. Technical Report. [Google Scholar]
- 52.Ferragina M, Manzini G. Opportunistic data structures with applications; Foundations of Computer Science, 2000. Proceedings. 41st Annual Symposium on; IEEE; 2000. pp. S.390–8. [Google Scholar]
- 53.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lippert RA. Space-efficient whole genome comparisons with Burrows-Wheeler transforms. J Comput Biol. 2005;12:407–15. doi: 10.1089/cmb.2005.12.407. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, Schmidt B. Long read alignment based on maximal exact match seeds. Bioinformatics. 2012;28(18):i318–24. doi: 10.1093/bioinformatics/bts414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Popp B, Schmidt B. CUSHAW3: sensitive and accurate base-space and color-space short-read alignment with hybrid seeding. PLoS One. 2014;9(1):e86869. doi: 10.1371/journal.pone.0086869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–4. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 58.Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 59.Liu CM, Wong T, Wu E, et al. SOAP3: ultra-fast GPU-based parallel alignment tool for short reads. Bioinformatics. 2012;28(6):878–9. doi: 10.1093/bioinformatics/bts061. [DOI] [PubMed] [Google Scholar]
- 60.Luo R, Wong T, Zhu J, et al. SOAP3-dp: fast, accurate and sensitive GPU-based short read aligner. PLoS One. 2013;8(5):e65632. doi: 10.1371/journal.pone.0065632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cloonan N, Forrest AR, Kolle G, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–9. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 63.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. Tophat2: accurate alignment of transcriptomes in the presence of insertion, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hernandez D, François P, Farinelli L, Osterås M, Schrenzel J. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–9. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012;22(3):549–56. doi: 10.1101/gr.126953.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schatz M, Deicher A, Salzberg S. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20(9):1165–73. doi: 10.1101/gr.101360.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bao S, Jiang R, Kwan W, Ma X, Song Y. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011;56(6):406–14. doi: 10.1038/jhg.2011.43. [DOI] [PubMed] [Google Scholar]
- 70.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 71.Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–5. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koboldt DC, Ding L, Mardis ER, Wilson RK. Challenges of sequencing human genomes. Brief Bioinform. 2010;11:484–98. doi: 10.1093/bib/bbq016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Teer JK, Bonnycastle LL, Chines PS, et al. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–31. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next- generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotech. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 82.Cancer Genome Atlas Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cancer Genome Atlas Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Banerji S, Cibulskis K, Rangel-Escareno C, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lower M, Renard BY, de Graaf J, et al. Confidence-based somatic mutation evaluation and prioritization. PLoS Comput Biol. 2012;8:e1002714. doi: 10.1371/journal.pcbi.1002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zook JM, Chapman B, Wang J, et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol. 2014;32(3):246–51. doi: 10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
- 88.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 90.Sharp AJ, Cheng Z, Eichler EE. Structural variation of the human genome. Annu Rev Genomics Hum Genet. 2006;7:407–42. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- 91.Tuzun E, Sharp AJ, Bailey JA, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37(7):727–32. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 92.Korbel JO, Abyzov A, Mu XJ, et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hodis E, Watson IR, Kryukov GV, et al. Psoriasis is associated with increased bold beta-defensin genomic copy number. Nat Genet. 2008;40:23–5. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–8. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–55. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 96.Chen K, Wallis JW, McLellan MD, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6:S13–20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- 98.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abyzov A, Gerstein M. AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics. 2011;27:595–603. doi: 10.1093/bioinformatics/btq713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–9. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang Z, Hormozdiari F, Yang W-Y, Halperin E, Eskin E. Research in Computational Molecular Biology Chor B. Vol. 7262. Berlin; Heidelberg: Springer; 2012. CNVeM: copy number variation detection using uncertainty of read mapping; pp. 326–40. Lecture Notes in Computer Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sathirapongsasuti JF, Lee H, Horst BA, et al. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–54. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Campbell PJ, Pleasance ED, Stephens PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A. 2008;105:13081–6. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chiang DY, Getz G, Jaffe DB, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–92. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet. 2012;44:226–32. doi: 10.1038/ng.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nijkamp JF, van den Broek MA, Geertman JM, Reinders MJ, Daran JM, de Ridder D. De novo detection of copy number variation by co-assembly. Bioinformatics. 2012;28(24):3195–202. doi: 10.1093/bioinformatics/bts601. [DOI] [PubMed] [Google Scholar]
- 109.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s online Mendelian inheritance in man (OMIM) Nucleic Acids Res. 2009;37:D793–6. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sabeti PC, Varilly P, Fry B, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–8. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(D1):D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc Series B. 1996;58(1):267–88. [Google Scholar]
- 114.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guy L, Kultima JR, Andersson GE. genoPlotR: comparative gene and genome visualization in R. Bioinformatics. 2010;26(18):2334–5. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gu Z, Gu L, Eils R, Schlesner M, Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 117.Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32(8):894–9. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Makarov V, O’Grady T, Cai G, Lihm J, Buxbaum JD, Yoon S. AnnTools: a comprehensive and versatile annotation toolkit for genomic variants. Bioinformatics. 2012;28:724–5. doi: 10.1093/bioinformatics/bts032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–9. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Johansen CT, Wang J, Lanktree MB, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–7. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gonzalez-Perez A, Deu-Pons J, Lopez-Bigas N. Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med. 2012;4:89. doi: 10.1186/gm390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kaminker JS, Zhang Y, Watanabe C, Zhang Z. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res. 2007;35:W595–8. doi: 10.1093/nar/gkm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Capriotti E, Altman RB. A new disease-specific machine learning approach for the prediction of cancer-causing missense variants. Genomics. 2011;98:310–7. doi: 10.1016/j.ygeno.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maerkl SJ, Quake SR. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007;315(5809):233–7. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 128.Badis G, Berger MF, Philippakis AA, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324(5935):1720–3. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kamburov A, Lawrence MS, Polak P, et al. Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc Natl Acad Sci U S A. 2015;112(40):E5486–95. doi: 10.1073/pnas.1516373112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dees ND, Zhang Q, Kandoth C, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22(8):1589–98. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Reimand J, Bader GD. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013;9:637. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schwender H, Ruczinski I. Logic regression and its extensions. Adv Genet. 2010;72:25–45. doi: 10.1016/B978-0-12-380862-2.00002-3. [DOI] [PubMed] [Google Scholar]
- 135.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 136.Tian L, Alizadeh AA, Gentles AJ, Tibshirani R. A simple method for estimating interactions between a treatment and a large number of covariates. J Am Stat Assoc. 2014;109(508):1517–32. doi: 10.1080/01621459.2014.951443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Matsui S, Simon R, Qu P, Shaughnessy JD, Barlogie B, Crowley J. Developing and validating continuous genomic signatures in randomized clinical trials for predictive medicine. Clin Cancer Res. 2012;18(21):6065–73. doi: 10.1158/1078-0432.CCR-12-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Werner HM, Mills GB, Ram PT. Cancer systems biology: a peek into the future of patient care? Nat Rev Clin Oncol. 2014;11(3):167–76. doi: 10.1038/nrclinonc.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 140.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chasman DI. On the utility of gene set methods in genomewide association studies of quantitative traits. Genet Epidemiol. 2008;32:658–68. doi: 10.1002/gepi.20334. [DOI] [PubMed] [Google Scholar]
- 144.Ritchie MD. Using prior knowledge and genome-wide association to identify pathways involved in multiple sclerosis. Genome Med. 2009;1:65. doi: 10.1186/gm65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Peng G, Luo L, Siu H, et al. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010;18:111–7. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gortzak-Uzan L, Ignatchenko A, Evangelou AI, et al. A proteome resource of ovarian cancer ascites: integrated proteomic and bioinformatic analyses to identify putative biomarkers. J Proteome Res. 2008;7:339–51. doi: 10.1021/pr0703223. [DOI] [PubMed] [Google Scholar]
- 147.Rual J-P, Venkatesan K, Hao T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 148.Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–68. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 149.Li J, Mansmann UR. A molecular signaling map and its application. Cell Signal. 2014;26(12):2834–42. doi: 10.1016/j.cellsig.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 150.Durate NC, Becker SA, Jamshidi N, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104:1777–82. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Krzywinski M, Birol I, Jones SJ, Marra MA. Hive plots – rational approach to visualizing networks. Brief Bioinform. 2012;13(5):627–644. doi: 10.1093/bib/bbr069. [DOI] [PubMed] [Google Scholar]
- 153.Vastrik I, D’Eustachio P, Schmidt E, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mi H, Thomas P. Protein Networks and Pathway Analysis. Humana Press; 2009. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools; pp. 123–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Sys Biol. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bordbar A, Mo LM, Nakayasu ES, et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol Syst Biol. 2012;8:558. doi: 10.1038/msb.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Notebaart RA, Teusink B, Siezen RJ, Papp B. Co-regulation of metabolic genes is better explained by flux coupling than by network distance. PLoS Comput Biol. 2008;4:e26. doi: 10.1371/journal.pcbi.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wessely F, Bartl M, Guthke R, Li P, Schuster S, Kaleta C. Optimal regulatory strategies for metabolic pathways in Escherichia coli depending on protein costs. Mol Syst Biol. 2011;7:515. doi: 10.1038/msb.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sadeh MJ, Moffa G, Spang R. Considering unknown unknowns: reconstruction of nonconfoundable causal relations in biological networks. J Comput Biol. 2013;20(11):920–32. doi: 10.1089/cmb.2013.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Merkel D. Docker: lightweight linux containers for consistent development and deployment. Linux J. 2014;239:2. [Google Scholar]
- 162.Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342(6250):705–8. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 163.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120(19):3327–35. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- 164.Jančík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. Biomed Res Int. 2010;2010:150960. doi: 10.1155/2010/150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hall MJ. Conflicted confidence: academic oncologists’ views on multiplex pharmacogenomic testing. J Clin Oncol. 2014;32:1290–2. doi: 10.1200/JCO.2013.54.8016. [DOI] [PubMed] [Google Scholar]
- 166.Gray SW, Hicks-Courant K, Cronin A, et al. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32:1317–23. doi: 10.1200/JCO.2013.52.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schwaederle M, Parker BA, Schwab RB, et al. Molecular tumor board: the University of California San Diego Moores cancer center experience. Oncologist. 2014;19(6):631–6. doi: 10.1634/theoncologist.2013-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tafe LJ, Gorlov IP, de Abreu FB, et al. Implementation of a molecular tumor board: the impact on treatment decisions for 35 patients evaluated at Dartmouth-Hitchcock Medical Center. Oncologist. 2015;20(9):1011–8. doi: 10.1634/theoncologist.2015-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Begley CG, Ioannis J. Reproducibility in science: improving the standard for basic and preclinical research. Circ Res. 2015;116:116–26. doi: 10.1161/CIRCRESAHA.114.303819. [DOI] [PubMed] [Google Scholar]
- 170.Goecks J, Nekrutenko A, Taylor J, The Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hadd AG, Houghton J, Choudhary A, et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15:234–47. doi: 10.1016/j.jmoldx.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 172.Kerick M, Isau M, Timmermann B, et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics. 2011;4:68. doi: 10.1186/1755-8794-4-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23:843–54. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lassmann T, Hayashizaki Y, Daub CO. SAMStat: monitoring biases in next generation sequencing data. Bioinformatics. 2011;27:130–1. doi: 10.1093/bioinformatics/btq614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Taub MA, Corrada Bravo H, Irizarry RA. Overcoming bias and systematic errors in next generation sequencing data. Genome Med. 2010;2:87. doi: 10.1186/gm208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nakamura K, Oshima T, Morimoto T, et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina genome analyzer using machine learning strategies. Genome Biol. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36(16):e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Meacham F, Boffelli D, Dhahbi J, Martin DI, Singer M, Pachter L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12:451. doi: 10.1186/1471-2105-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bainbridge MN, Wang M, Burgess DL, et al. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 2010;11(6):R62. doi: 10.1186/gb-2010-11-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer. 2010;103:1139–43. doi: 10.1038/sj.bjc.6605912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Campbell PJ, Stephens PJ, Pleasance ED, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–9. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sottoriva A, Spiteri I, Piccirillo SG, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009–14. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang J, Fujimoto J, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346(6206):256–9. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 191.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26(11):1293–300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rozowsky J, Euskirchen G, Auerbach RK, et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009;27(1):66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein–DNA binding sites from ChIP-seq data. Nucleic Acids Res. 2008;36(16):5221–31. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27(11):1571–2. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chen PY, Cokus SJ, Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics. 2010;11(1):203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xi Y, Li W. BSMAP: whole genome bisulfite sequence mapping program. BMC Bioinformatics. 2009;10(1):1. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jünemann S, Prior K, Albersmeier A, et al. GABenchToB: a genome assembly benchmark tuned on bacteria and benchtop sequencers. PLoS One. 2014;9(9):e107014. doi: 10.1371/journal.pone.0107014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Farrer RA, Kemen E, Jones JD, Studholme DJ. De novo assembly of the Pseudomonas syringae pv. syringae B728a genome using Illumina/Solexa short sequence reads. FEMS Microbiol Lett. 2009;291(1):103–11. doi: 10.1111/j.1574-6968.2008.01441.x. [DOI] [PubMed] [Google Scholar]
- 202.Lin Y, Li J, Shen H, Zhang L, Papasian C, Deng HW. Comparative studies of de novo assembly tools for next-generation sequencing technologies. Bioinformatics. 2011;27(15):2031–7. doi: 10.1093/bioinformatics/btr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.DiGuistini S, Liao NY, Platt D, et al. De novo genome sequence assembly of a filamentous fungus using Sanger, 454 and Illumina sequence data. Genome Biol. 2009;10(9):1–12. doi: 10.1186/gb-2009-10-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Liu Y, Schmidt B, Maskell D. Parallelized short read assembly of large genomes using de Bruijn graphs. BMC Bioinformatics. 2011;12:354. doi: 10.1186/1471-2105-12-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yi M, Zhao Y, Li J, He M, Kebebew E, Stephens R. Performance comparison of SNP detection tools with illumina exome sequencing data – an assessment using both family pedigree information and sample-matched SNP array data. Nucleic Acids Res. 2014;42(12):e101. doi: 10.1093/nar/gku392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Khatkar MS, Moser G, Hayes BJ, Raadsma HW. Strategies and utility of imputed SNP genotypes for genomic analysis in dairy cattle. BMC Genomics. 2012;13(1):1. doi: 10.1186/1471-2164-13-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1. Comparision of variant calling tools for five colon-cancer patients.
Supplementary File 2. Visualization of variants from cancer genes.