

Abstract
Type 2 diabetes is one of the leading causes of morbidity and mortality, consuming a significant proportion of public health spending. Oral hypoglycemic agents (OHAs) are the frontline treatment approaches after lifestyle changes. However, huge interindividual variation in response to OHAs results in unnecessary treatment failure. In addition to nongenetic factors, genetic factors are thought to contribute to much of such variability, highlighting the importance of the potential of pharmacogenetics to improve therapeutic outcome. Despite the presence of conflicting results, significant progress has been made in an effort to identify the genetic markers associated with pharmacokinetics, pharmacodynamics, and ultimately therapeutic response and/or adverse outcomes to OHAs. As such, this article presents a comprehensive review of current knowledge on pharmacogenetics of OHAs and provides insights into knowledge gaps and future directions.
Keywords: pharmacogenetics, type 2 diabetes, oral hypoglycemic agents, pharmacokinetics, pharmacodynamics, response
Introduction
Type 2 diabetes (T2D) is a complex disease characterized by persistent hyperglycemia as a result of insufficient insulin secretion, usually in the context of reduced insulin action. Frightening trends in morbidity and mortality of the disease are being observed. According to a recent estimate, some 382 million people between the ages of 20 years and 79 years live with diabetes, increasing to 592 million by 2035.1 T2D accounts for 85%–95% of the cases. According to the International Diabetes Federation, diabetes is the fifth leading cause of death, and it consumes ~11% of the global health care spending.2
Following initial dietary and lifestyle changes, the most common treatment for T2D is the addition of oral hypoglycemic agents (OHAs), with a progressive addition of agents over time before insulin treatment is required to maintain glycemia at target. Currently available treatments include biguanides, sulfonylureas (SUs), meglitinides (glinides), thiazolidinediones (TZDs), α-glucosidase inhibitors, glucagon-like peptide (GLP)-1 receptor (GLP-1R) agonists, dipeptidyl peptidase (DPP)-4 inhibitors, and sodium glucose transporter (SGLT)-2 inhibitors. Despite the availability of several OHAs, only 53% of Diabetes Mellitus patients achieve a target glycated hemoglobin (HbA1c) of <7.0%.3
There is a considerable interindividual variability in drug response, measured in terms of efficacy or adverse drug outcomes, in T2D. A complex interaction of biological and nonbiological factors could explain this variability. While adherence to prescribed treatment, access to health care, and prescribing practice are some of the nonbiological factors,4 biological factors could be either genetic or nongenetic. Nongenetic biological factors influencing response to OHAs are related to intestinal, hepatic, or renal function in addition to age, sex, and body weight. Pharmacogenomics is the study of genetic factors affecting efficacy or undesired effects of drugs. In this review, we assess the published evidence for the presence of gene–drug interactions in T2D and appraise the usage of such evidence to understand pharmacokinetics (PK) and/or pharmacodynamics (PD) of diabetes drugs and predict therapeutic response or adverse drug outcomes.
Challenges in the study of drug response in diabetes
In designing, conducting, and interpreting pharmacogenetics studies, there are a number of factors that should be considered, including how drug response is defined, what covariates are included in the model, and how to account for difference in baseline HbA1c, the need for large sample size, comorbidities, and drug interactions. The field of pharmacogenetics is plagued with many positive but very small studies that cannot be replicated, with only a few consistent pharmacogenetic findings. For SUs, the most robust findings are for cytochrome P450, family 2, subfamily C, polypeptide 9 (CYP2C9), ATP-binding cassette, sub-family C, member 8 (ABCC8), and transcription factor 7-like 2 (TCF7L2), and for metformin, they are for ataxia telangiectasia mutated (ATM) and possibly for multidrug and toxin extrusion (MATE) 1, with no consistent variants associated with response to glitazones, DPP-4 inhibitors, SGLT-2 inhibitors, or GLP-1R agonists – in part because no large studies have been done in these areas. In this review, we will first highlight the challenges in the study of drug response in diabetes before reviewing the literature in relation to genetic variation in PK and PD of all commonly used diabetes treatments. We highlight the key findings and whether they replicate or not.
Study design and confounders
Prospective genotype blind studies are optimal for pharmacogenetic studies. However, they require a large sample size and are costly and time consuming. Therefore, the majority of published studies are either retrospective or case–control in design and therefore at risk of selection bias and confounding.
The association between genetic variants and drug response may be confounded by multiple factors. Baseline HbA1c has a strong effect on response and should be considered in any model of glycemic response.5 Other factors, such as dose, drug group, and kidney and liver function tests, may alter magnitude and direction of reported effect sizes. Furthermore, adherence, estimated to range from 36%–93% in diabetic patients, could also be an important confounder.6 While it is a reasonable assumption that most covariates that alter response are not correlated with genotype, care should be taken to evaluate these covariates in any pharmacogenetic response models.
Selection of genes/SNPs
To date, most pharmacogenetic studies of OHAs adopted a candidate gene approach. Based on the PK and PD knowledge of the agents, genetic polymorphisms in transporter genes, metabolizing enzyme genes, and target genes were investigated. Apart from the largely consistent associations observed between CYP2C9*2/*3, Potassium Channel, Inwardly Rectifying Subfamily J, Member 11 (KCNJ11)/ABCC8, and TCF7L2 for response to SU, no other pharmacogenetic impact has been robustly established by these candidate gene studies. The existence of gene–gene interaction, as suggested by a few recent pharmacogenetic studies of metformin response, could be the explanation for some of the replication failure as the marginal impact of each individual variant would be much smaller and difficult to detect than in a true interaction model.
The genetic architecture of drug response, which encompasses the frequency, number, and effect size of genetic variants, has rarely been addressed for any commonly prescribed drug. A recent study showed that many common variants with small-to-moderate effect sizes together account for 20%–30% of variance in glycemic response to metformin.7 Given that these variants are likely to be distributed across the genome, a hypothesis-free Genome-Wide Association Study (GWAS) approach holds the potential to reveal more metformin response variants. Indeed, the only GWAS on OHAs published to date reported a robust association between glycemic response to metformin and variants at the ATM locus, which harbors no established candidate genes.8 With the ever-reducing cost of genotyping on microarrays, more drug response GWAS analyses are expected to reveal novel mechanistic insights and/or genetic markers that could predict an efficacy or safety of drugs in diabetes.
Sample size and power
When considering drug efficacy, the general disappointing lack of consistent replication in the candidate gene studies reviewed here suggests that none of the variants examined so far has a large impact on clinical outcomes. If the genetic architecture of treatment efficacy by other OHAs is similar to that of metformin, which is contributed by many common variants with small-to-moderate effect sizes, the large sample sizes will be essential to provide an adequate statistical power to uncover the variants. Moreover, when multiple variants are examined in a single study, such as the gene–gene interaction or GWAS analyses, even larger sample sizes, typically in the range of a few thousand, are required to compensate the statistical penalty associated with multiple testing. Most of the studies reviewed here used a few hundred individuals or less (column 4 or 6 in Tables S1–S5), which have probably resulted in the inconsistent reports, with an overrepresentation of positive results due to the winners’ curse and publication bias.9
However, it is worth noting that when considering more severe adverse reactions of drugs, such as metformin-induced lactic acidosis, a small sample size may be sufficient. This is seen most clearly in relation to drug-induced severe liver injury where the large impact causal variants were identified with just a few dozen samples.10,11 Therefore, genetic screening of rare severe adverse reactions with small samples is still warranted, provided that power calculations are presented to inform the range of effect sizes that could be excluded by the study design.
Choice and definition of end points
The phenotype for drug response is often variably defined depending on the available data that can make comparing the findings across the studies difficult. A linear term for HbA1c reduction or blood glucose reduction, or a dichotomous variable defined as achieving therapeutic target (HbA1c <7%) over a specified period of time, is the most commonly used end point in diabetes. Genetic determinants of safety and efficacy to the same drug might vary. However, some safety and efficacy measures may overlap and thus be associated with the same genes, for example, extreme response to SUs and hypoglycemia. The availability of multiple end points could increase the chance of selective outcome-reporting bias in pharmacogenetic studies. Therefore, consistent and functionally relevant response definitions where possible publishing a protocol in advance may be helpful.
Obesity and related comorbidities
Suboptimal glycemic control is usually associated with greater comorbidities, including hypertension and dyslipidemia. The fact that obesity and T2D are strongly linked led to the investigation of obesity as a clinical predictor of efficacy to OHAs. The first-line drug metformin showed similar efficacy in obese and nonobese T2D individuals.12,13 In another study, body mass index was not significantly associated with glycemic response to rosiglitazone, but responders had higher body fat percentage than nonresponders.14 Those with greater waist-to-hip ratio also showed a better reduction of fasting plasma glucose (FPG) and HbA1c when rosiglitazone was added to metformin and/or SUs.15
Drug–drug interactions
To achieve adequate glycemic control and treat concurrent pathologies, diabetic patients are often on polypharmacy, therefore there is a risk of drug–drug interactions.16 The concomitant administration of organic cation transporter (OCT) 1-inhibiting drugs with metformin is reported to increase the gut concentration of metformin and gastro intestinal (GI) side effects.17 Coadministration of CYP-inhibiting drugs with insulin secretagogues risks potentiating hypoglycemia. In healthy volunteers, simultaneous administration of gemfibrozil, a lipid-lowering drug that inhibits CYP2C8, and repaglinide resulted in an eightfold increase in the area under the concentration–time curve (AUC) of repaglinide that could prolong its hypoglycemic effect and warn precaution while prescribing.18 Therefore, in designing drug-response studies, common comorbidities and drug interactions should be considered.
Current state of evidence
Metformin
Metformin is the most widely prescribed first-line drug to treat T2D. There is a considerable interindividual variability in metformin’s glucose-lowering ability with approximately one-third of metformin users defined as poor responders.19 The mechanism for this variability, and indeed for the mechanism of action of metformin, remains uncertain. Metformin is also poorly tolerated by some individuals with up to 63% of patients experiencing metformin-induced gastrointestinal (GI) symptoms leading to 5%–10% premature discontinuation.20
Pharmacokinetics
Metformin is positively charged at physiological pH that renders it hydrophilic, resulting in limited passive diffusion. Therefore, metformin disposition is dependent on active transportation by OCTs (solute carrier family 22 [SLC22]) to cross the biological membranes. Plasma membrane monoamine transporter (PMAT), OCT1,21 and OCT3 are involved in the apical uptake of metformin into enterocytes (Figure 1). In addition, a recent in vitro study identified a possible role of serotonin and choline transporters.22 In the liver, OCT1 transports metformin to the hepatocytes23 with biliary excretion probably via MATE1.24 In the kidney, OCT2 is highly expressed in the basolateral membrane of the distal tubules of the kidney facilitating renal uptake,25 whereas MATE1 and MATE2, expressed in the apical membrane of the renal epithelial cells, are involved in the renal secretion of metformin.26
Figure 1.

Transport of metformin by organic cation transporters.
Abbreviations: MATE, multidrug and toxin extrusion antiporter; OCT, organic cation transporter; PMAT, plasma membrane monoamine transporter; Prox. Tub., proximal tubule.
Pharmacodynamics
Metformin is believed to lower blood glucose level by reducing hepatic glucose production and increasing insulin-mediated peripheral glucose utilization. Even though the molecular mechanisms of how metformin exerts its hypoglycemic action are still elusive, it inhibits mitochondrial complex I, resulting in decreased adenosine triphosphate (ATP), and increased adenosine monophosphate (AMP) levels. AMP activates adenosine monophosphate-activated protein kinase (AMPK),27 and this had been thought to mediate the suppression of gluconeogenesis. However, a preserved glucose-lowering effect of metformin has been reported in AMPK knockout mice studies.28 Recently, non-AMPK mechanisms have been proposed. One mechanism involves the inhibition of binding of AMP to adenylate cyclase by metformin, inhibiting its response to glucagon and disrupting downstream cAMP-PKA signaling. This inhibits enzymes of the gluconeogenic pathway in favor of glycolysis.29 An additional mechanism recently reported suggests that metformin inhibits mitochondrial glycerophosphate dehydrogenase enzyme with a subsequent augmentation of cytosolic redox state thereby reducing hepatic gluconeogenesis.30 Although AMPK is no longer believed to be required for glucose lowering, the lipid lowering and any potential cancer beneficial effects of metformin are probably mediated via this kinase.27
Pharmacogenetics
There is a considerable interindividual variation in metformin PK, PD, and adverse effects. The majority of genetic studies have focused on variation in the metformin transporters31 with more recent studies investigating the transcription factors (TCFs) that regulate these transporters19 and candidate genes in metformin PD. While substantial progress has been made to understand the detrimental effect of polymorphisms in transporter genes on PK, this does not robustly translate into drug response with inconsistent results being reported across many small studies (Table S1).
OCT1 and metformin efficacy
Nonsynonymous variants in the highly polymorphic SLC22A1 gene that encodes OCT1 have been reported to affect functionality.32 Studies in healthy and diabetic Caucasians showed the association of reduced function variants of OCT1 (G401S, R61C, 420del, and G465R) with a higher maximum plasma concentration (Cmax) and AUC, lower oral volume of distribution, increased pattern of renal secretary clearance,33 and decreased trough steady-state concentration.34
Several studies have been conducted in an effort to link OCT1 variants to the clinical efficacy of metformin. In an oral glucose tolerance test study carried out in 20 healthy volunteers (eight having reference OCT1 and 12 with at least one reduced-function OCT1 allele), subjects carrying OCT1 variants had a significantly higher (P=0.004) glucose AUC compared to those with the reference genotype after metformin treatment.35 A study carried out by Christensen et al showed individuals carrying the reduced function OCT1 alleles to have a significantly greater absolute HbA1c reduction during the initiation as well as maintenance period of treatment compared to carriers of the reference genotypes. However, the decrease became insignificant when adjusted for baseline HbA1c.34 The Genetics of Diabetes Audit and Research Tayside (GoDARTs) is the largest reported study to investigate OCT1 variants and glycemic response to metformin.36 The GoDARTs investigators studied the two most common loss-of-function OCT1 variants, R61C and 420del in 1,531 T2D patients treated with metformin. They showed no effect on a number of outcomes, including HbA1c reduction, odds of achieving treatment target, and hazard of monotherapy failure. Davis et al also showed no association of these variants with absolute change in HbA1c.37 The Rotterdam study that investigated eleven tagging single nucleotide polymorphisms in SLC22A1 gene could not find any significant association with response to metformin.38 The Diabetes Prevention Program study evaluated the protective role of metformin on the incidence of diabetes in 990 high-risk participants.39 The major allele of the missense SNP in OCT1, rs683369 encoding L160F, showed a significant protective effect (HR =0.69, 0.53–0.89, P=0.004). However, this variant is not reported to affect OCT1 functionality or in linkage disequilibrium (LD) with previously associated SNPs.
OCT1 and metformin intolerance: Tarasova et al screened the effect of seven variants in genes encoding transporter proteins in relation to GI side effects of metformin in 53 tolerant and 193 intolerant patients.40 Cases were defined as those with the presence of at least one of diarrhea, flatulence, abdominal pain, asthenia, and vomiting while being treated with metformin. Two OCT1 variants, rs628031 (M408V) and rs36056065 (8 bp insertion), that are in strong LD showed a protective effect (odds ratio [OR] =0.389 [95% confidence interval {CI} =0.186–0.815], P=0.012 and OR =0.405 [95% CI =0.226–0.724], P=0.002, respectively). In this same study, two of the loss-of-function OCT1 variants, rs12208357 (R61C) and rs34059508 (G465R), showed no significant association with intolerance. However, a recent GoDARTs study conducted on 2,166 (251 severely intolerant and 1,915 tolerant) T2D patients showed reduced activity OCT1 variants (rs12208357 [R61C], rs55918055 [C88R], rs34130495 [G401S], rs72552763 [M420del], and rs34059508 [G465R]) to be important determinants of metformin intolerance.17 Carriers of two reduced function alleles had 2.4 times higher odds (95% CI =1.48–3.93, P=0.001) of developing GI side effects. The concomitant use of other drugs known to inhibit OCT1 transport increased this risk to an OR of 4 (2.09–8.16, P<0.001). In this study, cases were patients who have been on immediate release metformin for <6 months and switched to another OHA (including modified release metformin) within 6 months after stopping the immediate release metformin; controls were defined as those patients who were on at least 2 g of metformin for >6 months.
OCT2: OCT2 is reported to account for ~80% of metformin’s renal clearance.25 Studies in healthy and diabetic individuals showed an association of reduced function OCT2 variants (T199I, T201M, and A270S) with an increased plasma concentration and a reduced renal clearance of metformin.41–44 However, other studies showed no association.33,45
Studies that aimed to link OCT2 variants with response to metformin have been focused on the A270S variant. Most of the reported studies do not show any association of this variant with response to metformin modeled as a dichotomous trait,42 linear HbA1c reduction,38 or GI side effect.40 However, a recent study in 209 newly diagnosed patients treated with 1,500 mg daily metformin for 1 year showed a greater HbA1c reduction (−2.2% vs −1.1%, P<0.05) in Chinese diabetic patients who were heterozygous for the minor allele than the wild type after adjusting for baseline HbA1c, exercise, and diet.43
MATEs: Nonsynonymous MATE1 and MATE2 variants with a reduced in vitro transport function have been reported.46,47 In a study of Chinese patients, homozygous carriers of the intronic MATE1 variant (rs2289669 G>A) had a greater AUC and a lower clearance (P<0.01) of metformin than carriers of the wild type.48 Several studies reported a link between this SNP with HbA1c reduction by metformin. Carriers of the minor allele at rs2289669 showed a significantly greater HbA1c reduction in both the dominant and the recessive models.48–50 In the Diabetes Prevention Program study, the T allele of a SNP (rs8065082 C>T) in LD with rs2289669 (r2=0.8) showed a protective effect against the incidence of diabetes in high-risk individuals.39 Finally, rs12943590, a promoter variant for MATE2, has been associated with PK of metformin in healthy individuals.51 This difference has also been seen in HbA1c reduction37 and successfully replicated in another study.47
In conclusion, while a number of variants have been reported in the metformin transporter genes, on the whole, there have been no definitive signals for these variants on glycemic response to metformin. However, MATE1 and MATE2 variants show some promise and larger studies, or meta-analysis of existing studies, are required to establish how much of these results are biased by small sample size and publication bias.
Gene–gene interaction: Although variants in transporter genes showed an association with metformin response, individual variants explain only a small fraction of the variation. Given that multiple transporters are involved in the disposition of metformin and localization of uptake and efflux transporters in the same organ (Figure 1), joint investigation could give a better understanding. Interaction between the OCT2 variant, c.808 G>T (rs316019), and MATE1 variant, g.−66 T>C (rs2252281), in relation to the PK of metformin was reported by Christensen et al.45 The c.808 G>T alone showed no effect on either secretory or renal clearance of metformin. However, an increased clearance was observed with carriers of the c.808 G>T variant that are homozygous for g.−66 T>C. In the Rotterdam study, interaction between the intronic MATE1 and OCT1 SNPs, rs2289669 and rs622342, respectively, was investigated. A more pronounced glucose-lowering effect of rs2289669 in MATE1 was reported in metformin users with CC genotype than AA genotype for OCT1.52 Gene–gene interaction between g.−66 T>C/rs2252281 and g.−130 G>A/rs12943590 promotor variants of MATE1 and MATE2, respectively, was reported by Stocker et al.51 Carriers of both variants showed a greater renal and secretary clearance. This clearly signifies the role of gene–gene interaction and the importance of complex network/pathway analysis to better understand the PK and PD of metformin.
TCF variants: Rather than studying transporter variants per se, an elegant study explored variants in TCFs that potentially regulate the expression of these transporter genes.19 They studied variants in specificity protein (SP) 1, which regulates the expression of a number of these transporter genes; activating enhancer-binding protein 2, which represses MATE1 expression; and the TCFs, hepatocyte nuclear factor 4, alpha (HNF4α) and peroxisome proliferator-activated, alpha (PPARα). They reported five variants in or near SP1 and one variant in activating enhancer-binding protein 2 that showed association with metformin elimination and HbA1c change. Of these, those homozygous GG at rs784892 (intronic SNP of AMHR2, downstream gene to SP1) achieved a 1.1% lower HbA1c and 98 mL/min lower secretory clearance of metformin than AA homozygotes. Up to 24% reduction in apparent clearance was also reported in patients’ homozygous GG at rs784888, a downstream variant to SP1. This SNP was associated with HbA1c reduction with β coefficient of −0.36% per G allele (P=0.01 before Bonferroni correction). Variants in HNF4α and PPARα were associated with HbA1c reduction, but their effect could not be explained by the PK of metformin suggesting that further investigation of other mechanisms is required.
Polymorphisms in the PD pathway: Genetic variants affecting the PD of metformin are not well studied. There are few candidate gene studies that reported nominal associations with metformin efficacy (Table S1). The GWAS on metformin response in 1,024 T2D incident users revealed an association of rs11212617, a SNP near ATM gene, with glycemic response to metformin as a linear reduction in glycated hemoglobin or achieving treatment target (HbA1C <7%).8 This finding was further replicated in two independent cohorts from Scotland and the UK with sample sizes of 1,783 and 1,113, respectively.53 A meta-analysis of three other studies separately or in combination with previous bigger studies confirmed the association of the variant with treatment success.53 This finding was also replicated in a Chinese population.54 However, the Diabetes Prevention Program could not find any effect of rs11212617 on the efficacy of metformin in delaying progression to diabetes.55
Sulfonylureas
SUs were first introduced into clinical practice in the 1950s and have long been a cornerstone of treatment in T2D. Currently, they are used as the first-line agents or an add-on therapy to other OHAs, usually metformin. About a quarter of newly diagnosed patients initiate therapy with SUs.56 Each drug in the group varies in their PK parameters, insulin secretory potency, and onset and duration of action.
PK and PD
The polymorphic CYP2C9 isoenzyme catalyzes the biotransformation of SUs in the liver. Catalytic function of the enzyme is reported to be affected by the type of inherited amino acid substitution.57 Substitution of arginine with cysteine at amino acid position 144 (Arg144Cys) and isoleucine with leucine at position 359 (Ile359Leu) gives rise to mutant alleles, CYP2C9*2 and CYP2C9*3, respectively. The mutant alleles are known to have a reduced catalytic activity than the wild-type CYP2C9*1. Involvement of CYP2C19 in the metabolism of SUs is also reported.58 CYP2C19*2 (681 G>A) and CYP2C19*3 (636 G>A) are variants that encode a nonfunctional CYP2C19 enzyme. Individuals with either of the variants are labeled as poor metabolizers.58 The *3 variant is most common in Asians with a frequency of 10%–25% compared to that of 2%–6% in Caucasians.
SUs induce glucose-independent insulin release from the pancreatic β-cells by binding to the ATP-sensitive potassium (KATP) channel (Figure 2).59 The channel is composed of four subunits of the SU receptor (SUR) 1 and four subunits of the potassium inward rectifier channel (Kir) 6.2. Two SU-binding sites have been reported in the channel. The A site resides exclusively on the SUR1 and the B site is available on both subunits of the KATP channel. Binding of SUs to these receptors induces the closure of KATP channels and increases intracellular K+ ion and hence membrane depolarization with subsequent opening of voltage-gated Ca2+ channels that increase intracellular Ca2+ followed by the release of insulin-containing granules.
Figure 2.
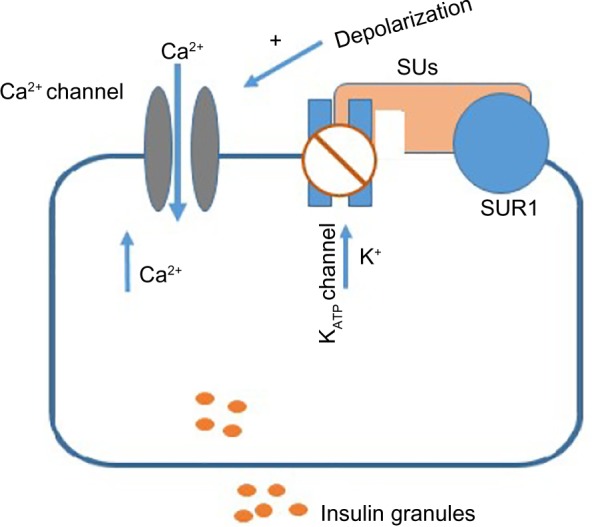
Action of sulfonylureas on β-cells.
Abbreviations: SUs, sulfonylureas; SUR1, sulfonylurea receptor 1.
Pharmacogenetics
Interindividual variability in SU response exists. Following SU initiation, an estimated 10%–20% of patients will have <20 mg/dL FPG reduction.20 Clinical factors, such as baseline glucose, duration of diabetes, β-cell function, and degree of insulin resistance, affect response to SUs.20 Variants in genes encoding proteins involved in the PK and PD are widely reported to influence therapeutic outcome of SUs.
CYPs: Several studies investigated the effect of mutant alleles of the rate-limiting CYP2C9 on the PK and PD and the safety of SUs in healthy and T2D individuals. Reduced drug-metabolizing activity has been reported in individuals carrying either the CYP2C9*2 or the CYP2C9*3 variants, *3 being more profound.60 Compared to wild-type carriers, healthy male volunteers homozygous for CYP2C9*3 and CYP2C9*2 had a 50% and 10% lower oral clearance of glyburide, respectively.61 In line with this, a significant increase in AUC and plasma half-life (t1/2) of glyburide has been reported for heterozygous CYP2C9*3 than *1/*1 carriers.62 Similar result was also reported for tolbutamide.63 The impact of CYP2C9 on the PK of the second generation SUs was also studied. Wang et al reported 40% and 30% more mean AUC of glimepiride for *3/*3 and *3/*1 carriers, respectively, compared to the wild type.64
Reduced function CYP2C19 variants also influence metabolism of SUs. More than threefold increase in AUC and prolonged half-life of gliclazide were reported among male healthy Chinese volunteers with reduced CYP2C19 variants compared to carriers of the wild type.65
Influence of CYP variants on efficacy to SUs has been widely studied. The largest study based on retrospective data on 1,073 incident users of SUs from the GoDARTs showed that carriers of loss-of-function CYP2C9*2 or CYP2C9*3 alleles were 3.4-fold more likely to achieve therapeutic target than carriers of the wild type, resulting in 0.5% greater HbA1c reduction.66 In the Rotterdam study, Becker et al defined response in terms of maintenance dose achieved among the incident SU users.67 In a subgroup of 172 patients who were on tolbutamide, a lower dose was needed to regulate glucose in the carriers of CYP2C9*3 than in the carriers of the wild type. A reduction in HbA1c in carriers of CYP2C9*1/*3 was also reported among Japanese patients who have been on glimepiride.68 These consistent findings are some of the most robust pharmacogenetic findings in the diabetes field and could potentially translate into genotype-guided therapy in SUs. However, prospective studies in T2D patients are required before translating into clinical practice. The role that CYP2C19 could play in the metabolism of gliclazide is documented.58 However, studies to link this with glycemic response are lacking.
Polymorphisms in mechanistic targets
Following the identification of SU-binding sites SUR1 and Kir6.2 (encoded by ABCC8 and KCNJ11, respectively), variants in these genes have been the subjects of many pharmacogenetic investigations. Rare pathogenic mutations in these genes lead to neonatal monogenic diabetes.69 Due to the low levels of insulin and ketoacidosis, insulin has been the typical treatment in neonatal diabetes. Successful transition from long-term insulin to SU treatment has been reported by Pearson et al in 2006.70 Following this, a number of studies investigated two strongly linked nonsynonymous common variants in the ABCC8 (S1369A, rs757110) and KCNJ11 (E23K, rs5219) in patients with T2D.
ABCC8/KCNJ11: The E23K and S1369A variants form a haplotype. While KATP channels containing the K23/A1369 haplotype are more sensitive to inhibition by gliclazide, they are less sensitive to inhibition by tolbutamide, chlorpropamide, and glimepiride.71 Association of S1369A with glycemic control in 115 Chinese patients who have been on gliclazide for 8 weeks was reported.72 Carriers of minor allele had a greater HbA1c reduction than carriers of the wild type (1.60%±1.36% vs 0.76%±1.70%, P=0.04). Another larger study also showed an association of the minor allele with a greater reduction in fasting (P<0.001) and 2-hour (P<0.003) glucose levels.73 Association between the KCNJ11 E23K and efficacy to SUs was reported in Chinese patients by Li et al.74 In this study, 108 newly diagnosed T2D individuals have been treated with gliclazide MR for 16 weeks. Homozygous KK carriers had a lower FPG and were more likely to achieve the target FPG of 7.0 mmol/L (Plog rank =0.03) than E allele carriers. In another study carried out in 101 T2D Caucasians treated with SUs after metformin therapy, homozygous KK carriers showed a greater HbA1c reduction than EE homozygous after 6 months of therapy (1.04%±0.10% vs 0.79%±0.12%, P=0.04).75
However, other studies could not replicate the above findings. The UK Prospective Diabetes Study investigated response to SUs in 363 individuals based on FPG measured at two time points in a 1-year period.76 No significant association between E23K and FPG was observed. This finding could probably be confounded by continual dose adjustment carried out in the UK Prospective Diabetes Study. In a study conducted on 525 Caucasians who have been on glibenclamide and metformin, carriers of the K allele showed 1.69 ([95% CI =1.02–2.74], P=0.04) times higher odds of secondary treatment failure, defined as FPG >300 mg/dL (16.7 mmol/L), than those who were homozygous for the reference allele.77 Since metformin was used as an add-on therapy, the failure is for the combination rather than SU alone. Moreover, this secondary failure phenotype is more likely to reflect diabetes progression associated with the K allele than SU response. Another study carried out in 176 (92 experienced hypoglycemia and 84 not) T2D patients showed no association of the E23K variant with a mild hypoglycemia.78 This study might be confounded by an incomplete definition of mild hypoglycemia as it relies on patients’ self-report.
TCF7L2: TCF7L2 harbors the strongest T2D risk variants among the 120 GWAS-established loci. It encodes T-cell TCF4, an important downstream target of the WNT signaling pathway.79 Reduced insulin secretion has been reported in relation to two intronic variants, rs7903146 and rs12255372, in the TCF7L2 gene and hence hypothesized to affect SU response.80
GoDARTs is the largest study conducted on 901 Scottish patients to link TCF7L2 variants with SU response.81 Patients homozygous for the minor allele of rs12255372 G>T were nearly twofold less likely to achieve therapeutic target after 3–12 months of SU treatment than homozygous carriers of the reference allele. Similar result was reported for rs7903146. Three other independent groups also showed a consistent result (Table S2).82–84
In conclusion, notable findings have been reported in the pharmacogenetics of SUs. Robust associations between variants in the CYP2C9, ABBC8/KCNJ11 and TCF7L2, are reported. More comprehensive assessments of these associations will be necessary to translate this genetic information into clinical utility.
Meglitinides
Meglitinides (glinides) are short acting non-SU secretagogues that lower postprandial glucose excursions preferentially by stimulating early phase insulin secretion. They act by regulating potassium channels in the pancreatic β-cells via a distinct mechanism from that of SUs. They are not commonly used in the UK.
After oral administration, glinides are absorbed rapidly with the peak plasma drug levels reached within 1 hour. Organic anion transporting polypeptide 1B1 (OATP1B1), encoded by Solute Carrier Organic Anion Transporter Family, Member 1B1 (SLCO1B1), mediates their transport into the liver,85 where >95% of the oral dose get metabolized by the CYP family isozymes.57
Association of genetic variants in the SLCO1B1, CYP2C9, and CYP2C8 genes with the PK and/or efficacy of glinides has been reported. In healthy individuals, carriers of the variant allele c.521 T>C in the SLCO1B1 had a reduced transport and an increased plasma concentration of repaglinide and nateglinide.86–89 The *1B/*1B haplotype in the same gene was also associated with a reduced transport of glinides.90 Association of the *3 variant in CYP2C8 and CYP2C9 with the PK of nateglinide and repaglinide has also been reported.86,91,92
In a study carried out in 100 Chinese patients, He et al investigated the effect of KCNJ11 genotype on the efficacy of repaglinide after 24 weeks of treatment.93 Carriers of the K allele of E23K showed a greater HbA1c reduction (EE: 1.52%±1.03%, EK: 2.33%±1.53%, and KK: 2.65%±1.73%, P=0.02). However, this result could be confounded by baseline effect as carriers of the variant allele had higher HbA1c at baseline than carriers of the wild type.
Studies pertaining to the pharmacogenetics of glinides are confounded by small sample sizes (most of them <100). PD investigations are available for repaglinide only, and most of the PK studies are limited to healthy volunteers (Table S3). Therefore, further studies with bigger sample sizes, methodological diversities, and replication are required.
Thiazolidinediones
TZDs, also known as glitazones, are OHAs that act as insulin sensitizers in different tissues, including the liver, muscle, and adipose tissue. Glitazones act via the activation of PPAR-γ that regulates the transcription of multiple downstream genes involved in glucose and lipid metabolism. They reduce HbA1c by ~0.5%–1.4%.94 There are three glitazones that have been licensed: rosiglitazone, pioglitazone, and troglitazone. While troglitazone was withdrawn from the global market in 2000 due to idiosyncratic hepatotoxicity, marketing authorization for roziglitazone has been withdrawn in Europe and put under restrictions in the USA due to the potential cardiovascular risks. France and Germany have suspended pioglitazone due to an increased risk of bladder cancer. However, recent mul-tipopulation studies showed no association of pioglitazone or rosiglitazone with the risk of bladder cancer.95,96
Pharmacogenetics
CYP2C8 and SLCO1B1: Hepatic uptake of TZDs is mediated by OAT1B197 with metabolism mostly by CYP2C8. Genetic variants in genes encoding these proteins have been investigated for their possible impact on the PK of TZDs in healthy volunteers. The homozygote carriers of CYP2C8*3 had 36% lower rosiglitazone plasma concentration and 39% higher weight-adjusted oral clearance compared to carriers of the wild type.98,99 Similar trends have been reported for pioglitazone in two other studies.100,101 For SLCO1B1, despite in silico modeling, PK studies in healthy Caucasians found no association between loss-of-function 521 T>C variant of SLCO1B1 and plasma concentrations of rosiglitazone and pioglitazone.99,102 It is worth noting that these studies had small samples that could limit statistical power to detect moderate genetic effects.
PPARG: PPARG, the mechanistic target of TZDs, is an obvious candidate for pharmacogenetic investigations. Association of the common variant rs1801282 P12A with risk of T2D has been reported.103 The most robust study that showed an association between P12A and response to pio-glitazone was carried out in 250 Chinese patients.104 Carriers of the minor allele (Ala) showed 2.32 ([95% CI =1.10–4.87], P=0.03) times higher odds of being a responder than carriers of the wild type. In this study, responders were defined as those with >15% decrease in HbA1c levels or >20% decrease in FPG levels (or both) after 24 weeks of pioglitazone treatment. Association of the same variant with a linear reduction in HbA1c and FPG after pioglitazone therapy was replicated in an independent cohort of 67 patients.105 Similar trend has been reported in 198 Korean patients treated with 4 mg rosiglitazone daily for 3 months.106
Adverse outcomes: Adverse effects induced by TZD therapy have been investigated in relation to genetic variants. Watanabe et al studied association of troglitazone-induced hepatotoxicity with the 68 polymorphic sites of 51 candidate genes in 110 Japanese patients (25 cases and 85 controls).10 The strongest correlation was observed for combined null genotype of glutathione S-transferase theta-1 and glutathione S-transferase mu-1 (OR =3.7 [95% CI =1.4–10.1], P=0.008). In another Japanese study, association of troglitazone-induced liver injury with mutations in CYP2C19 was reported.11 In the Diabetes REduction Assessment with ramipril and rosiglitazone Medication (DREAM) trial, a higher rate of roziglitazone-induced edema (OR =1.89 [95% CI =1.47–2.42], P=0.017) was reported for patients’ homozygous CC for rs6123045, a variant at the Nuclear Factor of Activated T-cells, Cytoplasmic, Calcineurin-Dependent 2 (NFATC2) locus (Table S4).107
Incretins
There is a greater insulin secretory response to oral than intravenous glucose load despite the same glucose concentrations at the level of the β-cell; this is termed as the incretin effect and has been attributed to the incretin peptides: GLP-1 and gastric inhibitory polypeptide.108 GLP-1 is a glucoincretin hormone secreted from enteroendocrine L cells within the crypts of the intestinal mucosa. It has a t1/2 of 1–2 minutes due to rapid degradation by the enzyme DPP-4 and thus limited therapeutic potential.109 Two therapeutic strategies were developed to overcome this rapid degradation – oral agents that inhibit DPP-4 (known as gliptins) and injectable agents that are resistant to breakdown by DPP-4 (GLP-1R agonists).
Pharmacogenetic studies on GLP-1R agonists are limited. A pilot study on healthy Caucasians showed differences in the insulinotropic response to exogenous GLP-1 in relation to two common variants (rs6923761 G>A and rs3765467 C>T) in the GLP-1R gene.110 ’t Hart et al reported significant association between a variant near the Chymotrypsinogen B1/2 (CTRB1/2) gene (rs7202877) and glycemic response to gliptins. CTRB1/2 encodes chymotrypsin, and the G allele at rs7202877 variant was associated with an increased fecal chymotrypsin activity. Carriers of the G allele at this SNP showed 0.51%±0.16% lower HbA1c reduction compared to TT genotype (P=0.0015) after being on gliptins for at least 3 months.111 Association of variants in other T2D-related genes, such as Potassium Channel, Voltage Gated KQT-Like Subfamily Q, Member 1 (KCNQ1), TCF7L2, and Wolfram Syndrome 1 (WFS1), with GLP-1R agonist response has also been reported (Table S5).
SGLT-2 inhibitors
SGLT-2 inhibitors are a new class of OHAs that inhibit SGLT-2-mediated renal reabsorption of glucose thereby increase glycosuria, resulting in a reduction of hyperglycemia independent of β-cell function.112 After oral administration, SGLT-2 inhibitors show a bioavailability of 65%–78% with a t1/2 ranging 10–13 hours allowing once a day administration.112 They are mainly eliminated through O-glucuronidation by uridine diphosphate glucuronosyl-transferases (UGTs).113 A recent study carried out in 134 healthy and T2D subjects showed involvement of UGT1A9 and UGT2B4 in the metabolism of canagliflozin.113 Carriers of reduced function variants, UGT1A9*3 and UGT2B4*2, had an increased plasma concentration of canagliflozin than carriers of the parent allele.
SGLT-2 inhibitors reduce HbA1c by 0.58%–1% when used as a mono- or an add-on therapy.114 Individual variation in response to SGLT-2 inhibitors has been reported, and part of this variation could be attributable to genetic variation. Nonsense and missense mutations in the SLC2A5 gene that result in the loss of SGLT-2 function cause familial renal glycosuria and are associated with the reduced circulating glucose levels.115,116
Conclusion and future directions
More than 120 studies pertaining gene–drug interaction in diabetes have been investigated for this review (Tables S1–S5). Even though small studies that lack replication predominate, well-powered, and successfully replicated findings are emerging. Promising advances in the pharmacogenomics of T2D have already been made. Genetic-guided therapy is now mainstream in the case of maturity onset diabetes of the young and neonatal diabetes.70,117 To further translate pharmacogenomics research into clinical practice, more well-designed studies with sufficiently large sample size and well-characterized phenotype should be conducted, and where possible meta-analysis across studies should be undertaken to provide robust evidence for an association. In addition, data from high-throughput sequencing of rare variants, noncoding regions, and multilevelomics, including transcriptomics, proteomics, metabolomics, and metagenomics, may yield greater mechanistic insights and possibly biomarkers with a larger clinical effect.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87(1):4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.International Diabetes Federation (IDF) IDF Diabetes Atlas. 6th ed. Brussels: International Diabetes Federation; 2013. [Google Scholar]
- 3.Casagrande SS, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care. 2013;36(8):2271–2279. doi: 10.2337/dc12-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker ML, Pearson ER, Tkáč I. Pharmacogenetics of oral antidiabetic drugs. Int J Endocrinol. 2013;2013:686315. doi: 10.1155/2013/686315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeFronzo RA, Stonehouse AH, Han J, Wintle ME. Relationship of baseline HbA1c and efficacy of current glucose-lowering therapies: a meta-analysis of randomized clinical trials. Diabetic Med. 2010;27(3):309–317. doi: 10.1111/j.1464-5491.2010.02941.x. [DOI] [PubMed] [Google Scholar]
- 6.Ho PM, Rumsfeld JS, Masoudi FA, et al. Effect of medication nonadherence on hospitalization and mortality among patients with diabetes mellitus. Arch Intern Med. 2006;166(17):1836–1841. doi: 10.1001/archinte.166.17.1836. [DOI] [PubMed] [Google Scholar]
- 7.Zhou K, Donnelly L, Yang J, et al. Heritability of variation in glycaemic response to metformin: a genome-wide complex trait analysis. Lancet Diabetes Endocrinol. 2014;2(6):481–487. doi: 10.1016/S2213-8587(14)70050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou K, Bellenguez C, Spencer CC, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43(2):117–120. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vella A. Pharmacogenetics for type 2 diabetes: practical considerations for study design. J Diabetes Sci Technol. 2009;3(4):705–709. doi: 10.1177/193229680900300415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe I, Tomita A, Shimizu M, et al. A study to survey susceptible genetic factors responsible for troglitazone-associated hepatotoxicity in Japanese patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2003;73(5):435–455. doi: 10.1016/s0009-9236(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 11.Kumashiro R, Kubota T, Koga Y, et al. Association of troglitazone-induced liver injury with mutation of the cytochrome P450 2C19 gene. Hepatol Res. 2003;26(4):337–342. doi: 10.1016/s1386-6346(03)00165-7. [DOI] [PubMed] [Google Scholar]
- 12.Donnelly LA, Doney AS, Hattersley AT, Morris AD, Pearson ER. The effect of obesity on glycaemic response to metformin or sulphonylureas in Type 2 diabetes. Diabet Med. 2006;23(2):128–133. doi: 10.1111/j.1464-5491.2005.01755.x. [DOI] [PubMed] [Google Scholar]
- 13.Ong CR, Molyneaux LM, Constantino MI, Twigg SM, Yue DK. Long-term efficacy of metformin therapy in nonobese individuals with type 2 diabetes. Diabetes Care. 2006;29(11):2361–2364. doi: 10.2337/dc06-0827. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki Y, De Filippis E, Bajaj M, Wajcberg E, Glass LC, Triplitt C. Predictors of improved glycemic control with rosiglitazone therapy in type 2 diabetic patients: a practical approach for the primary care physician. Br J Diabetes Vasc Dis. 2005;5:28–35. [Google Scholar]
- 15.Kim YM, Cha BS, Kim DJ, et al. Predictive clinical parameters for therapeutic efficacy of rosiglitazone in Korean type 2 diabetes mellitus. Diabetes Res Clin Pract. 2005;67:43–52. doi: 10.1016/j.diabres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Scheen AJ. Drug interactions of clinical importance with antihyperglycaemic agents: an update. Drug Saf. 2005;28(7):601–631. doi: 10.2165/00002018-200528070-00004. [DOI] [PubMed] [Google Scholar]
- 17.Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CN, Pearson ER. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: a GoDARTS Study. Diabetes. 2015;64(5):1786–1793. doi: 10.2337/db14-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46:347–351. doi: 10.1007/s00125-003-1034-7. [DOI] [PubMed] [Google Scholar]
- 19.Goswami S, Yee SW, Stocker S, et al. Genetic variants in transcription factors are associated with the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2014;96(3):370–379. doi: 10.1038/clpt.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeFronzo RA. Pharmacologic therapy for type 2 diabetes mellitus. Ann Intern Med. 1999;131(4):281–303. doi: 10.7326/0003-4819-131-4-199908170-00008. [DOI] [PubMed] [Google Scholar]
- 21.Han TH, Everett RS, Proctor WR, et al. Organic cation transporter 1 (OCT1/mOct1) is localized in the apical membrane of Caco-2 cell monolayers and enterocytes. Mol Pharmacol. 2013;84(2):182–189. doi: 10.1124/mol.112.084517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han TK, Proctor WR, Costales CL, Cai H, Everett RS, Thakker DR. Four cation-selective transporters contribute to apical uptake and accumulation of metformin in Caco-2 cell monolayers. J Pharmacol Exp Ther. 2015;352(3):519–528. doi: 10.1124/jpet.114.220350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT) Biochem Pharmacol. 2005;70(12):1851–1860. doi: 10.1016/j.bcp.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Kimura N, Masuda S, Tanihara Y, et al. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005;20(5):379–386. doi: 10.2133/dmpk.20.379. [DOI] [PubMed] [Google Scholar]
- 26.Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci USA. 2005;102(50):17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pernicova I, Korbonits M. Metformin – mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 28.Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maruthur NM, Gribble MO, Bennett WL, et al. The pharmacogenetics of type 2 diabetes: a systematic review. Diabetes Care. 2014;37:876–886. doi: 10.2337/dc13-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;(201):105–167. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 33.Tzvetkov MV, Vormfelde SV, Balen D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. 2009;86(3):299–306. doi: 10.1038/clpt.2009.92. [DOI] [PubMed] [Google Scholar]
- 34.Christensen MM, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21(12):837–850. doi: 10.1097/FPC.0b013e32834c0010. [DOI] [PubMed] [Google Scholar]; Erratum in Pharmacogenet Genomics. 2015;25(1):48–50. [Google Scholar]
- 35.Shu Y, Brown C, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83(2):273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou K, Donnelly LA, Kimber CH, et al. Reduced-function SLC22A1 polymorphisms encoding organic cation transporter 1 and glycemic response to metformin: a GoDARTs study. Diabetes. 2009;58(6):1434–1439. doi: 10.2337/db08-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis R, Giacomini K, Yee SW, Jenkins G, McCarty CA, Wilke RA. PS1-10: response to metformin and genetic variants of organic cation and multidrug and toxin extrusion transporters. Clin Med Res. 2010;8(3–4):191. [Google Scholar]
- 38.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 2009;9(4):242–247. doi: 10.1038/tpj.2009.15. [DOI] [PubMed] [Google Scholar]
- 39.Jablonski KA, McAteer JB, de Bakker PI, et al. Diabetes Prevention Program Research Group Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes. 2010;59(10):2672–2681. doi: 10.2337/db10-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarasova L, Kalnina I, Geldnere K, et al. Association of genetic variation in the organic cation transporters OCT1, OCT2 and multidrug and toxin extrusion 1 transporter protein genes with the gastrointestinal side effects and lower BMI in metformin-treated type 2 diabetes patients. Pharmacogenet Genomics. 2012;22(9):659–666. doi: 10.1097/FPC.0b013e3283561666. [DOI] [PubMed] [Google Scholar]
- 41.Song IS, Shin HJ, Shim EJ, et al. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther. 2008;84(5):559–562. doi: 10.1038/clpt.2008.61. [DOI] [PubMed] [Google Scholar]
- 42.Shikata E, Yamamoto R, Takane H, et al. Human organic cation transporter (OCT1 and OCT2) gene polymorphisms and therapeutic effects of metformin. J Hum Genet. 2007;52(2):117–122. doi: 10.1007/s10038-006-0087-0. [DOI] [PubMed] [Google Scholar]
- 43.Hou W, Zhang D, Lu W, et al. Polymorphism of organic cation transporter 2 improves glucose-lowering effect of metformin via influencing its pharmacokinetics in Chinese type 2 diabetic patients. Mol Diagn Ther. 2015;19(1):25–33. doi: 10.1007/s40291-014-0126-z. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y, Li S, Brown C, et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics. 2009;19(7):497–504. doi: 10.1097/FPC.0b013e32832cc7e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christensen MM, Pedersen RS, Stage TB, et al. A gene-gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet Genomics. 2013;23(10):526–534. doi: 10.1097/FPC.0b013e328364a57d. [DOI] [PubMed] [Google Scholar]
- 46.Kajiwara M, Terada T, Ogasawara K, et al. Identification of multidrug and toxin extrusion (MATE1 and MATE2-K) variants with complete loss of transport activity. J Hum Genet. 2009;54(1):40–46. doi: 10.1038/jhg.2008.1. [DOI] [PubMed] [Google Scholar]
- 47.Choi JH, Yee SW, Ramirez AH, et al. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90(5):674–684. doi: 10.1038/clpt.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He R, Zhang D, Lu W, et al. SLC47A1 gene rs2289669 G>A variants enhance the glucose-lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res Clin Pract. 2015;109(1):57–63. doi: 10.1016/j.diabres.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Tkáč I, Klimčáková L, Javorský M, et al. Pharmacogenomic association between a variant in SLC47A1 gene and therapeutic response to metformin in type 2 diabetes. Diabetes Obes Metab. 2013;15(2):189–191. doi: 10.1111/j.1463-1326.2012.01691.x. [DOI] [PubMed] [Google Scholar]
- 50.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: a preliminary study. Diabetes. 2009;58(3):745–749. doi: 10.2337/db08-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stocker SL, Morrissey KM, Yee SW, et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2013;93(2):186–194. doi: 10.1038/clpt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Interaction between polymorphisms in the OCT1 and MATE1 transporter and metformin response. Pharmacogenet Genomics. 2010;20(1):38–44. doi: 10.1097/FPC.0b013e328333bb11. [DOI] [PubMed] [Google Scholar]
- 53.van Leeuwen N, Nijpels G, Becker ML, et al. A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologia. 2012;55(7):1971–1977. doi: 10.1007/s00125-012-2537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y, Guo Y, Ye W, et al. rs11212617 is associated with metformin treatment response in type 2 diabetes in Shanghai local Chinese population. Int J Clin Pract. 2014;68(12):1462–1466. doi: 10.1111/ijcp.12534. [DOI] [PubMed] [Google Scholar]
- 55.Florez JC, Jablonski KA, Taylor A, et al. Diabetes Prevention Program Research Group The C allele of ATM rs11212617 does not associate with metformin response in the diabetes prevention program. Diabetes Care. 2012;35(9):1864–1867. doi: 10.2337/dc11-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desai NR, Shrank WH, Fischer MA, et al. Patterns of medication initiation in newly diagnosed diabetes mellitus: quality and cost implications. Am J Med. 2012;125(3):302.e1–302.e7. doi: 10.1016/j.amjmed.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirchheiner J, Roots I, Goldammer M, et al. Effect of genetic polymorphisms in cytochrome p450 (CYP) 2C9 and CYP2C8 on the pharmacokinetics of oral antidiabetic drugs: clinical relevance. Clin Pharmacokinet. 2005;44(12):1209–1225. doi: 10.2165/00003088-200544120-00002. [DOI] [PubMed] [Google Scholar]
- 58.Xu H, Murray M, McLachlan AJ. Influence of genetic polymorphisms on the pharmacokinetics and pharmacodynamics of sulfonylurea drugs. Curr Drug Metab. 2009;10:643–658. doi: 10.2174/138920009789375388. [DOI] [PubMed] [Google Scholar]
- 59.Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol. 1997;110(6):655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldstein JA. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Br J Clin Pharmacol. 2001;52(4):349–355. doi: 10.1046/j.0306-5251.2001.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirchheiner J, Brockmöller J, Meineke I, et al. Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther. 2002;71(4):286–296. doi: 10.1067/mcp.2002.122476. [DOI] [PubMed] [Google Scholar]
- 62.Yin OQ, Tomlinson B, Chow MS. CYP2C9, but not CYP2C19, polymorphisms affect the pharmacokinetics and pharmacodynamics of glyburide in Chinese subjects. Clin Pharmacol Ther. 2005;78(4):370–377. doi: 10.1016/j.clpt.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Shon JH, Yoon YR, Kim KA, et al. Effects of CYP2C19 and CYP2C9 genetic polymorphisms on the disposition of and blood glucose lowering response to tolbutamide in humans. Pharmacogenetics. 2002;12(2):111–119. doi: 10.1097/00008571-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 64.Wang R, Chen K, Wen SY, Li J, Wang SQ. Pharmacokinetics of glimepiride and cytochrome P450 2C9 genetic polymorphisms. Clin Pharmacol Ther. 2005;78(1):90–92. doi: 10.1016/j.clpt.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Si D, Chen X, et al. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on pharmacokinetics of gliclazide MR in Chinese subjects. Br J Clin Pharmacol. 2007;64(1):67–74. doi: 10.1111/j.1365-2125.2007.02846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou K, Donnelly L, Burch L, et al. Loss-of-function CYP2C9 variants improve therapeutic response to sulfonylureas in type 2 diabetes: a Go-DARTS study. Clin Pharmacol Ther. 2010;87(1):52–56. doi: 10.1038/clpt.2009.176. [DOI] [PubMed] [Google Scholar]
- 67.Becker ML, Visser LE, Trienekens PH, Hofman A, van Schaik RH, Stricker BH. Cytochrome P450 2C9 *2 and *3 polymorphisms and the dose and effect of sulfonylurea in type II diabetes mellitus. Clin Pharmacol Ther. 2008;83(2):288–292. doi: 10.1038/sj.clpt.6100273. [DOI] [PubMed] [Google Scholar]
- 68.Suzuki K, Yanagawa T, Shibasaki T, Kaniwa N, Hasegawa R, Tohkin M. Effect of CYP2C9 genetic polymorphisms on the efficacy and pharmacokinetics of glimepiride in subjects with type 2 diabetes. Diabetes Res Clin Pract. 2006;72(2):148–154. doi: 10.1016/j.diabres.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 69.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54(9):2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 70.Pearson ER, Flechtner I, Njølstad PR, et al. Neonatal Diabetes International Collaborative Group Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir62 mutations. N Engl J Med. 2006;355(5):467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 71.Lang VY, Fatehi M, Light PE. Pharmacogenomic analysis of ATP-sensitive potassium channels coexpressing the common type 2 diabetes risk variants E23K and S1369A. Pharmacogenet Genomics. 2012;22(3):206–214. doi: 10.1097/FPC.0b013e32835001e7. [DOI] [PubMed] [Google Scholar]
- 72.Zhang H, Liu X, Kuang H, Yi R, Xing H. Association of sulfonylurea receptor 1 genotype with therapeutic response to gliclazide in type 2 diabetes. Diabetes Res Clin Pract. 2007;77(1):58–61. doi: 10.1016/j.diabres.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 73.Feng Y, Mao G, Ren X, et al. Ser1369Ala variant in sulfonylurea receptor gene ABCC8 is associated with antidiabetic efficacy of gliclazide in Chinese type 2 diabetic patients. Diabetes Care. 2008;31(10):1939–1944. doi: 10.2337/dc07-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Q, Chen M, Zhang R, et al. KCNJ11 E23K variant is associated with the therapeutic effect of sulphonylureas in Chinese type 2 diabetic patients. Clin Exp Pharmacol Physiol. 2014;41(10):748–754. doi: 10.1111/1440-1681.12280. [DOI] [PubMed] [Google Scholar]
- 75.Javorsky M, Klimcakova L, Schroner Z, et al. KCNJ11 gene E23K variant and therapeutic response to sulfonylureas. Eur J Intern Med. 2012;23(3):245–249. doi: 10.1016/j.ejim.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 76.Gloyn AL, Hashim Y, Ashcroft SJ, et al. UK Prospective Diabetes Study (UKPDS 53) Association studies of variants in promoter and coding regions of beta-cell ATP-sensitive K-channel genes SUR1 and Kir62 with Type 2 diabetes mellitus (UKPDS 53) Diabet Med. 2001;18(3):206–212. doi: 10.1046/j.1464-5491.2001.00449.x. [DOI] [PubMed] [Google Scholar]
- 77.Sesti G, Laratta E, Cardellini M, et al. The E23K variant of KCNJ11 encoding the pancreatic beta-cell adenosine 5′-triphosphate-sensitive potassium channel subunit Kir62 is associated with an increased risk of secondary failure to sulfonylurea in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91(6):2334–2339. doi: 10.1210/jc.2005-2323. [DOI] [PubMed] [Google Scholar]
- 78.Ragia G, Tavridou A, Petridis I, Manolopoulos VG. Association of KCNJ11 E23K gene polymorphism with hypoglycemia in sulfonylurea-treated type 2 diabetic patients. Diabetes Res Clin Pract. 2012;98(1):119–124. doi: 10.1016/j.diabres.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 79.Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57(3):645–653. doi: 10.2337/db07-0847. [DOI] [PubMed] [Google Scholar]
- 80.Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38(3):320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 81.Pearson ER, Donnelly LA, Kimber C, et al. Variation in TCF7L2 influences therapeutic response to sulfonylureas: a GoDARTs study. Diabetes. 2007;56(8):2178–2182. doi: 10.2337/db07-0440. [DOI] [PubMed] [Google Scholar]
- 82.Schroner Z, Javorsky M, Tkacova R, et al. Effect of sulphonylurea treatment on glycaemic control is related to TCF7L2 genotype in patients with type 2 diabetes. Diabetes Obes Metab. 2011;13(1):89–91. doi: 10.1111/j.1463-1326.2010.01324.x. [DOI] [PubMed] [Google Scholar]
- 83.Javorský M, Babjaková E, Klimčáková L, et al. Association between TCF7L2 genotype and glycemic control in diabetic patients treated with gliclazide. Int J Endocrinol. 2013;2013:374858. doi: 10.1155/2013/374858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holstein A, Hahn M, Körner A, Stumvoll M, Kovacs P. TCF7L2 and therapeutic response to sulfonylureas in patients with type 2 diabetes. BMC Med Genet. 2011;12:30. doi: 10.1186/1471-2350-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bachmakov I, Glaeser H, Fromm MF, König J. Interaction of oral antidiabetic drugs with hepatic uptake transporters: focus on organic anion transporting polypeptides and organic cation transporter 1. Diabetes. 2008;57(6):1463–1469. doi: 10.2337/db07-1515. [DOI] [PubMed] [Google Scholar]
- 86.Niemi M, Backman JT, Kajosaari LI, et al. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77(6):468–478. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 87.Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics and pharmacodynamics of repaglinide and nateglinide. J Clin Pharmacol. 2008;48(3):311–321. doi: 10.1177/0091270007311569. [DOI] [PubMed] [Google Scholar]
- 88.Zhang W, He YJ, Han CT, et al. Effect of SLCO1B1 genetic polymorphism on the pharmacokinetics of nateglinide. Br J Clin Pharmacol. 2006;62(5):567–572. doi: 10.1111/j.1365-2125.2006.02686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng Y, Wang G, Zhang W, Fan L, Chen Y, Zhou HH. Effect of CYP2C9 and SLCO1B1 polymorphisms on the pharmacokinetics and pharmacodynamics of nateglinide in healthy Chinese male volunteers. Eur J Clin Pharmacol. 2013;69(3):407–413. doi: 10.1007/s00228-012-1364-9. [DOI] [PubMed] [Google Scholar]
- 90.Kalliokoski A, Backman JT, Neuvonen PJ, Niemi M. Effects of the SLCO1B1*1B haplotype on the pharmacokinetics and pharmacodynamics of repaglinide and nateglinide. Pharmacogenet Genomics. 2008;18(11):937–942. doi: 10.1097/FPC.0b013e32830d733e. [DOI] [PubMed] [Google Scholar]
- 91.Kirchheiner J, Meineke I, Müller G, et al. Influence of CYP2C9 and CYP2D6 polymorphisms on the pharmacokinetics of nateglinide in genotyped healthy volunteers. Clin Pharmacokinet. 2004;43(4):267–278. doi: 10.2165/00003088-200443040-00005. [DOI] [PubMed] [Google Scholar]
- 92.Niemi M, Leathart JB, Neuvonen M, Backman JT, Daly AK, Neuvonen PJ. Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin Pharmacol Ther. 2003;74(4):380–387. doi: 10.1016/S0009-9236(03)00228-5. [DOI] [PubMed] [Google Scholar]
- 93.He YY, Zhang R, Shao XY, et al. Association of KCNJ11 and ABCC8 genetic polymorphisms with response to repaglinide in Chinese diabetic patients. Acta Pharmacol Sin. 2008;29(8):983–989. doi: 10.1111/j.1745-7254.2008.00840.x. [DOI] [PubMed] [Google Scholar]
- 94.Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140–149. doi: 10.2337/dc14-2441. [DOI] [PubMed] [Google Scholar]
- 95.Levin D, Bell S, Sund R, et al. Scottish Diabetes Research Network Epidemiology Group; Diabetes and Cancer Research Consortium Pioglitazone and bladder cancer risk: a multipopulation pooled, cumulative exposure analysis. Diabetologia. 2015;58(3):493–504. doi: 10.1007/s00125-014-3456-9. [DOI] [PubMed] [Google Scholar]
- 96.Lewis JM, Ferrara A, Peng T, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone interim report of a longitudinal cohort study. Diabetes Care. 2008;34:916–922. doi: 10.2337/dc10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kalliokoski A, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism and oral antidiabetic drugs. Basic Clin Pharmacol Toxicol. 2010;107(4):775–781. doi: 10.1111/j.1742-7843.2010.00581.x. [DOI] [PubMed] [Google Scholar]
- 98.Kirchheiner J, Thomas S, Bauer S, et al. Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther. 2006;80(6):657–667. doi: 10.1016/j.clpt.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 99.Aquilante CL, Bushman LR, Knutsen SD, Burt LE, Rome LC, Kosmiski LA. Influence of SLCO1B1 and CYP2C8 gene polymorphisms on rosiglitazone pharmacokinetics in healthy volunteers. Hum Genomics. 2008;3(1):7–16. doi: 10.1186/1479-7364-3-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aquilante CL, Kosmiski LA, Bourne DW, et al. Impact of the CYP2C8 *3 polymorphism on the drug-drug interaction between gemfibrozil and pioglitazone. Br J Clin Pharmacol. 2013;75(1):217–226. doi: 10.1111/j.1365-2125.2012.04343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tornio A, Niemi M, Neuvonen PJ, Backman JT. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos. 2008;36(1):73–80. doi: 10.1124/dmd.107.018010. [DOI] [PubMed] [Google Scholar]
- 102.Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. No significant effect of SLCO1B1 polymorphism on the pharmacokinetics of rosiglitazone and pioglitazone. Br J Clin Pharmacol. 2008;65(1):78–86. doi: 10.1111/j.1365-2125.2007.02986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26(1):76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 104.Hsieh MC, Lin KD, Tien KJ, et al. Common polymorphisms of the peroxisome proliferator-activated receptor-gamma (Pro12Ala) and peroxisome proliferator-activated receptor-gamma coactivator-1 (Gly482Ser) and the response to pioglitazone in Chinese patients with type 2 diabetes mellitus. Metabolism. 2010;59(8):1139–1144. doi: 10.1016/j.metabol.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 105.Pei Q, Huang Q, Yang GP, et al. PPAR-γ2 and PTPRD gene polymorphisms influence type 2 diabetes patients’ response to pioglitazone in China. Acta Pharmacol Sin. 2013;34(2):255–261. doi: 10.1038/aps.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kang ES, Park SY, Kim HJ, et al. Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor gamma2 gene on rosiglitazone response in type 2 diabetes. Clin Pharmacol Ther. 2005;78(2):202–208. doi: 10.1016/j.clpt.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 107.Bailey SD, Xie C, Do R, et al. Variation at the NFATC2 locus increases the risk of thiazolidinedione-induced edema in the Diabetes REduction Assessment with ramipril and rosiglitazone Medication (DREAM) study. Diabetes Care. 2010;33(10):2250–2253. doi: 10.2337/dc10-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 109.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 110.Sathananthan A, Man CD, Micheletto F, et al. Common genetic variation in GLP1R and insulin secretion in response to exogenous GLP-1 in nondiabetic subjects: a pilot study. Diabetes Care. 2010;33(9):2074–2076. doi: 10.2337/dc10-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.’t Hart LM, Fritsche A, Nijpels G, et al. The CTRB1/2 locus affects diabetes susceptibility and treatment via the incretin pathway. Diabetes. 2013;62(9):3275–3281. doi: 10.2337/db13-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scheen AJ. Pharmacokinetics, pharmacodynamics and clinical use of SGLT2 inhibitors in patients with type 2 diabetes mellitus and chronic kidney disease. Clin Pharmacokinet. 2015;54(7):691–708. doi: 10.1007/s40262-015-0264-4. [DOI] [PubMed] [Google Scholar]
- 113.Francke S, Mamidi RN, Solanki B, et al. In vitro metabolism of canagliflozin in human liver, kidney, intestine microsomes, and recombinant uridine diphosphate glucuronosyltransferases (UGT) and the effect of genetic variability of UGT enzymes on the pharmacokinetics of canagliflozin in humans. J Clin Pharmacol. 2015;55(9):1061–1072. doi: 10.1002/jcph.506. [DOI] [PubMed] [Google Scholar]
- 114.Scheen AJ. Pharmacodynamics, efficacy and safety of sodium-glucose co-transporter type 2 (SGLT2) inhibitors for the treatment of type 2 diabetes mellitus. Drugs. 2015;75(1):33–59. doi: 10.1007/s40265-014-0337-y. [DOI] [PubMed] [Google Scholar]
- 115.van den Heuvel LP, Assink K, Willemsen M, Monnens L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2) Hum Genet. 2002;111(6):544–547. doi: 10.1007/s00439-002-0820-5. [DOI] [PubMed] [Google Scholar]
- 116.Calado J, Soto K, Clemente C, Correia P, Rueff J. Novel compound heterozygous mutations in SLC5A2 are responsible for autosomal recessive renal glucosuria. Hum Genet. 2004;114(3):314–316. doi: 10.1007/s00439-003-1054-x. [DOI] [PubMed] [Google Scholar]
- 117.Rafiq M, Flanagan SE, Patch AM, et al. Neonatal Diabetes International Collaborative Group Effective treatment with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;32(2):204–209. doi: 10.2337/dc07-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]