

Abstract
The paper describes the combination of optical tweezers and single molecule fluorescence detection for the study of protein-DNA interaction. The method offers the opportunity of investigating interactions occurring in solution (thus avoiding problems due to closeby surfaces as in other single molecule methods), controlling the DNA extension and tracking interaction dynamics as a function of both mechanical parameters and DNA sequence. The methods for establishing successful optical trapping and nanometer localization of single molecules are illustrated. We illustrate the experimental conditions allowing the study of interaction of lactose repressor (lacI), labeled with Atto532, with a DNA molecule containing specific target sequences (operators) for LacI binding. The method allows the observation of specific interactions at the operators, as well as one-dimensional diffusion of the protein during the process of target search. The method is broadly applicable to the study of protein-DNA interactions but also to molecular motors, where control of the tension applied to the partner track polymer (for example actin or microtubules) is desirable.
Keywords: Bioengineering, Issue 90, Single molecule biophysics, Optical tweezers, fluorescence microscopy, DNA binding proteins, lactose repressor, microfluidics
Introduction
Single molecule (SM) techniques have greatly developed over the past thirty years to respond to the need of overcoming some of the limitations of traditional, bulk solution measurements 1-3. The manipulation of single biological molecules has created the opportunity to measure mechanical properties of biopolymers4 and control the mechanical parameters of protein-protein5 and protein-DNA interactions6,7. SM fluorescence detection, on the other hand, represents an incredibly versatile tool for studying protein activity in vitro and in vivo, leading to the possibility of localizing and tracking single molecules with nanometer precision. Through fitting of the instrument point-spread-function to the SM image, in fact, one can accomplish localization with a precision depending mainly on signal-to-noise ratio (SNR) and reaching a limit of about one nanometer8,9. These methodologies find powerful applications in the study of the dynamics of motor proteins, as well as of the diffusion processes underlying target search in DNA-binding proteins. The capability of determining diffusion constants as a function of the DNA sequence, residence time on the target and accurately measuring the DNA length explored during one-dimensional diffusion events, represent a powerful tool for the study of protein-DNA interaction dynamics and for the investigation of the mechanisms of specific target search.
Recently, the combination of these two techniques has produced a new generation of experimental setups10-14 enabling the simultaneous manipulation of a biological substrate (for example an actin filament or a DNA molecule) and detection/localization of an interacting partner enzyme (for example myosin or a DNA-binding protein). The advantages of these techniques mainly rest on the possibility of exerting mechanical control over the trapped polymer, thus enabling the study of interaction dynamics versus forces or torques. Also, the methodology allows measuring biochemical reactions far from the surface, avoiding one of the main limitations of classic SM methods, i.e., the need for immobilization of the molecules under study on a surface (glass slide or microspheres).
The combination of two single molecules techniques requires overcoming several technical difficulties, mainly arising from the requirements of mechanical stability and adequate SNR (especially when requiring localization with nm precision)15. Particularly, when coupling SM fluorescence detection with optical tweezers, the reduction of noise and photobleaching from the trapping infrared lasers16 and the control of biochemical buffers for assembly of the biological complexes and performance of the experimental measurements11 are of paramount importance. Here, we describe the methods for performing successful measurements in a dual trapping/SM Fluorescence localization setup. The methodology is illustrated with the example of lactose repressor protein (LacI) fluorescently labeled (with Atto532) and detected as it binds to a DNA molecule (trapped between two optical tweezers) containing specific LacI binding sequences (i.e., operators). We demonstrate the effectiveness of the method in detecting binding of LacI to DNA and diffusion along its contour in the target search process. The method is applicable to any combination of DNA sequence and DNA-binding protein, as well as to other systems (microtubules or actin filaments and the motor proteins interacting with them).
Protocol
1. Optical Tweezers Setup with Nanometer Stability
The experimental setup must provide two optical tweezers with pointing stability at the nanometer level and intensity fluctuations of the trapping laser below 1%. Combination of these conditions will assure nanometer stability of the dumbbell under typical tension (1 pN - few tens of pN), trap stiffness (0.1 pN/nm) and measurement bandwidth (image acquisition rate 20 sec-1). A scheme of the experimental setup is depicted in Figure 1.
- Optical tweezers design and construction15,17:
- Mount the experimental apparatus on an optical table with active isolators to reduce mechanical vibrations. Mount the microscope structure on elastomeric isolators to absorb acoustic noise and mechanical resonances of the microscope structure.
- Insert an optical isolator in the path of the trapping laser, near the laser source, to reduce random amplitude fluctuations due to optical feedback.
- Reduce the path length of the trapping laser as much as possible and enclose the whole path in a sealed box to reduce laser pointing fluctuations due to air currents and turbulence.
- Create double optical tweezers by splitting a single near-infrared laser source (Nd:YAG laser, 1,064 nm wavelength) into two beams with orthogonal polarizations. Do not use time-shared traps because they cause oscillation of the dumbbell under tension12.
- Use acousto-optic deflectors (AODs) driven by Direct Digital Synthesizers (DDSs) to allow fine movements of (at least) one trap and precise regulation of DNA tension. DDSs allow a better trap stability than Voltage-Controlled Oscillators (VCOs).
- Insert two quadrant detector photodiodes (QDPs) in the back focal plane of the condenser to detect the position of the trapped beads with sub-nm accuracy.
Check that intensity fluctuations of the trapping laser at the microscope entrance are below 1% by using a photodiode with a bandwidth larger than the rate of image acquisition.
- Check pointing stability of the two trapping lasers:
- Prepare silica beads in phosphate buffer: Dilute 20 μl of silica beads (1.54 μm, 10% solids) into 1 ml of acetone, sonicate 30 sec, vortex briefly, and centrifuge 2 min at 19,000 x g. Resuspend in 1 ml acetone and repeat wash. Resuspend in 1 ml of 50 mM phosphate buffer, wash 2 times, and finally resuspend in 100 μl of 50 mM phosphate buffer.
- Calibrate optical tweezers with silica beads: prepare a flow cell by attaching a coverslip to a microscope slide using double-sticky tape (about 60 μm thick). Flow 1 mg/ml BSA and wait 3 min. Flow silica beads diluted 1/1,000 in phosphate buffer and seal the flow chamber with silicon grease. Trap one bead in each trap and calibrate the traps using the power spectrum method 18.
- Prepare silica beads in pentyl acetate and nitrocellulose: Dilute 20 μl of silica beads (1.54 μm, 10% solids) into 1 ml of acetone, sonicate 30 sec, vortex briefly, and centrifuge 2 min at 19,000 x g. Resuspend in 1 ml acetone and repeat wash. Resuspend in 1 ml of pentyl acetate and wash 2 times. Finally resuspend them in pentyl acetate with 0.1% nitrocellulose dissolved in pentyl acetate.
- Spread 2 μl of 1.54 μm diameter silica beads dissolved in pentyl acetate + 0.1% nitrocellulose onto the surface of a coverslip (24 x 24 mm) using a second coverslip (24 x 60 mm). Attach the coverslip to a microscope slide by using two pieces of double-sided tape to create a flow cell. Fill the flow-cell with 50 mM phosphate buffer and seal it with silicon grease.
- Image one silica bead in brightfield microscopy at 2,000X magnification. The field of view should be 7 x 5 μm acquired at 768 x 576 pixel, so that the bead image has a diameter of 190 pixels. Compensate thermal drifts with a feedback software that, as in ref. 17, calculates x-y position from the image centroid and z from the diffraction rings and corrects drifts moving a piezo stage with nm-accuracy or better.
- Overlap the center of left trap with the bead center (the x-y signal levels from the QDP must be the same measured during the calibration) and measure the position noise and its standard deviation from the QDP. Repeat the measurement for the right trap.
2. Single Molecule Localization with Nanometer Accuracy
- Fluorescence microscopy design and construction15,19
- Use a high quantum efficiency and electron-multiplied CCD to reach the high signal-to-noise ratios necessary for nanometer localization accuracy.
- Choose optics to get an image pixel size ~80 nm (for example, an image magnification ~200X with a camera pixel size of 16 μm). This is sufficiently small to neglect localization error due to pixelation8, but sufficiently large to collect a considerable number of photons per pixel.
- As excitation source, use laser light that is unpolarized (by passing through a non-polarization-maintaining optical fiber) or circularly polarized (by passing through a λ/4 waveplate) to maximize excitation of single chromophores, regardless of their orientation.
- Choose emission bandpass filters that efficiently cut-off both excitation and trapping laser lights. Note: In our experiments we labeled our protein with Atto532 dye and used a bandpass emission filter centered at 600 nm with 100 nm bandwidth.
3. Microfluidics
To achieve a precise control of buffer exchange and 'dumbbell' assembly, i.e., the anchoring of a single DNA molecule between two optically trapped microspheres, a custom-built laminar multichannel flow system must be developed. This is composed by a flow chamber, a pressure-reservoir and a pressure control system (Figure 2).
- Flow cell preparation NOTE: The flow chamber used for single-molecule experiments on protein-DNA interaction has preferably 4 inlets and 1 outlet, to allow up to 4 parallel buffer flows, each one containing one of the different components required for the experiment: beads, DNA, fluorescently-labeled protein, and imaging buffer. Separation of the components in the flow-channels allows efficient assembly of the dumbbell. Moreover, the imaging channel is useful to avoid background fluorescence from diffusing proteins and attain the high signal-to-noise ratio required for the localization of the DNA-binding protein with a precision of the order of few nanometers.
- Drill 1 mm diameter holes in the microscope slide (76 x 76 x 1 mm) with a diamond-tip, in order to create 4 inlets and one outlet.
- Clean the microscope slides with ethanol and center the port with the O-ring inserted and the adhesive ring in the slide surrounding the access holes. It is fundamental to wear gloves during this step to avoid fingerprints, which would disturb the gluing process.
- Clamp the NanoPorts to the slide, and place it in the oven preheated at 180 °C for 1 hr to allow complete attachment.
- Draw the outline pattern of the flow cell on a piece of paper. The channels should be ~3 mm wide and match the holes position on the microscope slide.
- Place the drawing on top of two pieces of Parafilm, and with a scalpel make sharp and continuous cuts along the drawn outline. Remove any protuberance formed. Discard the lower Parafilm layer, which is just used to guarantee a clean support.
- Place the microscope slide containing the attached NanoPorts upside down in the metallic support.
- Carefully align the Parafilm chamber outline with the holes, making sure that the parafilm does not obtrude the inlets and outlet of the chamber.
- Cover with a cleaned microscope coverslip (62 x 56 x 0.15 mm) and carefully remove excess Parafilm surrounding the chamber.
- Place two heat blocks, previously heated at 120 °C, on top of the flow cell to apply constant pressure on it. Let the Parafilm melt for approximately 25 min.
- Pressure reservoir and buffer containers The pressure reservoir should be made of Plexiglas (or similar) and allow accommodation of six buffer containers. The possibility of having at least two extra buffer containers improves the instrument flexibility. Note that transparent containers and tubes considerably assist in flow-system handling and troubleshooting of trapped air bubbles. Sterile and disposable luer lock-tip syringes (2.5 ml), from which the plungers have been previously removed, are good buffer containers and allow an easy connection with the inlet tubing. Make sure that the total fluid volume is small compared with the air volume inside the pressure reservoir, a condition essential to obtain smooth and stable flow.
- Connect shut-off valves at the exit of the buffer containers for turning the buffer flow on or off during the experiments.
- Mount the flow cell on the microscope stage. Connect the shut-off valves to the flow chamber inlets with polyetheretherketone tubing (inner diameter ~150 µm) and flangeless fittings. Add ~3 cm of fluorinated ethylene propylene tubing (inner diameter ~500 µm) as a linkage between the tubing and the NanoPort assemblies of the flow cell. This step is essential to reduce the buffer flow at the chamber entrance to avoid breakage of the flow cell or NanoPort detachment from the glass. The use of large inner diameter tubing alone, on the other hand, would require huge volumes of biological samples.
- Connect the flow chamber outlet to an open Falcon tube at atmospheric pressure. This tube serves as disposal for the buffer flowing out of the flow chamber.
- Pressure control system
- Build a pressure control system capable of finely adjusting the air pressure inside the pressure reservoir. This can be done by using two computer-controlled solenoid valves, one connected to a +0.5 atm pressure source (compressor) and the second one to atmospheric pressure. Solenoid valves are repeatedly opened for approximately 7 msec to increase or decrease the pressure in the line until the desired value is reached. NOTE: this approach was adapted from Carlos Bustamante20, and it yields a smoother flow than using a stepping motor syringe pump21.
- Add a pressure transducer controlled by a custom-written software to monitor the effective pressure exerted on the pressure reservoir. Typical working pressures are on the order of 20 mBar for dumbbell assembly and 200 mBar for washing the flow system components.
4. DNA Tailing with Biotinylated Deoxycytidine Triphosphate (biotin-dCTP)
The DNA molecule should be longer than 1 μm to facilitate its extension under flow and anchoring to the trapped beads. Moreover, a long DNA will prevent the IR trapping light from illuminating the DNA binding protein. This is of special relevance because absorption of IR light from an excited chromophore results in increased photobleaching. NOTE: In the present study, the DNA molecule was 3.7 μm long. The molecule contained three copies of the primary operator O1 and one copy of each of the two auxiliary operators, O2 and O3. These sequences have been inserted in the middle of the molecule, thus resulting approximately equidistant from the trapped beads and IR laser beams.
Mix 1 pmol of linear DNA 3’-ends with 60 pmol of biotin-14-dCTP, 4 μl of 5x Terminal deoxynucleotidyl Transferase (TdT) buffer (1 M potassium cacodylate, 125 mM Tris, 0.05% (v/v) Triton X-100, 5 mM CoCl2 (pH 7.2 at 25°C)) and 1.5 μl of TdT and ddH2O to a total volume of 20 μl. Incubate the mixture at 37 °C for 15 min. This will result in the addition of up to 60 nucleotides per DNA extremity. Note that DNA 3’-overhangs are tailed with higher efficiency than recessed or blunt ends.
Purify tailed DNA with spin columns or phenol/chloroform extraction and subsequent ethanol precipitation to remove CoCl2 from the TdT reaction buffer.
After purification, DNA can be stored at 4 °C for several weeks or at -20 °C for longer periods but avoid repeated thawing and refreezing.
5. Labeling Protein Thiol Groups with ATTO532
Labeling through modification of cysteine residues must not alter the protein activity. To overcome this problem, we used a LacI mutant, LacIQ231C, which carries just one cysteine per monomer at position 231. This mutant preserves the same characteristics of the wild type and chemical modifications at position 231 have been shown to not interfere with protein stability 22. NOTE: We labeled the protein with ATTO532 maleimide.
Ensure that the protein is dissolved in a buffer with a pH ranging from 7.0 to 7.5 and with a concentration ranging from 2 to 10 mg/ml. NOTE: in these experiments, LacI was dissolved in potassium phosphate 0.3 M, 5% glucose, pH 7.5 (buffer A).
Dissolve Tris-(2-carboxyethyl)phosphine hydrochloride (TCEP) to a final concentration of 100 mg/ml in H2O. TCEP solution must be prepared fresh prior to use.
Mix 100 μl of the protein with 3 μl TCEP and vortex carefully.
Dissolve the dye in N,N-dimethylformamide (DMF) to a final concentration of 10 mg/ml working at low light condition. Vortex until completely dissolved.
Reaction mix: Add 33 μl of dye to the protein+TCEP and vortex carefully. Fast spin to collect as much solution as possible.
Incubate the reaction mix for 2 hr in a shaker (8,000 rpm) at 20 °C (or at 4 °C O/N), protected from light.
Dissolve L-Glutathione reduced (GSH) to a final concentration of 100 mg/ml in H2O. Add 3 μl to the reaction mixture and incubate on the shaker at 8,000 rpm for 15 min. GSH will stop the reaction by blocking the reactivity of thiol groups.
- Purify the labeled protein using standard gel filtration columns, dialysis or spin concentrators. NOTE: In these experiments, labeled LacI was purified using 10 kDa cutoff spin concentrators.
- Dilute the reaction mixture with up to 12 ml of buffer A, apply it to the concentrator device, and centrifuge it at 8,000 x g, 1 hr, 4 °C. The labeled LacI is thus concentrated to a final volume of about 500 μl and the excess dye passes the membrane in the flow-through. Discard the flow-through and repeat step 5.8.1 at least three times.
Divide the labeled protein into small aliquots and store at -80 °C. Avoid repeated freezing and thawing.
6. Combined Optical Trapping and Single-molecule Fluorescence Imaging Experiments
Add 1 ml of buffer A to the buffer containers and apply 200 mBar pressure to wash the connecting tubes and flow cell. To maximize diffusion of the binding protein on DNA, here and in the following steps, buffer A may be replaced with a low ionic strength buffer. NOTE: In these experiments, we used TBE 0.5x.
Check for the presence of air-bubbles, which might disrupt the flow smoothness. A gentle flick on the outlet tube will help to remove any remaining bubbles.
Decrease the pressure to 20 mBar and verify that all channels and tubes are clear from air bubbles and the buffer flows with similar velocities in all channels.
Prepare the samples to a final volume of 300 µl: streptavidin-coated polystyrene beads 1.87 µm; 20 pM of linear DNA, biotinylated on both extremities according to section 4; 300 pM of LacI tetramer labeled with ATTO532 according to section 5.
Add each sample to one of the syringe containers in the following order: beads in the first channel, DNA in the second, third channel with the imaging buffer only (buffer A + oxygen scavenger system) and the labeled protein in the fourth channel.
Turn the flow on at 200 mBar for 3 min to let the samples arrive inside the flow channel. Then reduce the pressure to 20 mBar.
While imaging the first channel under bright field illumination, switch on the two optical tweezers and catch a polystyrene bead in each trap.
Move quickly to the second channel to avoid that other beads fall into the traps. Note that the arrival in the DNA channel is easily noticeable by the end of the bead flow.
Using one AOD, move one trap (bead) back and forth in the proximity of the other bead to allow the attachment of the flow-extended DNA in between the two optically trapped beads. When a DNA dumbbell is formed, the static bead starts following the movement of the bead that is actively moved back and forth by the trap.
Move quickly to the buffer channel and turn off the buffer flow. Perform a force-extension curve analysis4,23-25 to verify the presence of a single DNA molecule. If more than one molecule is present (i.e., the enthalpic, raising phase of the force-extension curve occurs at a length shorter than the DNA contour length), discard the dumbbell and start again from step 6.7.
Once a single DNA dumbbell is obtained, turn on the flow again and move the dumbbell to the protein channel. Switch to wide field fluorescence microscopy to monitor the interaction of the labeled-LacI with DNA. Turn off the flow and start acquisition.
Representative Results
In a successful experiment, one (or more) labeled proteins undergo binding/unbinding and/or monodimensional diffusion along the DNA molecule (Figure 3A). Localization of proteins along the DNA molecule allows the quantification of kinetic parameters as a function of the DNA sequence. When buffer conditions causing 1D diffusion are applied, it is possible to follow protein trajectories, and determine, for example, the diffusion coefficient D1D.
The precise position of a single spot, in each frame, can be determined by fitting the Point Spread Function (PSF) with a bidimensional Gaussian function or, alternatively, when handling a large data set, with a more rapid, recently published algorithm (Radial Symmetry Center, RSC)26,27. The latter method determines the point of maximal radial symmetry of the image, attaining a localization precision that is typically comparable with Gaussian fitting. In both cases, tracking can be achieved through MATLAB routines that can be accessed freely27.
Figure 3A shows a kymogram of a typical experiment in which a single LacI molecule diffuses along non-specific DNA sequences while another LacI molecule is specifically bound to one operator, located in the center of the DNA molecule. Figure 3B shows the position of the two LacI molecules (obtained using RSC) along the direction connecting the centers of the two traps (x), as a function of time. From the position of the molecule diffusing along the DNA molecule, we calculated the Mean Square Displacement (MSD) at different time intervals according to the following equation:
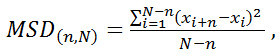 (1)
(1)
where N is the total number of positions measured and n is the measurement index going from 1 to N. In case of pure unidimensional Brownian diffusion, the MSD is directly proportional to nΔt (Δt is the time interval between two consecutive frames, 100 msec in our experiments), with a slope equal to twice the diffusion coefficient (MSD(n,N) = 2D1nΔt). Figure 4 shows the MSD vs nΔt plot for the molecule diffusing along DNA. The diffusion coefficient of the protein can be readily obtained from a linear fit of the MSD vs nΔt plot. Note that as n increases, the number of points for calculating MSD from Eq. 1 consequently decreases. The error in the evaluation of the MSD thus increases with n. For this reason, we chose to limit our analysis to n=N/4. This is anyway a quite large value compared to the N/10 used by many labs.
 Figure 1. The experimental setup consists of: halogen lamp (H), condenser (C), sample (S), piezo translators (x-y and z), objective (O), a low-magnification camera (CCD 200X) and a high-magnification camera (CCD 2,000X) used for the nm-stabilization feedback (BS is a 50:50 beam splitter cube). Double optical tweezers are inserted and extracted from the optical axis of the microscope through dichroic mirrors (D2 and D3) and comprise: Nd:YAG laser (1,064 nm), optical isolator (OI), λ/2 waveplates, polarizing beam splitter cubes (PBS), acousto-optic deflectors (AOD), 1,064 nm interferential filters (F1 and F2), quadrant detector photodiodes (QDP). Signals from QDPs were acquired through an analog-to-digital converter (ADC) and elaborated with a FPGA board. Two custom-built direct digital synthesizers (DDS) drove the AODs to control traps’ position. A digital input-output board (DIO) controlled the aperture of two solenoid valves (V1 and V2) connected to a compressor (+0.5 atm) and ambient pressure (+0 atm), respectively. A Labview software monitored the pressure inside the pressure reservoir (C1) through a pressure meter (PM) and automatically opened one of the two valves to reach the desired pressure level. Fluorescence excitation was provided by a duplicated Nd:YAG laser (532 nm) and the image projected on an electron multiplied camera (EMCCD). M is a movable mirror, F3 an emission filter. Please click here to view a larger version of this figure.
Figure 1. The experimental setup consists of: halogen lamp (H), condenser (C), sample (S), piezo translators (x-y and z), objective (O), a low-magnification camera (CCD 200X) and a high-magnification camera (CCD 2,000X) used for the nm-stabilization feedback (BS is a 50:50 beam splitter cube). Double optical tweezers are inserted and extracted from the optical axis of the microscope through dichroic mirrors (D2 and D3) and comprise: Nd:YAG laser (1,064 nm), optical isolator (OI), λ/2 waveplates, polarizing beam splitter cubes (PBS), acousto-optic deflectors (AOD), 1,064 nm interferential filters (F1 and F2), quadrant detector photodiodes (QDP). Signals from QDPs were acquired through an analog-to-digital converter (ADC) and elaborated with a FPGA board. Two custom-built direct digital synthesizers (DDS) drove the AODs to control traps’ position. A digital input-output board (DIO) controlled the aperture of two solenoid valves (V1 and V2) connected to a compressor (+0.5 atm) and ambient pressure (+0 atm), respectively. A Labview software monitored the pressure inside the pressure reservoir (C1) through a pressure meter (PM) and automatically opened one of the two valves to reach the desired pressure level. Fluorescence excitation was provided by a duplicated Nd:YAG laser (532 nm) and the image projected on an electron multiplied camera (EMCCD). M is a movable mirror, F3 an emission filter. Please click here to view a larger version of this figure.
 Figure 2. (A) Flow system diagram. The pressure-control system is composed by a pressure meter (PM) and two solenoid valves V1 and V2 connected to a compressor at +0.5 atm (PRS) and to ambient pressure (ATM), respectively. X1 and X2 represent 3-way connectors. The aperture of the valves is finely adjusted to reach the desired pressure in the pressure reservoir C1 with 4 buffer containers. Each inlet tube includes an independent on/off valve at the entrance of the multichannel flow chamber. The outlet tubing is connected to the waste container C2 placed at ambient pressure. Drawings are not to scale. (B) Schematic representation of the multichannel flow chamber. Four laminar flows separately translocate functionalized polystyrene beads, DNA molecules, imaging buffer and labeled proteins. Two beads are caught with dual optical tweezers and moved to the DNA channel to allow DNA anchoring in between them. A DNA force-extension analysis is performed in the buffer channel to verify the presence of a single DNA molecule and, just afterwards, the DNA is incubated in the protein channel. The inset shows a magnification of the final molecular construct, ready for single-molecule imaging in the buffer channel. Drawings are not to scale. Please click here to view a larger version of this figure.
Figure 2. (A) Flow system diagram. The pressure-control system is composed by a pressure meter (PM) and two solenoid valves V1 and V2 connected to a compressor at +0.5 atm (PRS) and to ambient pressure (ATM), respectively. X1 and X2 represent 3-way connectors. The aperture of the valves is finely adjusted to reach the desired pressure in the pressure reservoir C1 with 4 buffer containers. Each inlet tube includes an independent on/off valve at the entrance of the multichannel flow chamber. The outlet tubing is connected to the waste container C2 placed at ambient pressure. Drawings are not to scale. (B) Schematic representation of the multichannel flow chamber. Four laminar flows separately translocate functionalized polystyrene beads, DNA molecules, imaging buffer and labeled proteins. Two beads are caught with dual optical tweezers and moved to the DNA channel to allow DNA anchoring in between them. A DNA force-extension analysis is performed in the buffer channel to verify the presence of a single DNA molecule and, just afterwards, the DNA is incubated in the protein channel. The inset shows a magnification of the final molecular construct, ready for single-molecule imaging in the buffer channel. Drawings are not to scale. Please click here to view a larger version of this figure.
 Figure 3. Localization of single LacI molecules interacting with a stretched DNA molecule.(A) The vertical axis of the kymogram represents the x coordinate, parallel to the DNA molecule, while the horizontal axis is time (100 msec acquisition time). (B)
X position of the LacI molecule vs time. The position of the molecules was determined by RSC. Images were acquired at 100 msec exposure time and 1.25 x 10-2 W cm-2 excitation laser intensity. Please click here to view a larger version of this figure.
Figure 3. Localization of single LacI molecules interacting with a stretched DNA molecule.(A) The vertical axis of the kymogram represents the x coordinate, parallel to the DNA molecule, while the horizontal axis is time (100 msec acquisition time). (B)
X position of the LacI molecule vs time. The position of the molecules was determined by RSC. Images were acquired at 100 msec exposure time and 1.25 x 10-2 W cm-2 excitation laser intensity. Please click here to view a larger version of this figure.
 Figure 4. Plot of the MSD vs time for a single LacI molecule diffusing along a stretched DNA molecule. The MSD is calculated from the position record represented in Figure 3B (red). The diffusion coefficient D1D, obtained from linear regression of the data depicted in the figure, is 0.030 ± 0.002 μm2 sec-1.
Figure 4. Plot of the MSD vs time for a single LacI molecule diffusing along a stretched DNA molecule. The MSD is calculated from the position record represented in Figure 3B (red). The diffusion coefficient D1D, obtained from linear regression of the data depicted in the figure, is 0.030 ± 0.002 μm2 sec-1.
Discussion
In the last decade, single molecule manipulation and imaging techniques have seen great progress in terms of spatial and temporal resolution. The combination of manipulation and imaging techniques is at the base of powerful instruments that now allow the control of the mechanical conditions of a single biological polymer, such as DNA, RNA or cytoskeletal filaments, and the simultaneous localization of single proteins interacting with the same polymer. Controlling the mechanical conditions of the trapped polymer is of particular interest. In fact, in living cells, nucleic acids are continuously undergoing mechanical stress caused by interacting enzymes and proteins; similarly, a complex network of interacting proteins and molecular motors modulates tension on cytoskeletal filaments. On the other hand, the possibility to detect and precisely localize proteins interacting with nucleic acids, allows the measurement of molecular interactions as a function of their position along the DNA or RNA sequence.
Combining single-molecule manipulation and imaging techniques requires careful design of the experimental setup and the biological constructs. Optical traps must provide a stable support, ensuring nm-level stability of the suspended polymer. Such stability can be reached through careful isolation of the experimental apparatus from the numerous sources of mechanical noise and by minimizing laser pointing and amplitude fluctuations. Precise (sub-nm) movements of the optical traps are achieved using AODs driven by DDSs. Here, we illustrated a step-by-step protocol to optimize and check optical tweezers’ stability at the nm-level.
Simple wide-field epi-fluorescence microscopy coupled to an EMCCD camera is used to localize single chromophores interacting with the suspended polymer. Our experimental assay is limited to long DNA or RNA molecules (≥6 kbp) to ensure a spatial separation between the trapping laser beam and the fluorescent probe. In fact, superposition of the near-infrared trapping laser with the visible excitation laser would lead to enhanced photobleaching of the chromophores due to absorption of the near-infrared light while the chromophore is in the excited state16. Dumbbell assembly is another tricky procedure that is better obtained using a multichannel flow-cell, for which we provided a detailed description. We indicated how to optimize the buffer flow and how to avoid air bubbles, which can otherwise seriously compromise the experiments. We described protocols for labeling the DNA extremities with biotin, which is required for dumbbell assembly using streptavidin-coated beads, and protocols for protein labeling. Finally, we illustrated step-by-step operations to perform experiments in which the binding and diffusion of a single lactose repressor molecule on a stretched DNA molecule is quantified.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Gijs Wuite, Erwin J.G. Peterman, and Peter Gross for help with the microfluidics and Alessia Tempestini for help with sample preparation. This research was funded by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 284464 and from the Italian Ministry for Education, University and Research FIRB 2011 RBAP11X42L006, Futuro in Ricerca 2013 RBFR13V4M2, and in the framework of the Flagship Project NANOMAX.
References
- Monico C, Capitanio M, Belcastro G, Vanzi F, Pavone FS. Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level. International journal of molecular sciences. 2013;14:3961–3992. doi: 10.3390/ijms14023961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco I, Gonzalez RL. Biological mechanisms, one molecule at a time. Genes & development. 2011;25:1205–1231. doi: 10.1101/gad.2050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitanio M, et al. Exploring molecular motors and switches at the single-molecule level. Micr. Res. Tech. 2004;65:194–204. doi: 10.1002/jemt.20126. [DOI] [PubMed] [Google Scholar]
- Smith SB, Finzi L, Bustamante C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- Wang MD, et al. Force and velocity measured for single molecules of RNA polymerase. Science. 1998;282:902–907. doi: 10.1126/science.282.5390.902. [DOI] [PubMed] [Google Scholar]
- Capitanio M, et al. Ultrafast force-clamp spectroscopy of single molecules reveals load dependence of myosin working stroke. Nat Methods. 2012;9:1013–1019. doi: 10.1038/nmeth.2152. [DOI] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, et al. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Biebricher A, Wende W, Escude C, Pingoud A, Desbiolles P. Tracking of single quantum dot labeled EcoRV sliding along DNA manipulated by double optical tweezers. Biophys J. 2009;96:50–52. doi: 10.1016/j.bpj.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelli A, Wuite GJ, Peterman EJ. Combining optical trapping, fluorescence microscopy and micro-fluidics for single molecule studies of DNA-protein interactions. Physical chemistry chemical physics : PCCP. 2011;13:7263–7272. doi: 10.1039/c0cp02844d. [DOI] [PubMed] [Google Scholar]
- Capitanio M, Cicchi R, Pavone FS. Continuous and time-shared multiple optical tweezers for the study of single motor proteins. Optics and Lasers in Engineering. 2007;45:450–457. [Google Scholar]
- Harada Y, et al. Single-molecule imaging of RNA polymerase-DNA interactions in real time. Biophys J. 1999;76:709–715. doi: 10.1016/S0006-3495(99)77237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mameren J, et al. Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature. 2009;457:745–748. doi: 10.1038/nature07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitanio M, Maggi D, Vanzi F, Pavone F. Fiona in the trap: the advantages of combining optical tweezers and fluorescence. J Opt A: Pure Appl Opt. 2007;9:s157. [Google Scholar]
- Dijk MA, Kapitein LC, Mameren J, Schmidt CF, Peterman EJ. Combining optical trapping and single-molecule fluorescence spectroscopy: enhanced photobleaching of fluorophores. J Phys Chem B. 2004;108:6479–6484. doi: 10.1021/jp049805+. [DOI] [PubMed] [Google Scholar]
- Capitanio M, Cicchi R, Pavone FS. Position control and optical manipulation for nanotechnology applications. European Physical Journal B. 2005;46:1–8. [Google Scholar]
- Capitanio M, et al. Calibration of optical tweezers with differential interference contrast signals. Review of Scientific Instruments. 2002;73:1687–1696. [Google Scholar]
- Elangovan R, et al. An integrated in vitro and in situ study of kinetics of myosin II from frog skeletal muscle. J Physiol. 2012;590:1227–1242. doi: 10.1113/jphysiol.2011.222984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuite GJL, Davenport RJ, Rappaport A, Bustamante C. An integrated laser trap/flow control video microscope for the study of single biomolecules. Biophysical Journal. 2000;79:1155–1167. doi: 10.1016/S0006-3495(00)76369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer LR, Bianco PR. Laminar flow cells for single-molecule studies of DNA-protein interactions. Nat Methods. 2008;5:517–525. doi: 10.1038/nmeth.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkauskas D, Zhan HL, Matthews KS, Pavone FS, Vanzi F. Tetramer opening in LacI-mediated DNA looping. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16627–16632. doi: 10.1073/pnas.0904617106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko JF, Siggia ED. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- van Mameren J, et al. Unraveling the structure of DNA during overstretching by using multicolor, single-molecule fluorescence imaging. Proc Natl Acad Sci U S A. 2009;106:18231–18236. doi: 10.1073/pnas.0904322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MD, Yin H, Landick R, Gelles J, Block SM. Stretching DNA with optical tweezers. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Long F, Zeng S, Huang ZL. Fast and precise algorithm based on maximum radial symmetry for single molecule localization. Opt Lett. 2012;37:2481–2483. doi: 10.1364/OL.37.002481. [DOI] [PubMed] [Google Scholar]
- Parthasarathy R. Rapid, accurate particle tracking by calculation of radial symmetry centers. Nat Methods. 2012;9:724–726. doi: 10.1038/nmeth.2071. [DOI] [PubMed] [Google Scholar]