

Abstract
Echinoderms have long been a favorite model system for studies of reproduction and development, and more recently for the study of gene regulation and evolution of developmental processes. The sea star, Patiria miniata, is gaining prevalence as a model system for these types of studies which were previously performed almost exclusively in the sea urchins, Strongylocentrotus purpuratus and Lytechinus variegatus. An advantage of these model systems is the ease of producing modified embryos in which a particular gene is up or downregulated, labeling a group of cells, or introducing a reporter gene. A single microinjection method is capable of creating a wide variety of such modified embryos. Here, we present a method for obtaining gametes from P. miniata, producing zygotes, and introducing perturbing reagents via microinjection. Healthy morphant embryos are subsequently isolated for quantitative and qualitative studies of gene function. The availability of genome and transcriptome data for this organism has increased the types of studies that are performed and the ease of executing them.
Keywords: Developmental Biology, Issue 91, Embryology, Patiria miniata, sea star, echinoderm, development, gene regulatory networks, microinjection, gene expression perturbation, antisense oligonucleotide, reporter expression
Introduction
The sea star, Patiria miniata, (commonly known as the bat star) is emerging as an interesting and versatile model system for a variety of cellular1-3, developmental4,5, evolutionary6-8, and ecological studies9-11. Adult P. miniata are distributed along the pacific coast from Sitka, Alaska to Baja, California12 and are readily maintained in marine aquaria. Oocytes are obtainable year round and each female can shed tens of thousands of eggs. Oocytes are easily matured and fertilized externally13. The resulting embryos are transparent allowing for easy observation; they develop synchronously, and require only sea water for development. Whole genome assembly and multiple transcriptomes are also available for P. miniata (Echinobase.org). Such advantages make them ideal for a range of research and teaching purposes.
In recent years, P. miniata has become a model system for developmental gene regulatory network analyses14-16. The aim of such studies is to identify the entire compliment of regulatory genes and determine the network of their interactions. Much of this work entails perturbing gene expression through introduction of antisense oligonucleotides or in vitro synthesized mRNAs. Additionally, cis regulatory analyses are used to characterize the function of regulatory DNA15. These analyses require introduction of perturbation reagents and/or DNA reporter constructs into embryos. Furthermore, to characterize the downstream effects of these perturbations, one must assay many embryos for changes in gene expression of potential targets. Techniques for microinjection of many hundreds of zygotes are central for this work.
Echinoderms, including P. miniata, require many months to reach sexual maturity. Because of this, it is generally not practical to develop and maintain transgenic lines of these animals for experimentation. Therefore, breeding of transgenic adults cannot efficiently create modified embryos. Instead, perturbation must occur de novo through microinjection. Microinjection offers an opportunity to modify embryos with reagents that are not cell-permeable. The following protocol describes a method to introduce DNA, mRNA, cell tracers, and morpholino antisense oligonucleotides into hundreds of fertilized eggs in one 2-3 hr sitting through microinjection. This produces sufficient material for a variety of downstream experiments including, but not limited to, qPCR, in situ hybridization, RNA-Seq, and western blotting.
Protocol
Keep all sea water or artificial seawater (SW), adult animals, and cultures at 15 °C as much as is practical. Ensure eggs and zygotes are kept immersed in SW.
Commercially prepared sea salts reconstituted with distilled or reverse osmosis water serves well as a source of SW. Check salinity using a hydrometer and adjust salts or water to achieve optimum levels. Keep specific gravity levels between 1.020 to 1.025. Keep all glassware and plasticware separate from all other labware to avoid any contamination with chemicals. Clean embryo grade labware by rinsing with deionized water or occasional soaking in dilute sodium hypochlorite followed by rinsing several times in water.
1. Obtaining and Maturing Gametes from Patiria miniata Adults
Select an animal to excise gonads. Determine sex after excision by looking for the presence of ovaries or testes because P. miniata are not sexually dimorphic.
Cut a small opening, no larger than 1 cm, along the side of a sea star arm with a scalpel blade (Figure 1A). Using small blunt-ended forceps pull back edges of the incision to access the gonad tissue and pull gonad tissue out through the incision. NOTE: Avoid pulling out gut tissue, which is a loose gray or brown tissue (Figure 1B). Ovaries are typically orange, yellow, or light brown (Figure 1C), while testes are white or beige and when disrupted will cause sea water to become milky or cloudy in appearance (Figure 1D).
Return females to the aquaria. Isolate males for an hour or two in a container of SW since the stress from handling may induce spawning. Animals heal along the incision site and continue to provide gametes for several months.
Collect testes into a 1.5 ml Eppendorf tube with as little SW as possible and store on ice until use. Store any testes not used on the day of collection at 4 °C for up to a week.
Collect ovaries in a small glass culture dish in a several milliliters of SW. To release the oocytes from the ovary tissue, tease apart the tissue with two pairs of forceps until no large pieces remain. NOTE: Optimally, the majority of the oocytes are fully developed. Such oocytes are large, round, and clear yellow or light brown with a distinct germinal vesicle in the oocyte (Figure 2A), compared to the subset of undeveloped oocytes that are smaller, brown, grainy, and opaque (Figure 2B). If greater than around 25% of the oocytes are of the undeveloped variety, select a different female to obtain oocytes.
Pour the oocytes and remaining tissue through a mesh filter cup with a 200 μm pore size and collect flow through. This will contain all varieties of oocytes but remove ovarian tissue. Dislodge remaining oocytes from the tissue caught by the filter by spraying with SW from a squirt bottle. Filter cups are made using a 50 ml conical tube (Figure 3A-A’).
Pour the flow through from step 1.6 through a 100 μm mesh filter cup. Discard small oocytes in flow through. Large fully developed oocytes that are ready for maturation will stay on the filter. Rinse the oocytes into a clean 200 ml beaker.
Add 95 ml of SW to the 200 ml glass beaker of oocytes. Add 5 ml of 200 μM 1-methyladenine for a final concentration of 10 μM.
Transfer the oocytes to a large shallow culture dish and place at 15 °C. A high surface area to volume culture dish allows for oxygen exchange. The oocytes do not mature or may not develop well if they are overcrowded during this maturation phase. Split into several dishes of SW plus 10 μM 1-methlyadenine until culture is less than 200 oocytes/ml.
Allow the oocytes to mature for 45-90 min, but no longer as this will render the oocytes unable to be fertilized. To determine if maturation is complete, look at the oocytes under a dissecting scope. The germinal vesicle moves towards and fuses with the cell membrane during maturation. It then breaks down and therefore is no longer visible in mature oocytes (Figure 2C).
2. Fertilization of Mature Oocytes
Chose a small cluster of testis tissue, roughly pea-sized, and mince in a small glass culture dish with approximately 1 ml of SW.
Use a Pasteur pipette to transfer 2-3 drops of the resulting milky sea water to around 100 ml of the mature oocytes and swirl the culture dish to mix. Avoid transferring pieces of testis tissue. Avoid adding greater quantities of sperm as this may result in polyspermy and subsequent poor development.
After several minutes observe the oocytes under a dissecting scope. Fertilized eggs, or zygotes, have a fertilization envelope, which is a clear bubble that rises off the cell membrane (Figure 2D). NOTE: Do not proceed with the culture if less than around 90% of eggs fertilize since poor fertilization rates are an indication of an unhealthy culture that may also not develop well.
After fertilization is complete, change the SW on the zygotes by pouring through a 100 μm mesh filter and rinsing into a culture dish with fresh SW. It is important to remove the excess sperm to prevent oxygen starvation. Place the culture dish at 15 °C for 15-30 min.
3. De-jellying and Rowing Fertilized Eggs
- Prepare injection dishes by taking the lid from a 60 x 15 mm polystyrene Petri dish.
- Draw a permanent marker pen line on the back of the dish, around the area that matches the field of view on the microinjection microscope. This ensures that all rowed embryos are within the microscope field of view.
- Use a glass Pasteur pipette to score a line through the circle, preferably off to one side of the circle (Figure 3B). Use this line later to break injection needles (Step 4.5). NOTE: Reagents commonly used to adhere sea urchin embryos, e.g., protamine sulfate or poly-L Lysine, are unnecessary. De-jellied P. miniata zygotes stick very well to uncoated plastic. Note that they do not stick well to glass dishes.
Add around 10 ml SW to each dish so that the surface is covered, but the water will not splash excessively in the dish as this may perturb rowed embryos.
- Remove the P. miniata zygote jelly coat with acid SW prior to rowing. This jelly coat is more extensive than that observed for model sea urchin species.
- Prepare acid SW in the range of pH 4.0-4.2. The pH is critical to allow efficient removal of jelly coat while permitting normal development.
- Add single drops of 1 N (1 M) HCl in SW to a 1 L stock of SW. Allow each drop to mix completely and monitor pH before adding more HCl. NOTE: One to several ml of HCl results in the appropriate pH, but do not add the entire volume at once. NOTE: Prepare 1 N HCl in fume hood with SW and 12 N (12 M) HCl.
- Remove SW on zygotes by pouring them onto a 100 μm mesh filter. Rinse into a 200 ml beaker in the smallest amount of SW possible (approximately 10 ml). Fill the beaker up to 150 ml with acid SW (pH 4.0) and allow the zygotes to sit in the acid SW for 3 min.
Strip jelly coat from the zygotes by pouring them back and forth between two 200 ml beakers, each time passing the zygotes through a 200 μm mesh filter. Pour the zygotes through the filter around 5-10x over about a 2 min time window.
Remove acid SW by pouring the zygotes onto a 100 μm mesh filter. Rinse the zygotes into a small glass culture dish with SW. De-jellied zygotes may stick to plastic dishes. Row embryos immediately; they start to produce more jelly coat which leads to poor adhesion to plastic dishes over time.
- Use a mouth pipette (Figure 3C-C’) to pick up zygotes from the glass culture dish and place them into straight lines on the injection dishes. Melt the center of the thin side of a Pasteur pipette over a flame until the glass is soft. Quickly pull the ends of the glass apart to produce a very thin stretch of glass. Once the glass is cool, break off the small end of the Pasteur pipette (for additional information on preparing a mouth pipette see Wessel et al. 200417 or Sweet et al. 200418).
- Make a rowing pipette such that the diameter is just larger than the diameter of the embryos so that only a single zygote will enter and exit the pipette at a time. This will allow a single row to form. Smaller diameter pipettes may strip the fertilization envelope and disrupt the zygotes as P. miniata zygotes do not have a hyaline membrane and are quite soft.
- If the zygotes do not stick to the plastic dish, keep them for additional time in acid SW (up to several more minutes). Alternatively a smaller diameter pipette may aid in removing jelly coat to allow the zygotes to stick more effectively.
- The zygotes are slightly positively buoyant and tend to float or hover just above the bottom of the dish. In this case, position the rowing pipette so that the zygotes are gently pressed onto the surface of the dish as they exit the pipette (Figure 3D). NOTE: It is permissible to fit many rows on these dishes. P. miniata embryonic membranes do not become more difficult to penetrate over time and therefore one can inject hundreds of zygotes on a single dish.
4. Injection of Zygotes
- Prepare injection solutions with appropriate concentrations of morpholino antisense oligonucleotides (MASO), mRNA, or DNAs in a final concentration of 200 mM KCl. 400-800 μM MASO and 2.5 ng/μl of DNA tend to produce morphant phenotypes while minimizing toxicity. The final concentration in the egg will be 1/125th the starting concentration14. For injection solutions containing MASOs, heat to 65 °C for 5 min immediately prior to use and then store at room temperature.
- For ease of sorting injected embryos, add rhodamine dextran tracer to the injection solution to obtain a concentration of 0.1%.
Prepare injection needles by pulling glass capillary tubing (1.0 mm OD x 0.75 mm ID, 100 mm length) in a needle pulling instrument following manufacturer’s instructions. Needles suitable for injection in sea urchins also work well for sea stars, so use similar parameters19.
Load approximately 2 μl of injection solution into an injection needle using a microloader pipette tip.
Insert the blunt end of the needle into the manipulator. Set picospritzer to 100 msec pulse and an air pressure of 40 PSI.
Place a dish of rowed embryos onto the microscope stage and have all embryos in field of view. Orientate the dish such that the scored line is on the side closest to the needle. Position the needle at an approximately 60° angle so the tip is near the middle of the line and just above the surface of the water.
Focus on the scored line and lower the needle until the tip comes into focus. Lower the tip a little more and break it open by running it gently into the ridges of the scored line. Raise the tip off the surface of the dish and position it at the top of the first row of zygotes and focus on the zygotes.
Lower the tip such that it will impale the center of zygote coming in from around a 60° angle. The needle easily penetrates P. miniata zygotes. Step on the foot pedal (or other means of forcing material from the injection needle) to introduce a bolus of injection solution into the embryo.
Aim for a bolus that is 20-30% the diameter of the embryo, or between 25 and 80 pl. If the bolus is larger or smaller than this, adjust the duration of injection on the picospritzer and inject the next embryo. Continue to adjust the duration to obtain the desired bolus size. Start at 100 msec duration. Re-break the needle if greater than around 200 msec is needed to introduce the appropriate bolus. Discard the needle and prepare a new one if less than 15-20 msec is needed to obtain this bolus size as the needle size is too large and may disrupt normal development.
Proceed to inject the remaining zygotes on the injection dish. Embryos can readily be injected until cleavage starts and into single blastomeres after first cleavage. Periodically lift the needle above the surface of the water to remove zygotes that stick onto the needle. If necessary re-break the tip of the needle or load a new needle if a blockage occurs.
5. Collecting Injected Embryos for Downstream Analysis
Allow the embryos to develop at 15 °C in SW. Maintain high surface area to volume in the cultures to allow for oxygen exchange. Ensure embryos are not crowded; keep cultures at or below 200 embryos/ml. Depending upon the desired stage of embryo, plan to collect injected embryos at around 20-24 hr post-fertilization. NOTE: This is often an ideal time to sort and collect and because normally developing embryos are readily distinguished morphologically, but are not yet swimming.
If embryos have hatched, gently salt shock to prevent swimming. Add 200 μl of 5 M NaCl to 10 ml of SW in the injection dish while gently swirling the SW. After a few minutes, the embryos will fall to the bottom of the dish. Collect these and move them to normal salinity SW.
Collect injected embryos under an appropriate fluorescent light source compatible with the tracer used in the injection solution. If no tracer was used, sort embryos to select for normal morphology.
Using a mouth pipette, draw fluorescing embryos up with a minimal amount of SW and blow them out into a small culture dish filled with SW. Ideal mouth pipettes have a large enough opening for embryos to be pulled in and out without damaging them, but not so large that undesired embryos and extraneous SW are also pulled up.
Once all of the desired embryos have been collected into the new dish, observe the embryos under fluorescent light and remove any un-injected embryos or poorly developed embryos.
Culture sorted embryos to desired stages in an incubator and proceed with imaging or other desired protocols.
Representative Results
The goal of this protocol is to introduce reagents into embryos. We demonstrate the effectiveness of the protocol by injecting a DNA reporter construct that drives the expression of green fluorescent protein (GFP). Injected embryos express GFP in clonal patches (Figure 4A-B) as the DNA incorporates during early cleavage. Many reagents that are desirable to introduce into embryos are toxic in high quantities and to suboptimal batches of embryos. Toxicity manifests by delaying development, arresting development in early cleavage or at the fertilized egg stage, or by imposing aberrant development (Figure 5A-C).
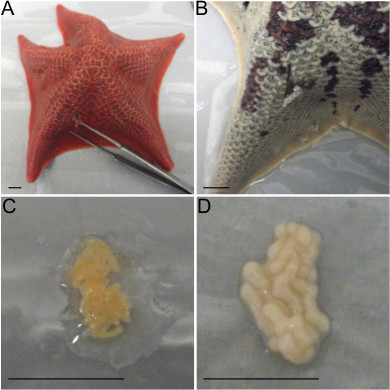 Figure 1. Obtaining gametes from P. miniata adults. A) To excise gonads make an incision on the side of an arm proximal to the middle of the animal. B) The dark brown material pulled from this incision is gut tissue. Avoid pulling gut tissue as it will injure the animal. C) Ovary tissue is typically orange in color as shown here, but may also be yellow or light brown. D) Testes are white or beige in color, as shown. All scale bars denote 1 cm.
Figure 1. Obtaining gametes from P. miniata adults. A) To excise gonads make an incision on the side of an arm proximal to the middle of the animal. B) The dark brown material pulled from this incision is gut tissue. Avoid pulling gut tissue as it will injure the animal. C) Ovary tissue is typically orange in color as shown here, but may also be yellow or light brown. D) Testes are white or beige in color, as shown. All scale bars denote 1 cm.
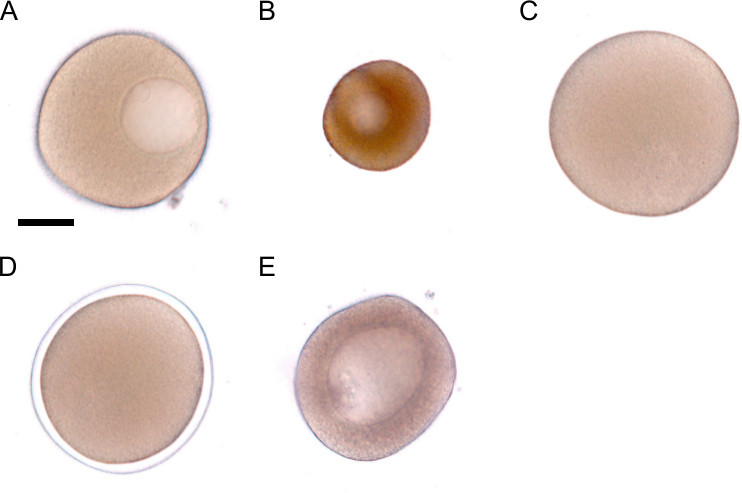 Figure 2. Maturation and fertilization of oocytes. A) Healthy oocytes that are ready for maturation are large and clear with a visible germinal vesicle. B) Oocytes that are not ready for maturation are smaller, brown, and grainy in appearance. C) An Oocyte that has been matured with 1-methyladenine. The germinal vesicle has broken down. D) A zygote surrounded by a fertilization envelope. E) Avoid cultures with large oocytes that cannot be matured because these oocytes will not be removed by filtration. Such oocytes are slightly smaller than the one in A. and are darker brown and grainy textured. The scale bar in A denotes 50 μm and represents the scale for images A-E.
Figure 2. Maturation and fertilization of oocytes. A) Healthy oocytes that are ready for maturation are large and clear with a visible germinal vesicle. B) Oocytes that are not ready for maturation are smaller, brown, and grainy in appearance. C) An Oocyte that has been matured with 1-methyladenine. The germinal vesicle has broken down. D) A zygote surrounded by a fertilization envelope. E) Avoid cultures with large oocytes that cannot be matured because these oocytes will not be removed by filtration. Such oocytes are slightly smaller than the one in A. and are darker brown and grainy textured. The scale bar in A denotes 50 μm and represents the scale for images A-E.
 Figure 3. Apparatuses for assembly. A-A’) A mesh filter fastened to a 50 ml conical tube. The tapered-end of the tube and the middle of the lid are cut out to allow cultures to be poured through the tube and filter. B) An injection dish constructed from the lid of a 60 x 15 mm polystyrene culture dish. A circle drawn around the center outlines the field of view of the injection microscope and serves as a guide for rowing embryos. A line scored into the dish is useful for breaking injection needles. C-C’) Mouth pipette set-up. Fit a mouthpiece into one end of a several foot long rubber tube. Fit the other end to a Pasteur pipette, which has been shaped by briefly melting and pulling the narrow end of the glass. D) Schematic demonstrating an ideal rowing Pasteur pipette shape and width to promote sticking without damaging embryos. When breaking the end off of the pulled pipette, it is helpful if the end is tapered to prevent emerging zygotes from floating away from the dish. All scale bars denote 3 cm.
Figure 3. Apparatuses for assembly. A-A’) A mesh filter fastened to a 50 ml conical tube. The tapered-end of the tube and the middle of the lid are cut out to allow cultures to be poured through the tube and filter. B) An injection dish constructed from the lid of a 60 x 15 mm polystyrene culture dish. A circle drawn around the center outlines the field of view of the injection microscope and serves as a guide for rowing embryos. A line scored into the dish is useful for breaking injection needles. C-C’) Mouth pipette set-up. Fit a mouthpiece into one end of a several foot long rubber tube. Fit the other end to a Pasteur pipette, which has been shaped by briefly melting and pulling the narrow end of the glass. D) Schematic demonstrating an ideal rowing Pasteur pipette shape and width to promote sticking without damaging embryos. When breaking the end off of the pulled pipette, it is helpful if the end is tapered to prevent emerging zygotes from floating away from the dish. All scale bars denote 3 cm.
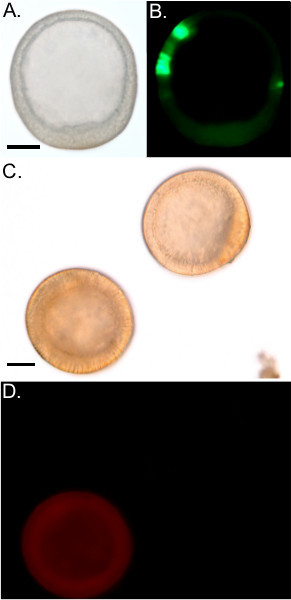 Figure 4. Reagents Successfully Introduced by Microinjection. A) DIC of a blastula stage embryo injected with a GFP reporter plasmid. The embryo has normal morphology. B) GFP expression in the embryo shown in A. C) DIC image of two morphologically normal blastula stage embryos. D) One embryo from C emits a red fluorescence from the introduction of Texas Red dextran. The other embryo is un-injected and exhibits no fluorescence. Scale bars denote 50 μm. Scale bar in A represents the scale for A and B. Scale bar in C represents the scale for C and D.
Figure 4. Reagents Successfully Introduced by Microinjection. A) DIC of a blastula stage embryo injected with a GFP reporter plasmid. The embryo has normal morphology. B) GFP expression in the embryo shown in A. C) DIC image of two morphologically normal blastula stage embryos. D) One embryo from C emits a red fluorescence from the introduction of Texas Red dextran. The other embryo is un-injected and exhibits no fluorescence. Scale bars denote 50 μm. Scale bar in A represents the scale for A and B. Scale bar in C represents the scale for C and D.
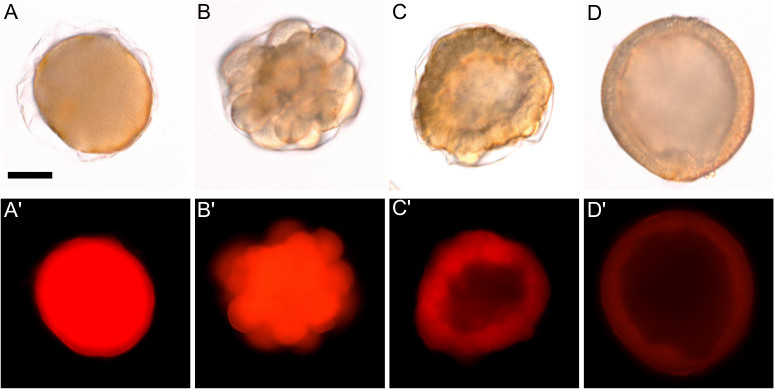 Figure 5. Overinjection and reagent toxicity result in arrested or aberrant development. All images are of embryos 24 hr post-injection from the same injection batch. A-A’) An overinjected zygote arrested at the one-cell stage. B-B’) An embryo arrested in early cleavage. A healthy embryo will divide symmetrically, while this embryo arrested due to abnormal cell divisions. C-C’) An embryo with wildly aberrant development due to toxicity. While this embryo is shaped roughly like a blastula embryo (D), it is asymmetrical and abnormally thickened. D-D’) A properly developing injected blastula stage embryo. Scale bar in A denotes 50 μm and represents the scale for all panels.
Figure 5. Overinjection and reagent toxicity result in arrested or aberrant development. All images are of embryos 24 hr post-injection from the same injection batch. A-A’) An overinjected zygote arrested at the one-cell stage. B-B’) An embryo arrested in early cleavage. A healthy embryo will divide symmetrically, while this embryo arrested due to abnormal cell divisions. C-C’) An embryo with wildly aberrant development due to toxicity. While this embryo is shaped roughly like a blastula embryo (D), it is asymmetrical and abnormally thickened. D-D’) A properly developing injected blastula stage embryo. Scale bar in A denotes 50 μm and represents the scale for all panels.
Discussion
There are two critical steps that are difficult for novice users of this technique but are essential for successfully creating morphant embryos. The first is selecting healthy oocytes that will mature and fertilize properly. The percentage of normal development in a culture depends on the season, the health of the animal, and the number of times that oocytes have been harvested from a single individual. Oocytes tend to be of better quality from April through October. It is important to look carefully at the oocytes before proceeding to ensure that the majority look like Figure 2A rather than Figure 2B. It is permissible to have a small number of oocytes that will not mature, particularly if they are small enough to filter out as the oocytes are processed. Occasionally, oocytes that are undeveloped are almost as large as fully developed oocytes that are ready for maturation. They are distinguishable by their grainy, brown coloring (Figure 2E). Do not use batches of oocytes of this type because filtration will not isolate fully developed oocytes from undeveloped oocytes and they often result in abnormal embryo cultures that are more likely to exhibit the defects seen in Figure 5.
The other critical step is injection bolus size. Introduce an amount of perturbing reagent that is sufficiently large to exert an effect, but not so large as to cause non-specific effects. The first time a perturbing reagent is used, it is helpful to perform a titration in which several concentrations of reagent are tried. Perform subsequent microinjections with the highest concentration that does not cause developmental abnormalities that are unrelated to the expected phenotype, or delay development (Figure 5A). Avoid injecting a bolus that is greater than 30% the diameter of the embryo as this may disrupt normal development. Overly small bolus sizes, by contrast, are difficult to visually inspect for consistent sizing which can lead to an excessive range of perturbation phenotypes across replicates.
Perturbing reagents may cause developmental delays or non-specific phenotypes at effective concentration ranges. It is very important, therefore, to inject sibling controls with a benign reagent such as a standard control MASO, standard DNA constructs, or rhodamine tracer. Assay morphant embryos only if these controls show normal development. Injected embryos, including controls, sometimes exhibit a wrinkled blastula phenotype but they will frequently proceed through the rest of development normally. Additionally, if a particular reagent consistently causes developmental delays in the resulting morphant embryos, compare these embryos to control embryos to determine how many hours of delay they are experiencing.
There are a few attributes of this method that may limit its usefulness in specific experimental scenarios. One such issue is the fact that one can only introduce reagents at fertilized egg or early cleavage stages. This is particularly problematic if the gene or pathway of interest is especially crucial in early development and resultant morphant embryos are inviable or too developmentally abnormal for meaningful study. Sometimes this issue is resolved by injecting fewer copies of the perturbing reagent, resulting in a gentler, incomplete knockdown. If an appropriate cell-permeable drug for the target gene or pathway exists, it is helpful to compare the phenotype of embryos injected with reagents at the fertilized egg stage with embryos treated with the drug at a later, more appropriate stage. Because injection of MASOs or mRNAs offers greater specificity, it is more powerful to use this method in conjunction with a drug-treatment study as opposed to abandoning microinjection in favor of drug treatment.
Finally, although this microinjection method will produce a sufficient amount of embryos for a wide variety of downstream applications, it will at best produce several hundred to one thousand healthy, successfully injected embryos. Some desired experiments may require considerably more material than this. Other methods successfully used in the sea urchin system may be modified to create even larger numbers of morphant sea star embryos, such as pantropic retroviruses20, although this method is novel and has not yet gained widespread use.
Disclosures
No conflicts of interest declared by the authors.
Acknowledgments
This work was supported by the National Science Foundation IOS 0844948 and IOS 1024811
References
- Terasaki M. Quantification of fluorescence in thick specimens, with an application to cyclin B-GFP expression in starfish oocytes. Biol. Cell Auspices Eur. Cell Biol. Organ. 2006;98:245–252. doi: 10.1042/BC20050040. [DOI] [PubMed] [Google Scholar]
- Terasaki M, Runft L. Two-stage dependence for 1-methyladenine induced reinitiation of meiotic maturation in starfish oocytes. Exp. Cell Res. 2010;316:2654–2663. doi: 10.1016/j.yexcr.2010.05.031. [DOI] [PubMed] [Google Scholar]
- O'Neill FJ, Gillett J, Foltz KR. Distinct roles for multiple Src family kinases at fertilization. J. Cell Sci. 2004;117:6227–6238. doi: 10.1242/jcs.01547. [DOI] [PubMed] [Google Scholar]
- McCauley BS, Akyar E, Filliger L, Hinman VF. Expression of wnt and frizzled genes during early sea star development. Gene Expr. Patterns GEP. 2013;13:437–444. doi: 10.1016/j.gep.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Yankura KA, Koechlein CS, Cryan AF, Cheatle A, Hinman VF. Gene regulatory network for neurogenesis in a sea star embryo connects broad neural specification and localized patterning. Proc. Natl. Acad. Sci. U. S. A. 2013;110:8591–8596. doi: 10.1073/pnas.1220903110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley BS, Wright EP, Exner C, Kitazawa C, Hinman VF. Development of an embryonic skeletogenic mesenchyme lineage in a sea cucumber reveals the trajectory of change for the evolution of novel structures in echinoderms. EvoDevo. 2012;3:17. doi: 10.1186/2041-9139-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziata R, Martinez P, Arnone MI. Intact cluster and chordate-like expression of ParaHox genes in a sea star. BMC Biol. 2013;11:68. doi: 10.1186/1741-7007-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morino Y, et al. Heterochronic activation of VEGF signaling and the evolution of the skeleton in echinoderm pluteus larvae. Evol. Dev. 2012;14:428–436. doi: 10.1111/j.1525-142X.2012.00563.x. [DOI] [PubMed] [Google Scholar]
- Hart MW. Structure and evolution of the sea star egg receptor for sperm bindin. Mol. Ecol. 2013;22:2143–2156. doi: 10.1111/mec.12251. [DOI] [PubMed] [Google Scholar]
- McGovern TM, Keever CC, Saski CA, Hart MW, Marko PB. Divergence genetics analysis reveals historical population genetic processes leading to contrasting phylogeographic patterns in co-distributed species. Mol. Ecol. 2010;19:5043–5060. doi: 10.1111/j.1365-294X.2010.04854.x. [DOI] [PubMed] [Google Scholar]
- Keever CC, et al. Discordant distribution of populations and genetic variation in a sea star with high dispersal potential. Evol. Int. J. Org. Evol. 2009;63:3214–3227. doi: 10.1111/j.1558-5646.2009.00801.x. [DOI] [PubMed] [Google Scholar]
- Lambert P. Sea Stars of British Columbia, Southeast Alaska, and Puget Sound. UBC Press; 2000. [Google Scholar]
- Dorée M, Guerrier P, Leonard NJ. Hormonal control of meiosis: specificity of the 1-methyladenine receptors in starfish oocytes. Proc. Natl. Acad. Sci. U. S. A. 1976;73:1669–1673. doi: 10.1073/pnas.73.5.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman VF, Nguyen AT, Cameron RA, Davidson EH. Developmental gene regulatory network architecture across 500 million years of echinoderm evolution. Proc. Natl. Acad. Sci. 2003;100:13356–13361. doi: 10.1073/pnas.2235868100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman VF, Nguyen A, Davidson EH. Caught in the evolutionary act: precise cis-regulatory basis of difference in the organization of gene networks of sea stars and sea urchins. Dev. Biol. 2007;312:584–595. doi: 10.1016/j.ydbio.2007.09.006. [DOI] [PubMed] [Google Scholar]
- McCauley BS, Weideman EP, Hinman VF. A conserved gene regulatory network subcircuit drives different developmental fates in the vegetal pole of highly divergent echinoderm embryos. Dev. Biol. 2010;340:200–208. doi: 10.1016/j.ydbio.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Wessel GM, Voronina E, Brooks JM. Obtaining and handling echinoderm oocytes. Methods Cell Biol. 2004;74:87–114. doi: 10.1016/s0091-679x(04)74005-4. [DOI] [PubMed] [Google Scholar]
- Sweet H, et al. Blastomere isolation and transplantation. Methods Cell Biol. 2004;74:243–271. doi: 10.1016/s0091-679x(04)74011-x. [DOI] [PubMed] [Google Scholar]
- Cheers MS, Ettensohn CA. Rapid microinjection of fertilized eggs. Methods Cell Biol. 2004;74:287–310. doi: 10.1016/s0091-679x(04)74013-3. [DOI] [PubMed] [Google Scholar]
- Core AB, Reyna AE, Conaway EA, Bradham CA. Pantropic retroviruses as a transduction tool for sea urchin embryos. Proc. Natl. Acad. Sci. U. S. A. 2012;109:5334–5339. doi: 10.1073/pnas.1117846109. [DOI] [PMC free article] [PubMed] [Google Scholar]