

Abstract
Protein arginine methylation is one of the most abundant post-translational modifications in the nucleus. Protein arginine methylation can be identified and/or determined via proteomic approaches, and/or immunoblotting with methyl-arginine specific antibodies. However, these techniques sometimes can be misleading and often provide false positive results. Most importantly, these techniques cannot provide direct evidence in support of the PRMT substrate specificity. In vitro methylation assays, on the other hand, are useful biochemical assays, which are sensitive, and consistently reveal if the identified proteins are indeed PRMT substrates. A typical in vitro methylation assay includes purified, active PRMTs, purified substrate and a radioisotope labeled methyl donor (S-adenosyl-L-[methyl-3H] methionine). Here we describe a step-by-step protocol to isolate catalytically active PRMT1, a ubiquitously expressed PRMT family member. The methyl transferase activities of the purified PRMT1 were later tested on Ras-GTPase activating protein binding protein 1 (G3BP1), a known PRMT substrate, in the presence of S-adenosyl-L-[methyl-3H] methionine as the methyl donor. This protocol can be employed not only for establishing the methylation status of novel physiological PRMT1 substrates, but also for understanding the basic mechanism of protein arginine methylation.
Keywords: Genetics, Issue 92, PRMT, protein methylation, SAMe, arginine, methylated proteins, methylation assay
Introduction
Protein methylation was first described in 19681. It was not until the first cloning of PRMT1 in 1996, that researchers began to appreciate the importance of this post-translational modification2. Interestingly, about 2% of arginine residues in the proteins of nuclear extracts are methylated3, indicating the abundance of this modification. Arginine is a positively charged amino acid with a basic side chain and the nitrogen/s within the side chains of arginine can be post-translationally modified via the addition of a methyl group, a process known as arginine methylation4-6. Arginine methylation is catalyzed by a class of enzymes viz., protein arginine methyl transferases (PRMTs). Arginines can either be monomethylated or dimethylated and the latter can be either symmetric or asymmetric depending on the type of PRMTs catalyzing the process4-6.
Proteins that display arginine-glycine rich motifs are potential targets for PRMT-mediated catalysis. PRMT-mediated methylation of substrates has been shown to modulate protein-protein interaction, protein-nucleic acid interaction, protein function, gene expression, and/or cellular signaling, all of which are critical for normal cellular homeostasis7-9. In order to understand the biological role of protein arginine methylation, precise, efficient, and reproducible assays are required to establish the methylation status of the identified PRMT substrates.
In vitro methylation assay, which evaluates the abilities of purified PRMTs to catalyze methylation of their substrates, is a well-accepted assay for studying arginine methylation10-12. The overall success of this assay largely depends upon the activity of the purified PRMTs. PRMTs can be expressed and purified from bacteria, or mammalian cells10,11. This in vitro methylation assay, as detailed in the protocol section, is based upon a method originally described by Tini and colleagues10. In this protocol, we show in detail the steps involved in the expression and purification of PRMT1 in mammalian cells. The ability of the purified PRMT1 to catalyze methylation on Ras-GTPase activating protein binding protein 1 (G3BP1), a known PRMT substrate8, was later evaluated in the presence of S-adenosyl-L-[methyl-3H] methionine as the methyl donor. Using this assay, we can reliably define the abilities of the PRMTs to methylate novel or known substrates, which is a primary step in the study of protein arginine methylation.
Protocol
1. Preparation of Expression Constructs
Before starting the protocol, clone PRMTs into mammalian expression vectors with an N-terminal epitope tag (Myc or HA), and the substrate in-frame with glutathione-S-transferase (GST) for high-level protein expression and purification from bacterial cell lysates, using standard molecular biological techniques.
2. Purification of PRMTs
Use 100 mm culture dishes to maintain human bronchial epithelial cells (Beas2B). Passage the cells every 3 days and do not let them grow to confluence.
On the day before transfection, seed 7.5 x 105 cells in 10 ml culture medium in a 100 mm culture dish. Swirl to evenly disperse the cells, and incubate at 37 °C in a 5% humidified CO2 incubator.
The next day, aspirate the media and add 5 ml of culture media without serum and antibiotics. Swirl and aspirate the medium. Repeat this step one more time, and finally add 5 ml of serum/antibiotic free medium and proceed to transfection.
Dilute 6 μg of plasmid DNA into 750 μl of serum free medium in a sterile 1.5 ml plastic centrifuge tube.
In another 1.5 ml plastic centrifuge tube, dilute 30 μl of transfection reagent into 750 μl of serum free medium.
Transfer the diluted transfection reagent solution (step 2.5) to the tube containing diluted DNA (step 2.4). Mix gently by pipetting up and down several times.
Incubate the transfection mixture at room temperature for 15 min.
After 15 min, pipette the transfection mixture gently onto the cells, swirl to mix evenly, and incubate at 37 °C in a 5% humidified CO2 incubator.
After 3 hr, aspirate the medium containing transfection mixtures from the cells. Add fresh cell culture medium supplemented with serum and antibiotics.
Incubate at 37 °C in a 5% humidified CO2 incubator for 48 hr.
After 48 hr, lyse the cells in 1 ml of lysis buffer (1x phosphate buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 1 µg/ml phenlymethyl sulphonyl fluoride). Collect the lysates into a plastic centrifuge tube with a cell lifter and incubate the lysates on ice for 30 min.
Centrifuge the lysates at 15,000 x g for 10 min on a bench-top centrifuge to clear the lysates from cell debris. [* It is highly recommended to confirm the expression of HA-PRMT1 via immunoblotting of an aliquot of the lysates with anti-HA antibodies, before proceeding to purification of the enzymes for methylation assay.]
For purification of PRMTs, incubate 3 mg of cell lysates with 30 μl of 50:50 suspension of anti-HA affinity matrix in phosphate buffered saline (PBS), and rotate overnight at 4 °C in a tube rotator.
Next day, spin the tubes at 14,000 x g on a bench-top centrifuge for 2 min to collect the beads. Discard the supernatant and wash the beads 3x with 1 ml of RIPA buffer [20 mM Tris (pH 8.0), 150 mM NaCl, 5 mM EDTA and 1% Triton-X-100] in each wash followed by centrifugation at 14,000 x g for 2 min. After the third wash, wash the beads once with 100 μl of methylation buffer (50 mM Tris pH 8.5, 20 mM KCl, 10 mM MgCl2, 1 mM β-mercaptoethanol, 100 mM sucrose). After the centrifugation, remove the supernatant. The beads containing the immobilized PRMTs are ready to be used in the methylation assay. [* It is highly recommended to use the immobilized PRMTs immediately in the methylation assay.]
3. Purification of Substrate
Inoculate a single colony of GST-G3BP1 transformed BL21 into 5 ml of Luria-Bertani (LB) medium containing 100 μg/ml ampicillin. Grow the cultures overnight at 37 °C with shaking at 250 rpm.
Next day, transfer the overnight culture to 50 ml of fresh LB medium and continue to grow to an OD600 of 0.5-0.7 (It takes about 2 hr).
When the OD600 reaches 0.5-0.7, add isopropyl-β-D-thiogalactoside (IPTG) to a final concentration of 1 mM and continue to culture for additional 4 hr at 37 °C with shaking at 250 rpm.
Harvest the cells by centrifugation at 1,500 x g for 10 min, and process the pellet for GST purification or store at -80 °C.
Re-suspend the bacterial pellet in 5 ml of lysis buffer (1x PBS with protease inhibitors, 100 μg/ml of lysozyme, and 0.1% Triton-X-100) and incubate the lysates at 37 °C for 30 min. At this time the lysates will appear turbid and viscous due to the release of bacterial genomic DNA.
Shear the genomic DNA by sonication (10 pulses of 1 sec each with an amplitude of 30%).
Clear the lysates by centrifugation at 15,000 x g for 10 min and transfer the clear supernatant to a 15 ml tube. If the supernatant is not clear repeat the centrifugation process one more time.
Meanwhile, wash the GST-sepharose beads twice with ice cold PBS and make a 50:50 suspension of GST-sepharose in PBS. Add 200 μl of the GST-sepharose suspension to the cleared bacterial cell lysate and rotate at 4 °C for 1 hr.
After 1 hr, centrifuge the tubes at 500 x g for 3 min to collect the beads. Wash the beads 3x with ice cold PBS.
After the final wash, resuspend the beads in 100 μl of elution buffer (50 mM Tris pH 8.0, 150 mM NaCl, 30 mM glutathione) and rotate at 4 °C for 10 min.
Centrifuge the tubes at 15,000 x g for 2 min and transfer the supernatant containing the purified protein into another plastic centrifuge tube. Carry out SDS polyacrylamide gel electrophoresis (SDS-PAGE) analysis on 5 μl of the purified protein along with known amounts of bovine serum albumin (BSA) to estimate the concentration of the purified protein.
4. Methylation Assay
NOTE: Perform following steps in a separate area designated for radioisotope work and in compliance with the institution’s protocols on performing experiments with radioactive isotopes.
Add 2 μg of purified GST-substrate and a master mix containing 1x methylation buffer (50 mM Tris pH 8.5, 20 mM KCl, 10 mM MgCl2, 1 mM β-mercaptoethanol, 100 mM sucrose), 1 μCi of S-adenosyl-L-[methyl-3H] methionine, and water to a final volume of 10 μl to the immobilized PRMTs (step 2.13) and incubate the reaction at 30 °C for 1 hr.
After 1 hr, stop the reaction by adding 10 μl of 2x SDS sample buffer. After denaturation of the samples at 95 °C for 10 min, separate the samples on an SDS-PAGE gel.
Later, transfer the separated proteins from the SDS gel onto nitrocellulose membrane by using a semi-dry electrophoretic transfer.
After the transfer, cover the membrane with autofluor (an autoradiographic image intensifier), and gently rock for 2 hr at room temperature. After 2 hr, pour off the autofluor and retain for later use. [*Label the tube as radioactive material]
Quickly air dry the membrane on a filter paper, seal it in a plastic bag and expose the membrane to an X-ray film at -20 °C. Sufficient exposure time ranges between 5-30 days.
5. Confirmation of PRMT Expression and Equal Loading of Substrate
After achieving sufficient exposures, transfer the membrane to a plastic box containing blocking buffer [Tris-buffered saline with Tween-20 (TBST + 3% non-fat milk)] and gently rock for 1 hr.
Discard the blocking buffer as radioactive waste. Incubate the membrane with anti-HA antibody (1:1,000 dilution) in TBST overnight at 4°C, with gentle rocking.
Wash the membrane with TBST 3x and incubate the membrane with HRP-labeled anti-mouse secondary antibody (1:5,000 dilution) in TBST at room temperature for 1 hr with gentle rocking.
After 1 hr, wash the membrane with TBST 3x, and perform chemiluminiscence detection of proteins with an HRP substrate.
After detection of the HA-tagged PRMTs, incubate the membrane with Ponseau-S staining solution for 5 min at room temperature. Quickly wash the membrane 3x with water to detect the GST-tagged substrates. Scan the image.
Representative Results
The abilities of HA-PRMT1 to methylate GST-G3BP1 were determined by an in vitro methylation assay. HA-tagged PRMT1 purified from Beas2B cell lysates was employed in a methylation reaction containing GST-G3BP1, and 1 μCi of S-adenosyl-L-[methyl-3H] methionine, as a methyl donor. PRMT1 could efficiently catalyze the methylation of GST-G3BP1 (Figure 1, upper panel). Methylation reactions using pull-downs performed on lysates of empty vector transfected Beas2B cells served as a negative control (Figure 1, upper panel, lane 1). Immunoblotting with anti-HA antibodies confirmed the expression of PRMT1 (Figure 1, middle panel). Ponseau-S staining was used to demonstrate equal loading of GST-G3BP1 in both the reactions (Figure 1, lower panel).
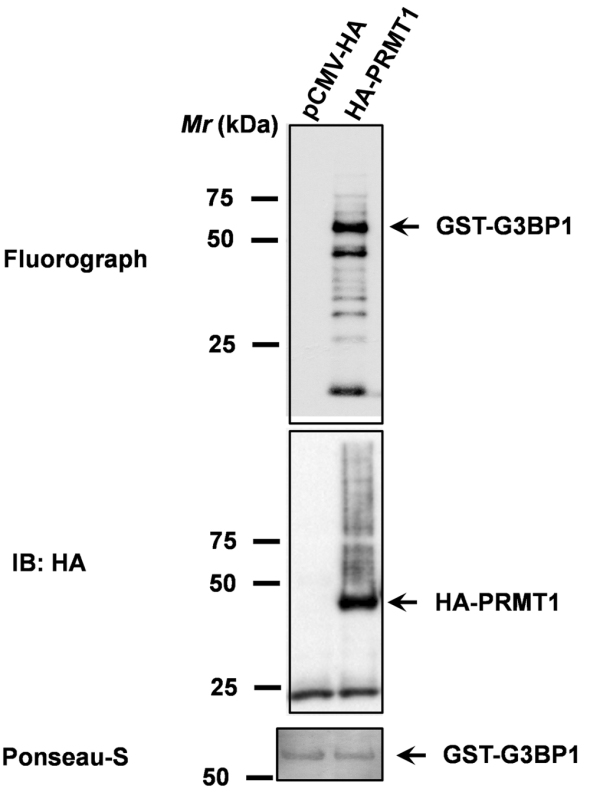 Figure 1. PRMT1 methylates G3BP1. PRMT1-mediated G3BP1 methylation was evaluated by using an in vitro methylation reaction that includes HA-tagged PRMT1 purified from Beas2B cell lysates, GST-G3BP1 purified from E. coli, and S-adenosyl-L-[methyl-3H] methionine, as a methyl donor. Upper panel represents the fluorograph for G3BP1 methylation. Middle panel represents the immunoblot confirming the expression of HA-tagged PRMT1. And, the lower panel represents Ponseau-S stained blot for determining equal loading of the substrate (GST-G3BP1).
Figure 1. PRMT1 methylates G3BP1. PRMT1-mediated G3BP1 methylation was evaluated by using an in vitro methylation reaction that includes HA-tagged PRMT1 purified from Beas2B cell lysates, GST-G3BP1 purified from E. coli, and S-adenosyl-L-[methyl-3H] methionine, as a methyl donor. Upper panel represents the fluorograph for G3BP1 methylation. Middle panel represents the immunoblot confirming the expression of HA-tagged PRMT1. And, the lower panel represents Ponseau-S stained blot for determining equal loading of the substrate (GST-G3BP1).
Discussion
The protocol described herein is routinely used to establish the methylation status of the identified PRMT substrates. This robust, and consistent assay will also provide definitive evidence on the specificity of PRMTs for their substrates. The key components for the success of this assay are: 1. Expression of PRMTs in mammalian cells, 2. Activity of purified PRMTs, 3. Expression and purification of substrate, and 4. Complete western transfer of the proteins to the nitrocellulose membrane.
Recombinant PRMTs expressed, and purified from bacteria can also be used in in vitro methylation reactions10,11,13. However, since each PRMT requires different purification strategies, storage conditions, and storage temperatures, the use of recombinant PRMTs in in vitro methylation reactions may not be ideal. Moreover, recombinant PRMTs are also short-lived11. Considering these challenges, we believe that purification of PRMTs from mammalian cells is convenient, and consistent to perform in vitro methylation reactions. If transient expression of PRMTs in mammalian cells appears to be inconvenient, alternatively, one can consider making stable cell lines for PRMTs. Most importantly, expression of PRMTs in mammalian cells is the only way available to determine PRMT catalytic activities in response to stimuli e.g., growth factors, cytokines, etc.
The major limitation of using PRMTs purified from mammalian sources is the contamination with other PRMTs during the purification process. However, one could use mutant constructs of the PRMTs that are devoid of methyl transferase activities as additional controls. If the PRMTs under investigation can form homodimers, mutant constructs might not be good controls. Alternatively, one can consider using PRMTs purified from cells depleted of target PRMTs as a negative control or use recombinant PRMTs from bacterial sources. The presented protocol describes a simple, convenient, and consistent assay to probe protein arginine methylation. The described method is not only essential for the identification and establishment of the methylation status of novel PRMT substrates, but also for our basic understanding of the mechanism of protein arginine methylation.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This study was supported by a Merit Award from the U.S. Department of Veterans Affairs, and an NIH grant R01CA138528 to RW.
References
- Paik WK, Kim S. Protein methylase I. Purification and properties of the enzyme. J Biol Chem. 1968;243:2108–2114. [PubMed] [Google Scholar]
- Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- Boffa LC, Karn J, Vidali G, Allfrey VG. Distribution of NG, NG,-dimethylarginine in nuclear protein fractions. Biochem Biophys Res Commun. 1977;74:969–976. doi: 10.1016/0006-291x(77)91613-8. [DOI] [PubMed] [Google Scholar]
- Bedford MT. Arginine methylation at a glance. J Cell Sci. 2007;120:4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- Lee YH, Stallcup MR. Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol Endocrinol. 2009;23:425–433. doi: 10.1210/me.2008-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikkavilli RK, et al. Dishevelled3 is a novel arginine methyl transferase substrate. Scientific reports. 2012;2:805. doi: 10.1038/srep00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikkavilli RK, Malbon CC. Arginine methylation of G3BP1 in response to Wnt3a regulates {beta}-catenin mRNA. J Cell Sci. 2011;124:2310–2320. doi: 10.1242/jcs.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikkavilli RK, Malbon CC. Wnt3a-stimulated LRP6 phosphorylation is dependent upon arginine methylation of G3BP2. J Cell Sci. 2012. [DOI] [PMC free article] [PubMed]
- Tini M, Naeem H, Torchia J. Biochemical analysis of arginine methylation in transcription. Methods Mol Biol. 2009;523:235–247. doi: 10.1007/978-1-59745-190-1_16. [DOI] [PubMed] [Google Scholar]
- Lee J, Cheng D, Bedford MT. Techniques in protein methylation. Methods Mol Biol. 2004;284:195–208. doi: 10.1385/1-59259-816-1:195. [DOI] [PubMed] [Google Scholar]
- Cheng D, Vemulapalli V, Bedford MT. Methods applied to the study of protein arginine methylation. Methods Enzymol. 2012;512:71–92. doi: 10.1016/B978-0-12-391940-3.00004-4. [DOI] [PubMed] [Google Scholar]
- Zurita-Lopez CI, Sandberg T, Kelly R, Clarke SG. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J Biol Chem. 2012;287:7859–7870. doi: 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]