

Abstract
The ability to migrate is a hallmark of various cell types and plays a crucial role in several physiological processes, including embryonic development, wound healing, and immune responses. However, cell migration is also a key mechanism in cancer enabling these cancer cells to detach from the primary tumor to start metastatic spreading. Within the past years various cell migration assays have been developed to analyze the migratory behavior of different cell types. Because the locomotory behavior of cells markedly differs between a two-dimensional (2D) and three-dimensional (3D) environment it can be assumed that the analysis of the migration of cells that are embedded within a 3D environment would yield in more significant cell migration data. The advantage of the described 3D collagen matrix migration assay is that cells are embedded within a physiological 3D network of collagen fibers representing the major component of the extracellular matrix. Due to time-lapse video microscopy real cell migration is measured allowing the determination of several migration parameters as well as their alterations in response to pro-migratory factors or inhibitors. Various cell types could be analyzed using this technique, including lymphocytes/leukocytes, stem cells, and tumor cells. Likewise, also cell clusters or spheroids could be embedded within the collagen matrix concomitant with analysis of the emigration of single cells from the cell cluster/ spheroid into the collagen lattice. We conclude that the 3D collagen matrix migration assay is a versatile method to analyze the migration of cells within a physiological-like 3D environment.
Keywords: Bioengineering, Issue 92, cell migration, 3D collagen matrix, cell tracking
Introduction
Like cell fusion (for an overview see 1,2) cell migration is another biological phenomenon that is involved in a plethora of physiological processes including embryonic development, wound healing and immune responses (for review see 3). However, the ability to migrate is also a prerequisite for tumor cells to metastasize (for review see 3,4).
Cell migration is a complex, not yet fully understood process that is directed by the interplay of several signal transduction pathways initiated by various ligand (e.g., cytokines, chemokines, growth factors, hormones, extracellular matrix components) receptor (e.g., receptor tyrosine kinases, chemokine receptors, integrins) interactions5 ultimately causing the reorganization of the actin cytoskeleton concomitant with de- and reassembly of focal adhesion complexes and integrin-mediated signaling6.
To analyze cell migration several in vitro and in vivo cell migration assays have been developed in the past decades, including the Boyden chamber/transwell assay7, scratch assay/wound healing assay8-10, three-dimensional (3D) collagen matrix migration assay11 as well as intravital imaging/microscopy (for review see 12). Each of these cell migration assays has pros and cons, e.g., concerning costs and need of equipment, handling or reliability of obtained data.
Both the Boyden chamber/transwell assay and the scratch assay/wound healing assay are easy, low-cost and well-developed assays to measure cell migration in vitro7-10. In the Boyden chamber/transwell assay cells are seeded on top of an insert containing pores (about 8 µm in diameter) - the so-called upper compartment7. Optional, the insert could be coated with extracellular matrix components, e.g., fibronectin, collagen, etc., to mimic a more physiological environment. Likewise, endothelial cells could be grown on top of the insert, thereby mimicking an endothelial cell barrier13. Those cells that have passed through the pores during a defined time interval into the lower compartment harboring media and supplements, such as growth factors and chemokines, are used as a read-out to quantify cell migration (or extravasation).
In the scratch assay/wound healing assay cells are seeded in plates and are grown to confluency10. In dependence of the experimental setting plates could be pre-coated with extracellular matrix components, such as fibronectin. After creating a scratch/wound by scraping the cell monolayer single cells from each side of the scratch/wound can migrate into the gap, thereby filling/healing it10. The distance between the two sides of the scratch/wounds is determined in dependence of time and is used as a read-out for the migratory activity of the cells10. However, to discriminate between cell proliferation (which could also result in filling/healing of the scratch/wound) and cell migration it is recommended to combine the assay with time-lapse video microscopy and single cell tracking10.
However, both the Boyden chamber/transwell assay and scratch assay/wound healing assay, are rather imperfect concerning a physiological-like cellular environment. In the Boyden chamber/transwell assay cells have to migrate through a plastic pore, whereas in the scratch assay/wound healing assay cells are seeded on a two-dimensional pre-coated plastic plate. Likewise, it is well recognized that the migratory behavior differs markedly between a two-dimensional and 3D environment3. For instance, three-dimensional-matrix adhesions of fibroblasts differ from focal and fibrillar adhesions characterized on two-dimensional substrates in their content of α5β1 and αvβ3 integrins, paxillin, other cytoskeletal components, and tyrosine phosphorylation of focal adhesion kinase14. Likewise, cells embedded within a 3D environment also displayed an altered migratory behavior15. Thus to analyze cell migration more accurately a migration assay is recommended allowing to measure the migration of single cells within a 3D physiological or physiological-like environment.
Intravital imaging/microscopy is the gold-standard for measuring cell migration within a 3D physiological context. This does not only belong to extracellular matrix-cell interactions, but also to the interactions among different cell types, such as tumor cells and endothelial cells during extravasation16 or lymphocyte trafficking within the lymph node17, which, to date, is possible due to improved fluorescence microscopy techniques, such as 2-photon confocal laser scanning microscopy, the use of vital fluorescent dyes and transgenic mouse strains expressing fluorescent proteins derivatives12,16,17. Additionally, intravital imaging/microscopy could be combined with manual and automated cell tracking18. However, because of the need of a 2-photon confocal laser scanning microscopy as well as animals (and appropriate transgenic animal models) intravital imaging/microscopy is a rather cost-intensive technique.
To overcome the limitations of the Boyden chamber/transwell assay and the scratch assay/wound healing assay and to analyze the migration of different cell types within a 3D environment the 3D collagen matrix migration assay was developed11,19. Thereby, migrating cells are embedded within a 3D collagen fiber network, which more resembles to the in vivo situation. Conjointly, due to time-lapse video microscopy real cell migration is measured allowing the determination of several migration parameters as well as their alterations in response to pro-migratory factors or inhibitors. Various cell types could be analyzed using this technique, including lymphocytes and leukocytes11,20, hematopoietic stem/progenitor cells21-24, and tumor cells5,25-29. In addition to single cells also cell clusters or spheroids could be embedded within the collagen matrix concomitant with analysis of the emigration of single cells from the cell cluster/ spheroid into the collagen lattice30,31.
This protocol presents an overview about a simple, but powerful technique to analyze the migratory behavior of different cell types within a 3D environment – an in vitro method yielding in results that are close to the in vivo situation.
Protocol
1. Preparation of Migration Chambers
Prepare a paraffin wax/petroleum jelly (1:1) mix and heat until the mixture has melted. Using a paint-brush and draw 2-3 layers of the paraffin wax/petroleum jelly (1:1) mix in the middle of the glass slide in accordance to Figures 1B-1D. NOTE: We are using common glass slides (76 x 26 x 1.0-1.5 mm (W/D/H))
Apply the melted paraffin wax/petroleum jelly mix rapidly on the glass slide to avoid solidification while drawing. Ensure the paraffin wax/petroleum jelly layer is about 2-2.5 cm in length and 0.3-0.5 cm in width. The thickness of the paraffin wax/petroleum jelly layer should be about 0.1-0.15 cm.
Add a coverslip to the solidified paraffin wax/petroleum jelly mix and seal it with two to three layers paraffin wax/petroleum jelly mix (Figures 1E,1F). Use 4-8 migration chambers for a common cell migration experiment. Place migration chambers in an upright position in a rack.
2. Preparation of the Collagen Suspension Cell Mix
Harvest (e.g., with 0.25% Trypsin/EDTA) and count cells of interest. Resuspend the cells in complete media containing fetal calf serum (FCS), antibiotics and recommended supplements. Prepare 4-8 1.5 ml reaction tubes and 20 µl cell suspension containing 4-6 x 104 cells (the total number of cells depends on the cell type to be analyzed) and fill up to 50 µl with complete media. NOTE: Additional compounds (e.g., growth factors, chemokines, inhibitors, etc.) are added to the cell suspension. Use appropriate stock solutions such that the final volume of cell suspension does not exceed 50 µl.
Prepare a total amount of 452 µl final collagen suspension for four cell migration experiments. Add 50 µl 10x MEM (pH 5.1-5.5) and 27 µl of 7.5% sodium bicarbonate solution (pH 9.0-9.5) to a 1.5 ml reaction tube and mix thoroughly. Observe the solution color turn from yellow-orange to intense purple (pH 9.0-9.5). Add 375 µl liquid collagen suspension to the 10x MEM/sodium bicarbonate solution and mix thoroughly. The pH of the final collagen suspension should be about 7.5 (indicated by a light purple color). NOTE: Check the pH of the 7.5% sodium bicarbonate solution. If the pH is about 7.5 place the sodium bicarbonate solution with an open lid in an incubator (37 °C, 5% CO2) to be saturated with CO2 and increase the pH (about 9.0 - 9.5). The pH of collagen suspension cell mix is crucial for the stability of the collagen network. If it is too low the collagen lattice will collapse during the cell migration experiment. NOTE: For this protocol, use liquid collagen (pH 1.9-2.2) from bovine hind (2.9-3.3 mg/ml collagen; 95% collagen type I, 5% collagen type IV). Examples of further collagen lattice preparation protocols using collagen type I from other sources, such as porcine or rat, are given in 32,33. Do not change the volumes of the used solutions as this will alter the ultimate collagen concentration of 1.67 mg/ml of the lattice. A higher or lower collagen concentration has an impact on the density of collagen fibers and the average distance between them concomitant with the cells migratory behavior34.
Add 100 µl of the final collagen suspension to 50 µl cell suspension and mix thoroughly.The final collagen suspension (1.67 mg/ml collagen) is slightly viscous. To transfer the correct amount (100 µl), pipette the final collagen solution once up and down before transferring it to the cell suspension. NOTE: Once the collagen suspension cell mix is combined with the 10x MEM/sodium bicarbonate solution and the pH value has changed to 7.5 the collagen fibers immediately start to polymerize.
Transfer the final collagen suspension cell mix from the reaction tubes to the migration chambers. Gently tap the migration chamber to equally distribute the collagen suspension cell mix on the bottom of the migration chamber (Figure 1H). Place the migration chambers in an upright position in a rack and incubate for about 30 min (37 °C, 5% CO2) to allow polymerization of the collagen fibers.
Notice that the polymerized collagen lattice is slightly turbid but still being light purple color. Fill up the migration chambers with complete medium or complete medium with supplements of interest and seal the migration chamber with paraffin wax/petroleum jelly mix (Figures 1I,1J).
3. Recording and Analysis of Cell Migration
This section describes the recording of cell migration by time-lapse video microscopy and analysis of cell migration by manual cell tracking.
Switch on the microscope and the microscope stage heater. Adjust the temperature to 37 °C. Use a 10X objective. Place migration chamber under a microscope and focus approximately 50 cells or more in the field of view.
Record cell migration in time-lapse mode using a multi-camera video surveillance software application. Save cell migration movies in an appropriate format, e.g., “*.avi” or “*.mov”. Link the cell migration movie files to a database containing supporting information, such as cell type and experimental conditions. NOTE: We are recording lymphocytes and HSPCs for up to 2 hr using a time-lapse factor of 1:80, which means that 0.75 sec in time-lapse is equal to 1 min in real-time. Tumor cells are slow moving cells and are thus recorded for at least 16 hr using a time-lapse factor of 1:1,800 (0.5 sec in time-lapse is equal to 15 min in real-time).
Analyze cell migration movies using an appropriate cell tracking software application, like ImageJ software plugins (an overview is given in 18). For an unbiased analysis it is of crucial importance that cells will be randomly selected without the knowledge whether cells are migratory active or not. NOTE: We are using a self-developed software application to manually track the paths of at least 30 cells per experiment by following the moving cells accurately with the mouse cursor (which is positioned to the nucleus). While tracking, the xy-coordinates of the tracked cells are automatically determined in accordance to the used time-lapse mode. For lymphocytes/HSPCs xy-coordinates are determined each 0.75 sec (equal to 1 min realtime), whereas for tumor cells the xy-coordinates are determined each 0.5 sec (equal to 15 min realtime).
Determine a total of 60 xy-coordinates per cell for a typical cell migration experiment. This is equal to 1 hr real-time for lymphocytes/HSPCs and 15 hr real-time for tumor cells. A “,” indicates that a cell has not moved between two time points, whereas a “numerical value” indicates the distance in “pixels” a cell has moved between 2 time points (Figure 2A). NOTE: Manual cell tracking requires experience. Particularly fast migrating cells are difficult to track and each cell type exhibit a distinct migratory behavior that might be triggered by supplemented factors. In case of cell division while tracking, randomly choose one daughter cell for continuing tracking. Cells that migrate out of the sight field or die while tracking are not neglected, but are considered for analysis.
4. Data Analysis
- Analyze cell tracking data by using an appropriate software application, e.g., a spreadsheet program.
- Analyze cell tracking data by copying the cell tracking raw data (Figure 2A), into a self-designed spreadsheet template. Multiply sum of the “numerical values”, representing the total distance a cell has migrated with a correction factor to convert “pixel” into “µm”.
- Determine two cell migration parameters: locomotory activity (which is equivalent to migration rate) and time of active movement (Figures 2C,2D).
- Identify the cells that have migrated lesser than 25 µm (threshold level to reduce the number of false-positive cells) as non-moving cells. Replace “numerical values” of these cells with “,”. NOTE: The parameter “locomotory activity” (or migration rate) represents the percentage of cells of the tracked cell population that have moved between two time points (Figure 2D). A “numerical value” is defined as a moving cell, whereas “,” is defined as a non-moving cell. The parameter “time of active movement” (time active) represents the percentage of the total time a cell has migrated in relation to the time frame of the observation period (Figure 2C). A “numerical value” indicates that a cell has moved, whereas “,” indicates that a cell has not moved. This parameter is further used to determine the number of cells that has not moved.
- Calculate statistical significance and display cell migration data.
- Calculate statistical significance of the mean locomotory activity of the cells (migration rate) using the Mann-Whitney U test. Consider p-value < 0.05 as significant.
- Display the mean locomotory activity of cells as xy-diagram, BoxPlot diagram, or as a bar chart diagram. Display single cell-based data, such as time of active movement or speed, as a bar chart diagram or as a histogram. NOTE: If the chosen spreadsheet software application does not contain a BoxPlot chart or histogram chart tool an online search should be performed for looking for suitable tutorials.
Representative Results
The used 3D-collagen matrix migration assay combined with time-lapse video-microscopy and computer-assisted cell tracking allows for the determination of various cell migration parameters including both population-based parameter (e.g., mean locomotory activity) and single cell-based parameters (e.g., time of active movement, speed, distance migrated). An example of the obtained cell tracking data sets, data processing and data presentation are given in Figure 2. A cell tracking data file resembles a table (Figure 2A). Each column stands for one tracked cell, whereby per cell migration experiment 30 cells are tracked. The rows contains the information whether a cell has moved or not between two time points. The sum of the “numerical values”, representing the distance in “pixel” a cell has moved between two time points, is the total distance this cell has migrated. It is multiplied by a correction factor to convert “pixel” into “µm”. Cells that have migrated lesser than 25 µm are defined as non-moving cells to reduce the number of false-positive cells. The speed (µm/min) of each cell is calculated by dividing “total distance migrated” by “total time (min) of the observation period”.
To characterize the locomotory behavior of cells commonly two parameters are used. Locomotory activity (Figure 2D) represents the mean locomotory activity (or migration rate) of the analyzed cell population, whereas time of active movement (Figure 2C) represents the total time a cell has migrated within the observation period. The latter parameter is further used to determine the number of cells that has not moved. The reason for using two cell migration parameters for analysis is attributed to the fact that the locomotory activity of cells can be triggered by different mechanisms. There are three possibilities how, e.g., a growth factor, can “induce” cell migration: a) in comparison to the control more cells are migrating, b) the number of moving cells in comparison to control has not changed, but growth factor treated cells exhibit an increased time of active movement, which means that these cells will migrate for a longer time, and c) a combination of a) and b).
The diagram in Figure 2B schematically illustrates how the locomotory activity and time of active movement is calculated. In this example the migratory activities of 120 cells (which is equal to four independent experiments) are displayed. Each diamond indicates that a cell has moved between at least two time points (irrespective of the distance the cell has migrated!). The gray lines were added to this diagram to show that the analyzed population of cells consists of moving and non-moving cells. The parameter “locomotory activity” represents the locomotory activity of the analyzed cell population in dependence of each time point (Figure 2D). For instance, a locomotory activity of 10% at t = 5 hr indicates that between t = 4.45 hr and t = 5 hr 10% of the analyzed cells have moved. When displayed as a xy-diagram the parameter locomotory activity indicates the dynamicity of the analyzed cell population. Another possibility to display the locomotory activity of a cell population is the BoxPlot diagram (Figure 2F).
The parameter “time of active movement” (time active) is correlated to the total time a cell has moved during the observation period, whereby a time active of “0” indicates that a cell has not moved (Figure 2C). The time active of the cells can be displayed as a histogram.
The interplay of EGFR/HER2/PLC-γ1 and HER2/HER3/PI3K signaling contributes to a migratory phenotype of EGFR/HER2/HER3 positive breast cancer cells25. PI3K signaling is required for generation of phosphatidyl-3,4,5-trisphosphate (PIP3), which acts as an anchor molecule for PLC-1, thereby recruiting this enzyme to the plasma membrane to be activated by receptor tyrosine kinases35. Stimulation of solely EGFR positive MDA-MB-468-NEO (MDA-NEO) breast cancer cells resulted in long-term PLC-γ1 tyrosine phosphorylation concomitant with sustained IP3 and DAG levels producing sinusoidal calcium oscillations27. By contrast, EGFR/HER2 positive MDA-MB-468-HER2 (MDA-HER2) breast cancer cells displayed baseline transient calcium oscillations after EGF treatment due to short-term PLC-γ1 tyrosine activation and short-term IP3 and DAG turnover27 indicating that HER2 expression was correlated to a differential PLC-γ1 kinetics and signaling.
Analysis of the migratory behavior of MDA-HER2 and MDA-NEO breast cancer cell lines revealed that both cell lines exhibited a similar spontaneous locomotory activity (MDA-HER2: 23.0 - 28.9%, median: 26.7% vs. MDA-NEO: 19.3 - 24.4%, median: 21.5%; Figures 3A-3D). The parameter time active, representing the time of active movement of single cells in relation to the observation period, revealed a slightly higher number of spontaneously moving MDA-HER2 cells (64%; Figure 3E) as compared to MDA-NEO breast cancer cells (53%; Figure 3F). Stimulation with 100 ng/ml EGF resulted in a significantly increased locomotory activity of both cell lines. The migratory activity of EGF treated MDA-HER2 raised to 30.4 - 35.2% (median 33.7%; Figures 3A,3C), which was attributed to both an increased number of moving cells (100 ng/ml EGF: 81% vs. control: 64%) and an increased time of active movement. For instance, the amount of MDA-HER2 cells exhibiting a time active of 40% was increased from 20% (control) to 28% (100 ng/ml EGF). Similar data were obtained for MDA-NEO breast cancer cells, which migratory activity was increased to 25.2 to 35.2 (median 30.4%; Figures 3B,3D) upon EGF stimulation. Conjointly, more MDA-NEO cells migrated in response to EGF stimulation (100 ng/ml EGF: 66% vs. control: 53%) and displayed a shifted time active pattern (Figure 3F). For instance, the amount of cells possessing a time active of 60% was markedly increased to 30% in the presence of EGF as compared to untreated MDA-NEO cells (13%).
Treatment of MDA-HER2 and MDA-NEO breast cancer cells with the PLC-γ1 inhibitor U73122 resulted in a inhibition of both the spontaneous and EGF induced migration of the cells (Figure 3A-D). However, compared to MDA-NEO breast cancer cells the inhibitory effect of U73122 on the spontaneous migration of MDA-HER2 cells was rather moderate, but nevertheless significant (control: 23.0-28.9%, median: 26.7% vs. 2 µM U73122: 17.8-21.5%, median: 20.0%; Figure 3C). Analysis of the parameter time active revealed a slightly increased amount of non-moving cells in the presence of U73122 (control: 36% vs. 2 µM U73122: 48%; Figure 3E). Similarly, U73122 treatment blocked the EGF induced migration of MDA-HER2 breast cancer cells (100 ng/ml EGF: 30.4-35.2%, median 33.7% vs. 100 ng/ml EGF + 2 µM U73122: 19.3-24.1%, median 22.6%; Figure 3C). In accordance to solely U73122 treated MDA-HER2 cells this inhibitory effect was rather attributed to an increased amount of non-moving cells, but not an altered time active pattern (Figure 3C). In fact, a slight shift towards an increased time of active movement was detectable in migrating MDA-HER2 cells being treated with EGF and U73122 (Figure 3C).
By contrast, treatment of MDA-NEO cells with 2 µM U73122 resulted in a markedly decreased locomotory activity of 5.6 to 10.7% (median: 9.3%; Figure 3F) that was primarily attributed to an increased amount of non-moving cells (control: 47% vs. 2 µM U73122: 79%; Figure 3F). Likewise, the EGF induced migration of MDA-NEO breast cancer cells was markedly blocked by U73122 to 1.1 to 7.4% (median: 4.8%), which was attributed to an increased amount of non-moving cells (100 ng/ml EGF: 34% vs. 100ng/ml EGF + 2 µM U73122: 70%) as well as to a decreased time active pattern (Figure 3F).
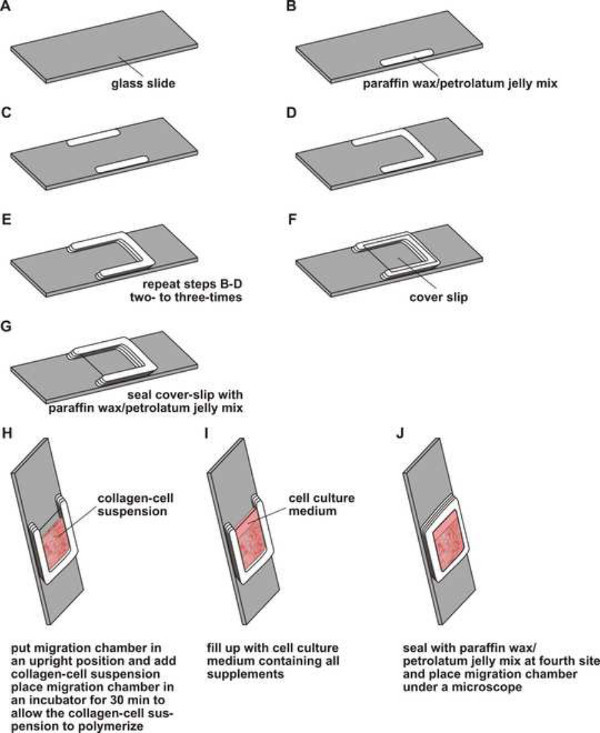 Figure 1. Schematic overview of preparation of cell migration chambers. (A-E) A cell migration chamber is prepared by drawing two to three layers of a melted paraffin wax/petroleum jelly mix onto a glass slide. (F,G) A coverslip is added and sealed with melted paraffin wax/ petroleum jelly mix. (H) The migration chamber is put in an upright position followed by adding the collagen-cell suspension. Subsequently, the migration chamber is placed in an incubator allowing the collagen-cell suspension to polymerize. (I,J) The migration chamber is filled up with media and sealed with melted paraffin wax/petroleum jelly mix at the fourth side. Thereafter, migration chambers are placed under a microscope. Please click here to view a larger version of this figure.
Figure 1. Schematic overview of preparation of cell migration chambers. (A-E) A cell migration chamber is prepared by drawing two to three layers of a melted paraffin wax/petroleum jelly mix onto a glass slide. (F,G) A coverslip is added and sealed with melted paraffin wax/ petroleum jelly mix. (H) The migration chamber is put in an upright position followed by adding the collagen-cell suspension. Subsequently, the migration chamber is placed in an incubator allowing the collagen-cell suspension to polymerize. (I,J) The migration chamber is filled up with media and sealed with melted paraffin wax/petroleum jelly mix at the fourth side. Thereafter, migration chambers are placed under a microscope. Please click here to view a larger version of this figure.
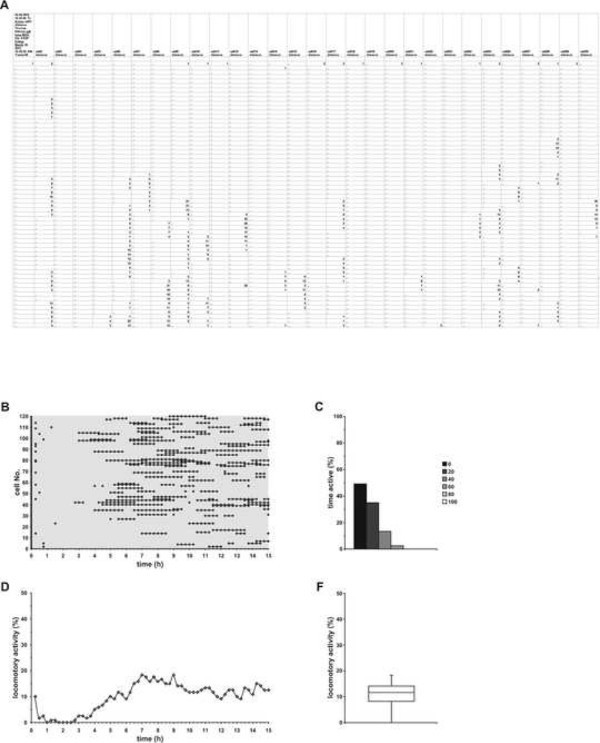 Figure 2. Schematic overview of cell migration data analysis. (A) Cell tracking data file. The migratory activities of 30 cells is determined per experiment, whereby 60 time points are analyzed. A “,” indicates that a cell has not moved between two point, whereas a “numerical value” indicates cell movement. (B) This diagram is a schematic view of data presented in (A). Each diamond indicates that a has moved between two time points; a line indicates longer movement. The gray lines were included to visualize that the cell population consists of non-moving cells and moving cells (which, however, make pauses). (C) Time of active movement (time active) represents the percentage of the total time a cell has migrated in relation to the total time of the observation period. A time active of 20% and a total time of 15 hr indicates that this cell has moved in total for 3 hr. This parameter is further used to determine the number of non-moving cells, which is equivalent to a time active of 0%. (D) Locomotory activity (%) (or migration rate) represents the locomotory activity of the analyzed cell population in dependence of time. It is calculated for each time point by calculating the number of cells that have moved between two time points in relation to the total number of analyzed cells. Displaying the locomotory activity as a xy-diagram reveals the dynamicity of the analyzed cell population. (E) Alternatively, the locomotory activity of the analyzed cells can be displayed as a BoxPlot diagram. Please click here to view a larger version of this figure.
Figure 2. Schematic overview of cell migration data analysis. (A) Cell tracking data file. The migratory activities of 30 cells is determined per experiment, whereby 60 time points are analyzed. A “,” indicates that a cell has not moved between two point, whereas a “numerical value” indicates cell movement. (B) This diagram is a schematic view of data presented in (A). Each diamond indicates that a has moved between two time points; a line indicates longer movement. The gray lines were included to visualize that the cell population consists of non-moving cells and moving cells (which, however, make pauses). (C) Time of active movement (time active) represents the percentage of the total time a cell has migrated in relation to the total time of the observation period. A time active of 20% and a total time of 15 hr indicates that this cell has moved in total for 3 hr. This parameter is further used to determine the number of non-moving cells, which is equivalent to a time active of 0%. (D) Locomotory activity (%) (or migration rate) represents the locomotory activity of the analyzed cell population in dependence of time. It is calculated for each time point by calculating the number of cells that have moved between two time points in relation to the total number of analyzed cells. Displaying the locomotory activity as a xy-diagram reveals the dynamicity of the analyzed cell population. (E) Alternatively, the locomotory activity of the analyzed cells can be displayed as a BoxPlot diagram. Please click here to view a larger version of this figure.
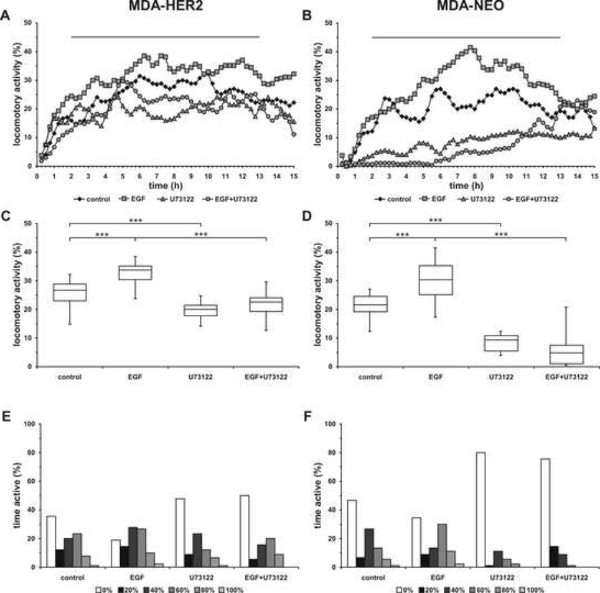 Figure 3. Representative cell migration data of MDA-HER2 and MDA-NEO breast cancer cells. Cells were treated with 100 ng/ml EGF, 2 µM U73122 or with a combination of both. (A,B) Mean locomotory activity of MDA-HER2 (A) and MDA-NEO (B) cells in dependence to time. (C,D) BoxPlot diagram of the mean locomotory activity, representing the 2 hr to 13 hr time interval (see bar in (A,B)) of MDA-HER2 (C) and MDA-NEO (D) cells. Statistical significance was calculated using the Mann-Whitney-U-test. *** = p < 0.001. (E,F) Histogram plots of the time active of MDA-HER2 (E) and MDA-NEO (F) cells. A time active of 0% indicates that cells have not moved during the observation period. Conjointly, a time active of 20% indicates that, considering an observation period of 15 hr, these cells have moved for up to 3 hr. Shown are the mean of at least three independent experiments. Please click here to view a larger version of this figure.
Figure 3. Representative cell migration data of MDA-HER2 and MDA-NEO breast cancer cells. Cells were treated with 100 ng/ml EGF, 2 µM U73122 or with a combination of both. (A,B) Mean locomotory activity of MDA-HER2 (A) and MDA-NEO (B) cells in dependence to time. (C,D) BoxPlot diagram of the mean locomotory activity, representing the 2 hr to 13 hr time interval (see bar in (A,B)) of MDA-HER2 (C) and MDA-NEO (D) cells. Statistical significance was calculated using the Mann-Whitney-U-test. *** = p < 0.001. (E,F) Histogram plots of the time active of MDA-HER2 (E) and MDA-NEO (F) cells. A time active of 0% indicates that cells have not moved during the observation period. Conjointly, a time active of 20% indicates that, considering an observation period of 15 hr, these cells have moved for up to 3 hr. Shown are the mean of at least three independent experiments. Please click here to view a larger version of this figure.
Discussion
The ability to migrate is a hallmark of tumor cells4. Without the ability to detach from the primary tumor and to migrate through the surrounding connective tissue tumor cells won’t be able to seed secondary lesions, which are the main cause of death of nearly all cancer patients. Because of this relationship many studies are focusing on cancer cell migration. The aim of these studies is the identification of novel target molecules and target pathways that efficiently block tumor cell migration, thereby impairing or slowing down metastasis formation. An example for such an effort might be β-blockers, which have been shown to inhibit the metastatic spreading of breast cancer cells in vitro and in vivo36 and which have been associated with an improved relapse-free survival in patients with triple-negative breast cancer cells37. We have recently demonstrated that hybrid cells, which derived from spontaneous fusion events between a human breast epithelial cell line and a human breast cancer cell line, but not the parental cells, responded to the chemokine CCL21 with an increased migratory activity26. CCL21 has been associated with lymph node metastasis formation of various cancer types, including breast38, suggesting that cell fusion could be a process how tumor (hybrid) cells could acquire metastatic properties39,40.
The 3D collagen matrix migration assay has several benefits as compared to the Boyden chamber/transwell assay and the scratch assay/wound healing assay. Firstly, cells are embedded within a 3D physiological environment. As stated out above, the migratory behavior of cells differs markedly between a two-dimensional and 3D environment3,14,15. Thus it can be concluded that the migratory behavior of cells embedded within a 3D collagen lattice resembles more to the in vivo situation than the locomotory activity of cells crawling on a two-dimensional surface. Secondly, due to time-lapse video recording it is possible to analyze the migration of several cells over a certain time frame, which allows the determination of various cell migration parameters. By contrast, in the Boyden chamber/transwell assay only those cells are considered for analysis that have moved to the lower compartment. Those cells that are migrating e.g., on top of the membrane are not included in the data analysis even though they are migratory active.
The 3D collagen matrix can be combined with microfluidic devices, which in addition to mimicking the extracellular matrix do also mimic the interstitial fluid, which has been shown to affect the morphology and migration of various cell types, such as fibroblasts, cancer cells, endothelial cells, and mesenchymal stem cells41. Data of Haessler et al. revealed that interstitial flow increases the percentage of cells that become migratory as well as increases the migrational speed in about 20% of the cells42. In addition to study the chemotactic behavior of cells, e.g., leukocytes/lymphocytes or tumor cells, a 3D microfluidic setting is also a suitable tool to study tumor cell intravasation and endothelial barrier function43.
Even though the 3D collagen matrix migration assay is a suitable tool to analyze the migration of cells in response to a stimulus or to an inhibitor or to a combination of both it has to be noted that this assay do also has some limitations. For instance, only collagen type I is used for mimicking the extracellular matrix, which, however, is a complex mixture of several components including proteoglycans, non-proteoglycans (e.g., hyaluronic acid), collagen fibers, as well as fibronectin and laminin and which composition further varies markedly between different connective tissues in different organs44. Likewise, cells exhibit various integrin molecules recognizing different extracellular matrix components, which may result in the activation of different signal transduction cascades45 concomitant with a differential outcome. For instance, β1-integrin stimulation caused activation of ERK in monocytes while β2-stimulation did not46. Likewise, PI3K was required for β1-integrin-, but not β2-integrin-, mediated NF-κB activation46. On the other hand, collagen type I is the most abundant collagen in the human body47 suggesting that the migration of cells is chiefly triggered by this collagen type.
Another critical point in deciphering the cells migratory activity is the cell tracking procedure. To obtain valid and objective data it is mandatory that the path of single cells is analyzed accurately. Cell tracking is performed manually by hand, which demands cell tracking experience to get objective data. To further improve the data and to avoid a critical number of false positive cells we are using a cut-off level of 25 µm, which means that cells, which have migrated lesser than 25 µm were excluded from data analysis. Likewise, cells are randomly chosen for tracking without the knowing whether they are migratory active or not to avoid biased data.
Within the past years several automated cell tracking software applications have been developed, whereby some of them are suitable to analyze cell and particle tracking in a 3D setting18. The advantage of such applications is that migrating cells are truly analyzed in an objective manner. Because of that it would be interesting to compare automated cell tracking data with manually-made cell tracking data.
As mentioned above, the gold-standard to analyze cell migration within a 3D physiological context is the usage of intravital imaging combined with 2-photon laser-scanning confocal microscopy. The advantage of this assay is that migrating cells are embedded within their physiological environment consisting of specific soluble factors, hormones, etc., extracellular matrix components as well as cell-to-cell interactions. It is remarkable to see movies showing how lymphocytes are trafficking within a lymph node17, how immune cells are communicating with each other48 or how drugs are distributed in vivo at a single cell level12,49.
However, intravital imaging combined with 2-photon laser-scanning confocal microscopy is a rather cost-expensive technique due to the need of an appropriate microscope and appropriate animal models. Likewise, it is not really suitable to study the influence of single soluble factors/inhibitors on the migration of particular cells or the analysis of cell migration related signal transduction cascades. For these purposes cell migration assays are recommended allowing the analysis of discrete cell types, which may be further modified, e.g. overexpression or knockdown of target molecules, in defined experimental conditions. Because of the pivotal relationship between a 3D environment and the locomotory behavior of cells we conclude that the 3D collagen matrix migration assay is thus a versatile method to study the migration of cells in an in vitro setting that is close to the in vivo situation.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by the Fritz-Bender-Foundation, Munich, Germany
References
- Dittmar T, Zänker KS. Cell Fusion in Health and Disease. Vol. 1. Springer; 2011. [DOI] [PubMed] [Google Scholar]
- Dittmar T, Zänker KS. Cell Fusion in Health and Disease. Vol. 2. Springer; 2011. [DOI] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heyder C, et al. Role of the beta1-integrin subunit in the adhesion, extravasation and migration of T24 human bladder carcinoma cells. Clin. Exp. Metastasis. 2005;22:99–106. doi: 10.1007/s10585-005-4335-z. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Boyden S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962;115:453–466. doi: 10.1084/jem.115.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudenschild CC, Schwartz SM. Endothelial regeneration. II. Restitution of endothelial continuity. Lab. Invest. 1979;41:407–418. [PubMed] [Google Scholar]
- Todaro GJ, Lazar GK, Green H. The initiation of cell division in a contact-inhibited mammalian cell line. J. Cell. Physiol. 1965;66:325–333. doi: 10.1002/jcp.1030660310. [DOI] [PubMed] [Google Scholar]
- Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- Friedl P, Noble PB, Zänker KS. Lymphocyte locomotion in three-dimensional collagen gels. Comparison of three quantitative methods for analysing cell trajectories. J. Immunol. Methods. 1993;165:157–165. doi: 10.1016/0022-1759(93)90341-4. [DOI] [PubMed] [Google Scholar]
- Pittet MJ, Weissleder R. Intravital imaging. Cell. 2011;147:983–991. doi: 10.1016/j.cell.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akedo H, Shinkai K, Mukai M, Komatsu K. Establishment of an experimental model for tumor cell invasion and potentiation and inhibition of the invasive capacity. Gan To Kagaku Ryoho. 1987;14:2048–2055. [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Friedl P, Brocker EB. The biology of cell locomotion within three-dimensional extracellular matrix. Cell. Mol. Life Sci. 2000;57:41–64. doi: 10.1007/s000180050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc. Natl. Acad. Sci. U. S. A. 2003;100:2604–2609. doi: 10.1073/pnas.2628040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Shields ED, Noble PB. Methodology for detection of heterogeneity of cell locomotory phenotypes in three-dimensional gels. Exp. Cell Biol. 1987;55:250–256. doi: 10.1159/000163423. [DOI] [PubMed] [Google Scholar]
- Entschladen F, Gunzer M, Scheuffele CM, Niggemann B, Zanker KS. T lymphocytes and neutrophil granulocytes differ in regulatory signaling and migratory dynamics with regard to spontaneous locomotion and chemotaxis. Cell. Immunol. 2000;199:104–114. doi: 10.1006/cimm.1999.1605. [DOI] [PubMed] [Google Scholar]
- Kasenda B, et al. The stromal cell-derived factor-1alpha dependent migration of human cord blood CD34 haematopoietic stem and progenitor cells switches from protein kinase C (PKC)-alpha dependence to PKC-alpha independence upon prolonged culture in the presence of Flt3-ligand and interleukin-6. Br. J. Haematol. 2008;142:831–845. doi: 10.1111/j.1365-2141.2008.07256.x. [DOI] [PubMed] [Google Scholar]
- Kassmer SH, et al. Cytokine combinations differentially influence the SDF-1alpha-dependent migratory activity of cultivated murine hematopoietic stem and progenitor cells. Biol. Chem. 2008;389:863–872. doi: 10.1515/BC.2008.099. [DOI] [PubMed] [Google Scholar]
- Seidel J, et al. The neurotransmitter gamma-aminobutyric-acid (GABA) is a potent inhibitor of the stromal cell-derived factor-1. Stem Cells Dev. 2007;16:827–836. doi: 10.1089/scd.2007.0004. [DOI] [PubMed] [Google Scholar]
- Weidt C, Niggemann B, Hatzmann W, Zanker KS, Dittmar T. Differential effects of culture conditions on the migration pattern of stromal cell-derived factor-stimulated hematopoietic stem cells. Stem Cells. 2004;22:890–896. doi: 10.1634/stemcells.22-6-890. [DOI] [PubMed] [Google Scholar]
- Balz LM, et al. The interplay of HER2/HER3/PI3K and EGFR/HER2/PLC-gamma1 signalling in breast cancer cell migration and dissemination. J. Pathol. 2012;227:234–244. doi: 10.1002/path.3991. [DOI] [PubMed] [Google Scholar]
- Berndt B, et al. Fusion of CCL21 non-migratory active breast epithelial and breast cancer cells give rise to CCL21 migratory active tumor hybrid cell lines. PLoS ONE. 2013;8:e63711. doi: 10.1371/journal.pone.0063711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar T, et al. Induction of cancer cell migration by epidermal growth factor is initiated by specific phosphorylation of tyrosine 1248 of c-erbB-2 receptor via EGFR. FASEB J. 2002;16:1823–1825. doi: 10.1096/fj.02-0096fje. [DOI] [PubMed] [Google Scholar]
- Ozel C, et al. Hybrid cells derived from breast epithelial cell/breast cancer cell fusion events show a differential RAF-AKT crosstalk. Cell Commun Signal. 2012;10 doi: 10.1186/1478-811X-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katterle Y, et al. Antitumour effects of PLC-gamma1-(SH2)2-TAT fusion proteins on EGFR/c-erbB-2-positive breast cancer cells. Br. J. Cancer. 2004;90:230–235. doi: 10.1038/sj.bjc.6601506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, et al. Migration of Coordinated Cell Clusters in Mesenchymal and Epithelial Cancer Explants in Vitro. Cancer Res. 1995;55:4557–4560. [PubMed] [Google Scholar]
- Keeve PL, et al. Characterization and analysis of migration patterns of dentospheres derived from periodontal tissue and the palate. J. Periodontal Res. 2013;48:276–285. doi: 10.1111/jre.12005. [DOI] [PubMed] [Google Scholar]
- Kreger ST, et al. Polymerization and matrix physical properties as important design considerations for soluble collagen formulations. Biopolymers. 2010;93:690–707. doi: 10.1002/bip.21431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan N, Habermehl J, Cote MF, Doillon CJ, Mantovani D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006;1:2753–2758. doi: 10.1038/nprot.2006.430. [DOI] [PubMed] [Google Scholar]
- Friedl P, et al. Migration of Highly Aggressive MV3 Melanoma Cells in 3-Dimensional Collagen Lattices Results in Local matrix Reorganization and Shedding of alpha2 and alpha1 Integrins and CD44. Cancer Res. 1997;57:2061–2070. [PubMed] [Google Scholar]
- Falasca M, et al. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998;17:414–422. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm D, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int. J. Cancer. 2006;118:2744–2749. doi: 10.1002/ijc.21723. [DOI] [PubMed] [Google Scholar]
- Melhem-Bertrandt A, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011;29:2645–2652. doi: 10.1200/JCO.2010.33.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar T, Heyder C, Gloria-Maercker E, Hatzmann W, Zanker KS. Adhesion molecules and chemokines: the navigation system for circulating tumor (stem) cells to metastasize in an organ-specific manner. Clin. Exp. Metastasis. 2008;25:11–32. doi: 10.1007/s10585-007-9095-5. [DOI] [PubMed] [Google Scholar]
- Berndt B, Zanker KS, Dittmar T. Cell Fusion is a Potent Inducer of Aneuploidy and Drug Resistance in Tumor Cell/ Normal Cell Hybrids. Crit. Rev. Oncog. 2013;18:97–113. doi: 10.1615/critrevoncog.v18.i1-2.60. [DOI] [PubMed] [Google Scholar]
- Dittmar T, Nagler C, Niggemann B, Zänker KS. The dark side of stem cells: triggering cancer progression by cell fusion. Curr.Mol. Med. 2013;13:735–750. doi: 10.2174/1566524011313050005. [DOI] [PubMed] [Google Scholar]
- Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. U. S. A. 2011;108:11115–11120. doi: 10.1073/pnas.1103581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haessler U, Teo JC, Foretay D, Renaud P, Swartz MA. Migration dynamics of breast cancer cells in a tunable 3D interstitial flow chamber. Integr. Biol. 2012;4:401–409. doi: 10.1039/c1ib00128k. [DOI] [PubMed] [Google Scholar]
- Zervantonakis IK, et al. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U. S. A. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B, et al. The extracellular matrix of animals. 4th. Garland Science; 2002. [Google Scholar]
- Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J. Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Reyes M, Mora N, Gonzalez G, Rosales C. beta1 and beta2 integrins activate different signalling pathways in monocytes. Biochem. J. 2002;363:273–280. doi: 10.1042/0264-6021:3630273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lullo GA, Sweeney SM, Korkko J, Ala-Kokko L, San Antonio JD. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002;277:4223–4231. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- Mempel TR, et al. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–141. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Reiner T, Earley S, Turetsky A, Weissleder R. Bioorthogonal small-molecule ligands for PARP1 imaging in living cells. Chembiochem. 2010;11:2374–2377. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]