

Abstract
We constructed linear covalently closed (LCC) DNA minivectors as a non-viral gene-delivery vector alternative produced via a simple platform in vivo. DNA ministrings possess a heightened safety profile and also efficiently deliver DNA cargo to targeted cells. Conventional DNA vectors carry undesirable prokaryotic sequences, including antibiotic resistance genes, CpG motifs, and bacterial origins of replication, which may lead to the stimulation of host immunological responses. The bioavailability of conventional DNA vectors is also compromised due to their larger molecular size. Their circular nature may also impart chromosomal integration, leading to insertional mutagenesis.
Bacterial sequences are excised from DNA minivectors, leaving only the gene of interest (GOI) and necessary eukaryotic expression elements. Our LCC DNA minivectors, or DNA ministrings, are devoid of immunogenic bacterial sequences; therefore improving their bioavailability and GOI expression. In the event of vector integration into the chromosome, the LCC DNA ministring will lethally disrupt the host chromosome, thereby removing the potentially dangerous mutant from the proliferating cell population. Consequently, DNA ministrings offer the benefits of 'minicircle' DNA while eliminating the potential for undesirable vector integration events. In comparison to conventional plasmids and their isogenic circular covalently closed (CCC) counterparts, DNA ministrings demonstrate superior bioavailability, transfection efficiency, and cytoplasmic kinetics - they thus require lower amounts of cationic surfactants for effective transfection of target cells.
We have constructed a one-step inducible in vivo system for the production of DNA ministrings in Escherichia coli that is simple to use, rapid, and scalable.
Keywords: Molecular Biology, Issue 108, DNA Ministring, DNA minivector, Plasmid, Tel Protelomerase, In vivo production system, Inducible vector processing, Gene therapy
Introduction
The goal is to produce scalable quantities of linear covalently closed (LCC) DNA minivectors using a simple and high efficiency one-step heat-inducible in vivo DNA ministring production system. DNA ministrings provide a safe and effective non-viral strategy to deliver DNA. They combine the safety of LCC vectors with the efficiency of DNA minicircles, and also offer a safer alternative to virus-derived vectors without compromising transfection efficiency.
In order for a transgene delivery system to be successful, the DNA vector must enter the target host cell and express the encoded transgene(s). There are several cellular barriers that need to be overcome in practice, particularly in mammalian systems. In order to avoid degradation by serum nucleases and immune detection by reticulo-endothelial system, the DNA vector must be bio- and immune-compatible with target system. Unprotected DNA is quickly digested by plasma nucleases and the plasmid membrane is composed of dense lipoprotein barriers so the DNA vector must be capable of rapidly crossing the plasma membrane of target cells. Once in the cell, vectors must traverse the cytoplasm and pass through the nuclear membrane to enter the nucleus for transgene expression. Non-viral gene delivery techniques focus on enhancement of tissue- and cell-targeting. However, the use of conventional plasmids with these techniques reduces transfection efficiency due to the presence of immunogenic bacterial sequences, which rapidly silences gene expression1,2. Conventional plasmids typically carry antibiotic resistance genes for maintenance in prokaryotic systems. However, these may produce potential adverse effects in human hosts or impart resistance to naturally occurring host flora via horizontal gene transfer effects. Conventional plasmids also contain dinucleotide CpG motifs2, which can trigger an unwanted immunostimulatory response, potentially reducing or silencing transgene expression.
As they are solely comprised of the eukaryotic expression cassette, DNA minivectors, such as DNA minicircles3, are a better alternative for gene delivery as they exhibit improved extracellular and intracellular bioavailability and improved gene expression due to their reduced size and absence of immunostimulatory prokaryotic elements4. The reduced vector size fares better with respect to resistance to shear forces associated with in vivo administration to a target site5. The higher copy number of the vector per unit mass requires less transfection reagent, thus decreasing toxicity. However, in the event of random vector integration into a host chromosome, circularly covalently closed (CCC) vectors, including both DNA minicircles and conventional plasmids, impart molecular continuity and therefore may lead to insertional mutagenesis, which can have devastating consequences6. Comparatively, integration of a linear DNA vector disrupts the chromosome and initiates cell death pathways, thereby removing the mutant cell from the proliferating cell population and preventing insertional mutagenesis7. Minimalistic immunologically defined gene expression (MIDGE) vectors8 and micro-linear vectors9 (MiLV) are LCC DNA vectors developed in vitro. MIDGE vectors have exhibited up to a 17-fold improved transgene expression in vivo compared to conventional plasmid DNA vectors11, and have demonstrated promising results in vaccine10 and cancer8 gene therapy. In addition, due to the torsion-free structure of the LCC minivector, less transfection reagent is required in comparison to the CCC supercoiled counterpart, thus reducing toxicity7.
Our enhanced LCC DNA vectors, DNA ministrings, have demonstrated superior expression efficiency and bioavailability7. Furthermore, DNA ministrings are produced on a one-step heat-inducible in vivo production system, an expedient and cost-effective alternative to MIDGE and MiLV LCC vectors, which require multiple steps in vitro. The DNA ministring production system to be demonstrated is a simple heat-inducible process performed in vivo, making it both cost-effective and easily scalable. This system exploits the Yersinia enterocolitica bacteriophage PY54-derived Tel/pal protelomerase recombination system12 to separate the minimal eukaryotic expression cassette from the prokaryotic plasmid backbone. We have engineered Escherichia coli cells (W3NN) to express Tel protelomerase under the control of the heat-inducible bacteriophage λ promoter, cI[Ts]85713. Upon expression, Tel protelomerase acts on pal target sites present within "Super Sequence" (SS) sites located on the precursor plasmid to yield LCC products, from which the DNA ministring can then be purified (Figure 1). The SS sites on the DNA ministring precursor plasmid also encode target sites for other recombinases including Cre recombinase (lox), TelN protelomerase (telRL) and Flp recombinase (FRT), thereby facilitating the production of isogenic LCC and CCC minivectors from one precursor plasmid. In addition, each SS site is flanked on both sides by SV40 enhancer (SV40e) sequences, which serve to improve nuclear translocation14. We have demonstrated elsewhere that successive addition of SV40e progressively confers corresponding increases in transfection efficiency7. The precursor plasmid (pDNA MiniString or pDMS) contains a polylinker (Figure 1) to facilitate insertion of any desired gene of interest in transcriptional fusion with the green fluorescent reporter (GFP).
The DNA minivector technology may be used in place of conventional methods for gene transfer, expression of reporter genes, and assessments towards the efficiency of transgene expression in eukaryotic systems. DNA ministrings combine the biocompatibility and increased transfection efficiency benefits of "mini" vectors with the superior safety profile of linear DNA vectors. Our robust one-step production platform rapidly and easily produces DNA ministrings for any gene transfer application and offers a higher safety profile.
Protocol
1. Preparation of Media and Cultures
For LB + Amp: Prepare 500 ml of Luria-Bertani broth (LB) sterilized using an autoclave at 121 °C for 30 min. Supplement with ampicillin to yield a final concentration of 100 µg/ml of ampicillin and store at 4 °C until required.
For TB + Amp: Prepare for large (500 ml or greater) volumes of ministring production. Prepare 600 ml of sterile Terrific Broth (TB) sterilized using an autoclave at 121 °C for 30 min. Supplement with ampicillin to yield a final concentration of 100 µg/ml of ampicillin and store at 4 °C until required.
For LB agar + Amp: Prepare sterile LB agar plates by preparing LB + Amp supplemented with 15 g/L of agar. Aliquot approximately 15 - 20 ml in sterile Petri dishes and store upside down at 4 °C until required.
Prepare a streak plate of W3NN[pDMS] by aseptically streaking W3NN[pDMS] cells on LB agar + Amp. Incubate the plate overnight at 30 °C, until single colonies can be observed. Do not incubate W3NN above this temperature to avoid derepressing protelomerase expression.
Aseptically transfer 5 ml of LB + Amp into a sterile test tube. Prepare an overnight culture of W3NN[pDMS] by inoculating this volume with a single colony from the streak plate. Incubate the culture overnight at 30 °C in a shaker at 230-250 rpm.
2. Growth Phase
- Aseptically transfer 10 ml of LB + Amp into a sterile 125 ml Erlenmeyer flask and inoculate with 100 µl of the overnight W3NN[pDMS] culture into the prepared LB + Amp, making a 1:100 dilution.
- For larger volumes, add 500 µl of overnight culture into 50 ml of TB + Amp in a 250 ml Erlenmeyer flask, instead of LB + Amp.
- Alternatively, use as the second "representative" culture in step 2.2.
- Incubate in a shaking incubator at 30 °C and 250 rpm until the culture reaches mid to late log phase, indicated by an optical density (OD) or absorbance: A600 = 0.8. After 1-2 hr, the medium should begin to appear turbid to the naked eye.
- Aseptically transfer 1 ml of culture to a clean spectrophotometer cuvette.
- Measure the light absorbance using a UV-vis spectrophotometer with the wavelength set to 600 nm. Use 1 ml of the media in which cells were grown to set the blank (LB or TB). Optionally, use the representative culture from step 2.1.3 for this step instead.
- If the culture has not yet reached the appropriate OD, return to the shaker and check once again every half hour. Check the OD more frequently as the A600 approaches 0.8.
Once the culture has reached late log phase, indicated by A600 = 0.8, aseptically transfer the culture into sterile 50 ml centrifuge tubes.
Collect cells by centrifuging the culture at 10,000 x g for 10 min. Inspect for a pellet at the bottom of the tube.
Decant the cleared supernatant into an appropriate biohazard waste container. Do not disturb the cell pellet.
Repeat steps 2.4-2.5 until all cells have been collected.
Re-suspend the pellet in 100 µl LB + Amp. Aspirate using a micropipette or use a vortex mixer. For greater ministring production, re-suspend the pellet from a 50 ml TB + Amp culture into 1 ml of fresh LB + Amp media.
3. Ministring Production
Aseptically add 20 ml of LB + Amp in a 125 ml Erlenmeyer flask and heat to 42 °C. For greater ministring production, prepare 500 ml of LB + Amp, preheated to 42 °C.
Transfer the resuspension into the preheated medium.
Incubate at 42 °C and 250 rpm until past log phase, indicated by A600 = 1.0. Do not leave 20 ml of culture at 42 °C past 1 hr.
Reduce the temperature to 37 °C and leave the culture for an additional 90 min. For 500 ml, leave the culture for an additional induction period of 90 - 120 min and do not decrease the temperature.
Reduce the temperature to 30 °C and leave the culture shaking at 200 rpm for an additional temperature downshift period of 2 hr. For 500 ml, extend the temperature downshift to 4 hr or overnight.
4. Harvesting Ministrings
Aseptically transfer the culture into sterile 50 ml centrifuge tubes. Centrifuge at 10,000 x g for 10 min and inspect for a pellet.
Decant the supernatant into an appropriate biohazard waste container without disturbing the pellet. Repeat until all the cells have been collected.
Retain the pellet for plasmid extraction with any commercial extraction and endotoxin removal kit or flash freeze in liquid nitrogen and store at -80 °C for later extraction.
Representative Results
After DNA extraction, residual precursor plasmid, LCC prokaryotic backbone, and the DNA ministring will be recovered. Overall plasmid yield and purity can be assessed using a spectrophotometer. Purity is estimated using the ratio of the absorbance at 260 nm and 280 nm (A260/A280) where a ratio of 1.9-2.0 is optimal. All LCC and CCC products can be separated through agarose gel electrophoresis (AGE) (Figure 2). AGE visualizes the results of ministring production after heat induction, prior to ministring purification. A qualitative assessment of ministring production can be made at this stage by comparing band intensity of the ministring (2.4 kb) to parent plasmid (5.6 kb) (Figure 2). Ministring DNA can be recovered via standard plasmid gel extraction protocols following AGE. Figure 3 summarizes ministring production efficiencies under various conditions likely to occur while carrying out the protocol. Ministring production efficiency was determined based on DNA band intensity using the manufacturer's analysis program. Any similar densitometry program can be used to analyze the results of AGE.
 Figure 1: Overview of ministring production. (A) The parent vector contains a polylinker in transcriptional fusion with a reporter gene, framed by the two SS sites. The backbone encodes beta-lactamase for ampicillin resistance and also contains several cut sites unique to the backbone for in vitro digestion. (B) Overview of the main protocol. Steps 2) Growth Phase and 3) Ministring Production are represented in a diagram to provide an overall schematic of the steps involved, highlighting the simplicity of the protocol. (C) Ministring induction. Heat induction inactivates the temperature sensitive repressor, allowing tel expression followed by Tel activity on SS sites in the parent plasmids. Please click here to view a larger version of this figure.
Figure 1: Overview of ministring production. (A) The parent vector contains a polylinker in transcriptional fusion with a reporter gene, framed by the two SS sites. The backbone encodes beta-lactamase for ampicillin resistance and also contains several cut sites unique to the backbone for in vitro digestion. (B) Overview of the main protocol. Steps 2) Growth Phase and 3) Ministring Production are represented in a diagram to provide an overall schematic of the steps involved, highlighting the simplicity of the protocol. (C) Ministring induction. Heat induction inactivates the temperature sensitive repressor, allowing tel expression followed by Tel activity on SS sites in the parent plasmids. Please click here to view a larger version of this figure.
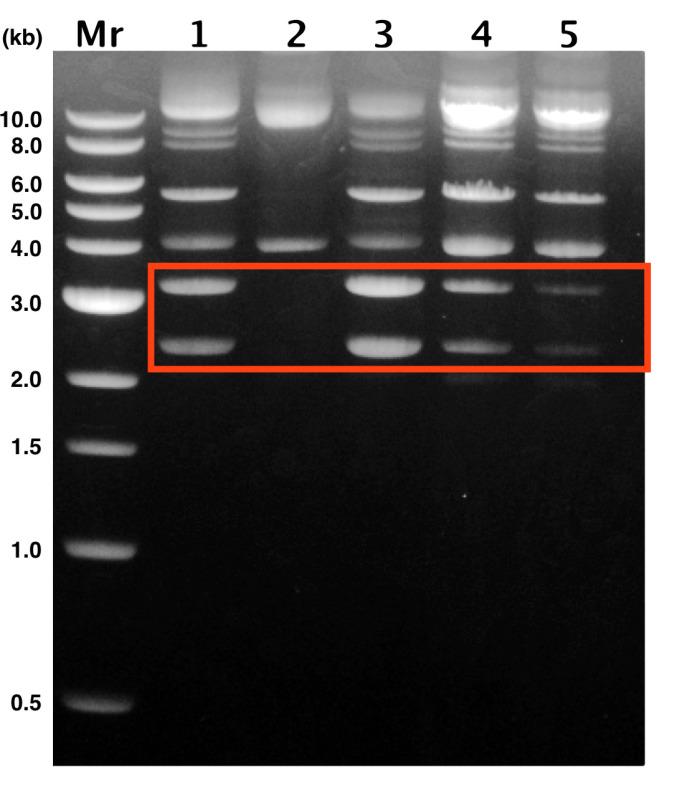 Figure 2: Ministring production after deviations from protocol. 1: Control, 2: Uninduced, 3: Overgrown, 4: Overinduced, 5: Early Removal. DNA ministring is approximately 2.4 kb, LCC backbone is 3 kb, and pNN9 parent plasmid is 5.6 kb. Band intensity of ministring and LCC backbone bands drop with deviations from protocol, indicating lower ministring yield. From left to right: 1 kb ladder from New England Biolabs; Control, following protocol; Uninduced, no temperature upshift; Overgrown, temperature upshift during stationary phase; Overinduced, extended temperature upshift; Early removal, no temperature downshift. Plasmids were extracted using the Omega-Biotek E.Z.N.A. Plasmid Mini Kit. Please click here to view a larger version of this figure.
Figure 2: Ministring production after deviations from protocol. 1: Control, 2: Uninduced, 3: Overgrown, 4: Overinduced, 5: Early Removal. DNA ministring is approximately 2.4 kb, LCC backbone is 3 kb, and pNN9 parent plasmid is 5.6 kb. Band intensity of ministring and LCC backbone bands drop with deviations from protocol, indicating lower ministring yield. From left to right: 1 kb ladder from New England Biolabs; Control, following protocol; Uninduced, no temperature upshift; Overgrown, temperature upshift during stationary phase; Overinduced, extended temperature upshift; Early removal, no temperature downshift. Plasmids were extracted using the Omega-Biotek E.Z.N.A. Plasmid Mini Kit. Please click here to view a larger version of this figure.
 Figure 3: Effects of deviations from protocol on ministring production efficiency (PE). Results are the mean of 3 trials. PE is estimated from relative band intensity measured using AlphaImager HP after AGE. The deviations occur at the following steps: Control, following protocol; Uninduced, no temperature upshift at step 2; Overgrown, temperature upshift began during stationary phase at step 1.6; Overinduced, extended temperature upshift at step 2.3; Early removal, cells removed and extracted without temperature downshift at step 2.3. Please click here to view a larger version of this figure.
Figure 3: Effects of deviations from protocol on ministring production efficiency (PE). Results are the mean of 3 trials. PE is estimated from relative band intensity measured using AlphaImager HP after AGE. The deviations occur at the following steps: Control, following protocol; Uninduced, no temperature upshift at step 2; Overgrown, temperature upshift began during stationary phase at step 1.6; Overinduced, extended temperature upshift at step 2.3; Early removal, cells removed and extracted without temperature downshift at step 2.3. Please click here to view a larger version of this figure.
Discussion
We describe here a simple system for the production of LCC DNA ministrings using a one-step heat-inducible protocol, which requires no special equipment aside from that which is used in typical bacterial growth and manipulation. DNA ministrings are torsion-free, stable linear DNA expression cassettes, devoid of prokaryotic genetic elements. They may be used in place of conventional methods for gene transfer, expression of reporter genes, as well as in assessments towards the efficiency of transgene expression in eukaryotic systems. DNA ministrings combine the biocompatibility and increased transfection efficiency benefits of "mini" vectors with the superior safety profile of linear DNA vectors. Our robust one-step in vivo production platform rapidly and easily produces DNA ministrings for any gene transfer application without the need for multiple costly in vitro processes.
There are several critical steps to ensure optimal ministring production (Figures 2 and 3). Heat induction is the most critical step to ministring production. Complete lack of ministring production is most likely due to lack of heat induction (cells retained at 30 °C), where repression of protelomerase expression prevents conversion of precursor plasmid to LCC ministring. Following the heat induction protocol, expect a production efficiency of approximately 75%. Heat induction is optimal while cells are in log phase. Though there is no statistically significant difference in ministring PE when using cells in stationary phase ("Overgrown", Figure 3), we did observe almost a 50% decrease in overall plasmid yield. Yields will depend on the plasmid extraction protocol used. For optimal ministring production, trigger heat induction while cells are in log phase. Poor ministring production will most likely be a result of prolonged temperature upshift or insufficient temperature downshift. Prolonged temperature upshift is discouraged as this activates the E. coli heat shock response15, which may inhibit protelomerase activity and interfere with plasmid stability. Therefore, timing the temperature downshift is another critical step for efficient ministring production. Without additional incubation time at 30 °C, ministring production efficiency was found to be very poor ("Early Removal", Figure 3). This may be attributed to the amount of time required for Tel expression and activity. The originating prophage PY54 encoding Tel protelomerase normally infects Yersinia, which optimally grows around 28 °C12, indicating that 30 °C may also be more optimal for Tel activity.
The DNA minivector production system utilizes an in vivo platform to generate high quality bacterial sequence-free DNA minivectors. Modifications to the protocol may include altering the growth media to optimize plasmid and protein production in order to promote cell growth during the growth phase (TB instead of LB), or increase ministring production and conversion efficiency. For larger culture volumes (200 ml and greater), temperature downshift and incubation at 30 °C may be extended overnight without severe negative impact on ministring PE. We have included modifications in our protocol for 500 ml cultures as an example. Purification of DNA ministrings can be expedited through in vitro endonuclease digestion of the prokaryotic backbone. There are a number of endonuclease target sites (e.g., PI-SceI, PvuI, ScaI) available on the prokaryotic backbone of the precursor plasmid, which can be cut to expose open linear ends, allowing for in vitro degradation of the prokaryotic backbone. Isolation of ministrings from other vector species can also be achieved through anion exchange membrane chromatography16.
One current limitation associated with the LCC DNA vector production system is the need for purification of the DNA ministring from other LCC and CCC products. Although easily purified using standard plasmid isolation methods, the isolation step can still be a disadvantage in a large-scale setting. Separation of the ministring can be time-consuming using AGE if the ministring and the LCC prokaryotic backbone are too similar in size. Alternatively, anion exchange membrane chromatography may provide better separation of ministrings from CCC vector species16. Temperature upshift can be another potential limitation as high temperatures also activate the heat shock response in E. coli, which negatively affects recombinant protein yields17 and consequently reduce rates of plasmid to ministring conversion. We report elsewhere optimizations of the production platform to circumvent and/or mitigate such effects of heat shock18. We are also currently optimizing methods to eliminate or reduce LCC prokaryotic backbone contamination in vivo.
Chromosomal integration of the LCC DNA ministring disrupts the host chromosome and subjects the cell to apoptosis. Not only does this reduce potential negative consequences of insertional mutagenesis such as oncogenesis and silencing of adjacent genes, thereby improving its safety; the lethal effect can also serve as an ideal method for knock-in and gene replacement studies without the associated side effects and damage of integration. DNA ministring vectors can be applied towards the transfer and expression of genes for therapeutic proteins, RNA, antibodies, or antigens into a given target in vivo or replace a mal- or non- functional allele. The simple one-step heat-inducible in vivo production system allows DNA ministring vectors to be easily produced for clinical or industrial applications.
Disclosures
The authors declare they have no competing financial interests.
Acknowledgments
The authors thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for funding this work.
References
- Klinman DM, Ylt AK, Beaucaget SL, Conover J, Kriegt AM. CpG motifs present in bacterial DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon γ. Proc Natl Acad Sci. 1996;93:2879–2883. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges BL, Taylor KM, Joseph MF, Bourgeois SA, Scheule RK. Long-term transgene expression from plasmid DNA gene therapy vectors is negatively affected by CpG dinucleotides. Mol Ther. 2004;10(2):269–278. doi: 10.1016/j.ymthe.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Darquet AM, et al. Minicircle: an improved DNA molecule for in vitro and in vivo gene transfer. Gene Ther. 1999;6(2):209–218. doi: 10.1038/sj.gt.3300816. [DOI] [PubMed] [Google Scholar]
- Gracey Maniar LE, et al. Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol Ther. 2012;21(1):131–138. doi: 10.1038/mt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese DJ, Fogg JM, Ii DES, Gilbert BE, Zechiedrich L. Supercoiled minivector DNA resists shear forces associated with gene therapy delivery. Gene Ther. 2011;19(1):94–100. doi: 10.1038/gt.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-bey-abina S, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafissi N, et al. DNA ministrings: highly safe and effective gene delivery vectors. Mol Ther Nucleic Acids. 2014;3(April) doi: 10.1038/mtna.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schakowski F, et al. A novel minimal-size vector (MIDGE) improves transgene expression in colon carcinoma cells and avoids transfection of undesired DNA. Mol Ther. 2001;3(5 Pt 1):793–800. doi: 10.1006/mthe.2001.0322. [DOI] [PubMed] [Google Scholar]
- Wang HS, et al. A novel micro-linear vector for in vitro and in vivo gene delivery and its application for EBV positive tumors. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Fuertes L, et al. DNA vaccination with linear minimalistic (MIDGE) vectors confers protection against Leishmania major infection in mice. Vaccine. 2002;21(3-4):247–257. doi: 10.1016/s0264-410x(02)00450-4. [DOI] [PubMed] [Google Scholar]
- Schakowski F, et al. Minimal size MIDGE vectors improve transgene expression in vivo. In Vivo. 2007;21(1):17–23. [PubMed] [Google Scholar]
- Hertwig S, Klein I, Lurz R, Lanka E, Appel B. PY54, a linear plasmid prophage of Yersinia enterocolitica with covalently closed ends. Mol Microbiol. 2003;48:989–1003. doi: 10.1046/j.1365-2958.2003.03458.x. [DOI] [PubMed] [Google Scholar]
- Nafissi N, Slavcev RA. Construction and characterization of an in-vivo linear covalently closed DNA vector production system. Microb Cell Fact. 2012;11(1):154. doi: 10.1186/1475-2859-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZL, et al. Optimization of transcriptional regulatory elements for constructing plasmid vectors. Gene. 2001;272(1-2):149–156. doi: 10.1016/s0378-1119(01)00550-9. [DOI] [PubMed] [Google Scholar]
- Hoffmann F, Rinas U. Stress induced by recombinant protein production in Escherichia coli. Adv Biochem Eng Biotechnol. 2004;89:73–92. doi: 10.1007/b93994. [DOI] [PubMed] [Google Scholar]
- Sum C, Chong JY, Wettig S, Slavcev RA. Separation and purification of linear covalently closed deoxyribonucleic acid by Q-anion exchange membrane chromatography. J Chromatogr A. 2014;1339:214–218. doi: 10.1016/j.chroma.2014.03.016. [DOI] [PubMed] [Google Scholar]
- Hoffmann F, Rinas U. Plasmid amplification in Escherichia coli after temperature upshift is impaired by induction of recombinant protein synthesis. Biotechnol Lett. 2001;23:1819–1825. [Google Scholar]
- Nafissi N, Sum CH, Wettig S, Slavcev RA. Optimization of a one-step heat-inducible in vivo mini DNA vector production system. PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0089345. [DOI] [PMC free article] [PubMed] [Google Scholar]