

Abstract
Human cardiac tissue engineering can fundamentally impact therapeutic discovery through the development of new species-specific screening systems that replicate the biofidelity of three-dimensional native human myocardium, while also enabling a controlled level of biological complexity, and allowing non-destructive longitudinal monitoring of tissue contractile function. Initially, human engineered cardiac tissues (hECT) were created using the entire cell population obtained from directed differentiation of human pluripotent stem cells, which typically yielded less than 50% cardiomyocytes. However, to create reliable predictive models of human myocardium, and to elucidate mechanisms of heterocellular interaction, it is essential to accurately control the biological composition in engineered tissues.
To address this limitation, we utilize live cell sorting for the cardiac surface marker SIRPα and the fibroblast marker CD90 to create tissues containing a 3:1 ratio of these cell types, respectively, that are then mixed together and added to a collagen-based matrix solution. Resulting hECTs are, thus, completely defined in both their cellular and extracellular matrix composition.
Here we describe the construction of defined hECTs as a model system to understand mechanisms of cell-cell interactions in cell therapies, using an example of human bone marrow-derived mesenchymal stem cells (hMSC) that are currently being used in human clinical trials. The defined tissue composition is imperative to understand how the hMSCs may be interacting with the endogenous cardiac cell types to enhance tissue function. A bioreactor system is also described that simultaneously cultures six hECTs in parallel, permitting more efficient use of the cells after sorting.
Keywords: Bioengineering, Issue 109, Cell therapy, tissue engineering, human pluripotent stem cell derived cardiac myocytes, mesenchymal stem cells, mesenchymal stromal cells, bioengineering, cellular microenvironment, cardiovascular, SIRPA, CD90
Introduction
Cardiac tissue engineering has advanced greatly in the last decade, with multiple groups publishing results of fully functional, beating tissues made from both murine cardiomyocytes1-6 and, more recently, human stem cell-derived cardiac myocytes7-12. The cardiac tissue engineering field is driven by two primary and essentially independent goals: 1) to develop exogenous grafts that can be transplanted into failing hearts to improve function4-6; and 2) to develop in vitro models for studying physiology and disease, or as screening tools for therapeutic development2,7.
Three-dimensional (3-D) cell culture is considered essential for developing next generation screening tools, as the 3-D matrix reflects a more natural cardiac microenvironment than traditional 2-D monolayer cell culture; indeed some aspects of cell biology are fundamentally different in 2-D vs. 3-D cultures13,14. Additionally, engineered cardiac tissues are constructed from completely defined components: an extracellular matrix, and a cell population. For traditional engineered human cardiac tissues, while the extracellular matrix composition (usually fibrin9 or collagen7,8,10) is strictly controlled, the input cell composition is less well defined, with the entire mixture of cells from a directed cardiac differentiation of either embryonic stem cells (ESC7,9) or induced pluripotent stem cells (iPSC10,12) being added to the tissues. Depending on the specific cell line and the efficiency of the differentiation protocol used, the resulting percentage of cardiomyocytes can range from less than 25% to over 90%, the specific cardiomyocyte phenotype (i.e., ventricular-, atrial-, or pacemaker-like) can also vary, even the non-cardiomyocyte fraction can be highly heterogeneous15,16 and alter the maturity of the differentiated cardiac myocytes17.
Recent cardiac tissue engineering work has attempted to control the input population of cells, with either a cardiac reporter human embryonic stem cell line8 or cell surface markers18 being used to isolate the cardiac myocyte component of the differentiation. While initially a tissue composed of only cardiac myocytes would seem to be the ideal, this is in fact not the case; hECTs composed solely of cardiac myocytes fail to compact into functional tissues, with some groups finding a 3:1 ratio of cardiac myocytes:fibroblasts producing the highest twitch force8. By using various cell selection methods, including surface markers for live cell sorting, it is possible to create hECTs with defined cell populations. While markers of non-cardiac stromal cells have been available for some time, such as the putative fibroblast marker CD9019,20, surface markers of cardiac myocytes have been more difficult to identify. SIRPα was among the first cardiac surface markers identified for human cardiac myocytes18 and has been shown to be highly selective for the cardiac lineage. Recently, we have found that double-sorting for SIRPα+ and CD90- cells yields nearly pure cardiomyocytes, with the CD90+ population exhibiting a fibroblast-like phenotype (Josowitz, unpublished observations). Based on these collected findings, herein we describe creating hECTs using a 3:1 combination of SIRPα+/CD90- cardiomyocytes and CD90+ fibroblasts.
The ability to engineer a completely defined human cardiac tissue is essential not only for creating robust screening tools, but also for developing model systems to investigate emerging cell- and gene-based cardiac therapies. In particular, numerous cell therapies for heart failure, utilizing cell types including mesenchymal stem cells (MSC)21, cardiac stem cells22 and bone marrow mononuclear cells23-25, have been tested in clinical trials. While many of the initial results have been promising21,23,25, the initial benefit often diminishes over time26-29. A similar trend has been reported in murine engineered cardiac tissues, which display a significant functional benefit due to MSC supplementation, but the benefit is not sustained during long-term culture1. Underlying the sub-optimal performance is our limited knowledge of the mechanisms governing cell therapies. A deeper understanding of how therapeutic cells exert their beneficial influence, as well as potential negative consequences of myocyte-nonmyocyte interactions, would enable the development of improved therapies yielding clinically significant and sustained benefits, with minimal side effects, for patients with heart failure.
Here, we describe the use of defined hECTs to interrogate mechanisms of cell-based therapy. The controlled tissue composition is essential to identify specific factors impacting cardiomyocyte performance. Directly supplementing hECTs with the therapeutic cell type of interest (e.g., MSCs), can reveal the effects on cardiac myocyte performance, as we have demonstrated in rat ECTs1.
The following multi-step protocol begins with directed cardiac stem cell differentiation, followed by fabrication of the multi-tissue bioreactor, and concluding with a description of tissue construction and functional analysis. Our experiments are performed using the NIH-approved H7 human embryonic stem cell (hESC) line. However, the following protocols have also been tested using an additional hESC line and three induced pluripotent stem cell (hiPSC) lines with similar results. We have found that efficiency in cardiomyocyte differentiation and success in hECT fabrication can be cell line dependent, particularly for hiPSC lines derived from individual patients. By following this protocol, two 6-well dishes are plated with a total of 1.68 million hESCs (140,000 cells per well), which yields approximately 2.5 million myocytes after differentiating for 20 days and sorting, enough to make six defined tissues.
Protocol
Note: Perform all cell manipulations in aseptic conditions using a HEPA-filtered class II biological safety cabinet and sterilize all solutions by filtering them through a 0.2 µm filter. Perform tissue construction and function testing in either the same aseptic conditions or a laminar flow hood.
1. Seeding of H7 hESCs in Preparation for Cardiac Differentiation
- (Day 1) Preparing the Basement Membrane Matrix
- Thaw 150 µl aliquot of hESC qualified basement membrane matrix on ice overnight at 4 °C.
- (Day 0-4) Plating of hESCs on Coated Plates
- Dilute the thawed matrix into 12 ml of ice-cold DMEM/F12 and mix well.
- Transfer 1 ml of the DMEM/F12-matrix solution into each well of a 6-well tissue culture treated dish. Each aliquot of matrix can coat two 6-well plates.
- Incubate the coated plates at room temperature for at least 1 hr. Note: In our experience, coated dishes sealed with paraffin can be stored in matrix solution at 4 °C for up to 10 days before use.
- At approximately 75% confluence, dissociate the H7 hESCs from the 10 cm dish using 6 ml of the non-enzymatic dissociation reagent. After 5 min, gently scrape the cells from the culture surface using a disposable cell scraper and transfer the cell suspension to a sterile 15 ml centrifuge tube.
- Remove 0.5 ml of the dissociation solution with the cells from the 15 ml tube and transfer to a new sterile 15 ml tube (leaving 5.5 ml of the dissociation reagent to further dissociate the hESCs) for stem cell line propagation.
- Pellet the 0.5 ml of cells at 300 x g for 5 min at 20 °C. Remove the supernatant and gently resuspend in 8 ml of pluripotent stem cell media containing 1% penicillin-streptomycin then transfer to a new, coated, 10 cm tissue culture dish and maintain 37 °C and 5% CO2 to maintain the stem cell line. Note: Keep pluripotent stem cell media on ice during media changes.
- Meanwhile, add 5.5 µl of 10 mM ROCK inhibitor Y-27632 to the remaining 5.5 ml of dissociation solution and continue to incubate at room temperature for another 5-10 min. Gently mix the cells with a 5 ml serological pipet. Continue to incubate until a single cell suspension is achieved, then pellet the cells at 300 x g for 5 min at room temperature.
- Resuspend the cells in 5 ml of pluripotent stem cell media and perform a cell count using a hemocytometer.
- Seed each well of the 6-well dish at a density of 140,000 cells per well. Seed two 6-well plates to create a total of six defined tissues. Dispose of any remaining cells after plating.
- Fill the well to 1 ml with pluripotent stem cell media and incubate at 37 °C, 5% CO2. Twenty-four hours later, remove the media and add 2 ml of fresh pluripotent stem cell media to each well (Figure 1A).
- Check the confluence of the plates each day and begin the differentiation once the cells reach approximately 75% confluence (Figure 1B-D).
2. (Day 4-24) Differentiation of Human Embryonic Stem Cells to Cardiomyocytes30,31
- (Day 4-7, Differentiation Day 0-3) Mesoderm Induction
- Prepare RPMI differentiation media I (Table 1) by combining 500 ml RPMI 1640 with 10 ml B27 supplement (without insulin) and 5 ml of penicillin-streptomycin stock solution (10,000 IU/ml penicillin; 10,000 µg/ml streptomycin). Aliquot, on ice, into 50 ml tubes and store at 4 °C.
- (Day 4, Differentiation Day 0) Prepare mesoderm induction media by adding 2.4 µl of the small molecule GSK3 inhibitor CHIR99021 (10 mM stock, 6 µM final concentration) to 12 ml of RPMI differentiation media I. Remove pluripotent stem cell media from each well and replace with 2 ml of the mesoderm induction media per well. Return the plate to the incubator. Note: Significant cell death typically occurs with the addition of CHIR99021 (Figure 1E). The monolayer will recover, but it is important to rinse the dead cells away with the DMEM/F12 rinse.
- (Day 5, Differentiation Day 1) Prepare fresh mesoderm induction media as described above. Remove the old media from each well and rinse once with 1 ml of DMEM/F12 per well. Add 2 ml of fresh mesoderm induction media to each well. Note: Rinsing each day from Day 0-10 greatly increases yield and purity of the cardiac myocytes.
- (Day 6, Differentiation Day 2) Remove the cardiac induction media and rinse once with 1 ml DMEM/F12 per well. Replace the rinse with 2 ml RPMI differentiation media I (no small molecules added but still with the B27 (without insulin) supplement) and return to the incubator.
- (Day 7-13, Differentiation Day 3-10) Cardiac Mesoderm Induction
- (Day 7, Differentiation Day 3) Prepare cardiac mesoderm induction media by adding 6 µl of the small molecule Wnt inhibitor IWR-1 (10 mM stock, 5 µM final) to 12 ml of RPMI differentiation media I.
- Remove the media from each well, rinse once with 1 ml DMEM/F12 per well and replace with 2 ml of cardiac mesoderm induction media per well.
- (Day 8, Differentiation Day 4) Prepare an additional 12 ml of cardiac mesoderm induction media as described above. Remove the media added the day prior, rinse once with 1 ml DMEM/F12 per well and replace with 2 ml of fresh cardiac mesoderm differentiation media per well and return to the incubator.
- (Day 9-10, Differentiation Day 5-6) On both days, remove the cardiac mesoderm induction media and rinse each well with 1 ml of DMEM/F12.
- Add 2 ml of fresh RPMI differentiation media I (no small molecules added but still with the B27 (without insulin) supplement) to each well and return the plate to the incubator.
- (Day 11-24, Differentiation Day 7-20) hES-derived Cardiac Myocyte Organization/Maturation
- Prepare the RPMI differentiation media II (Table 1) by combining 500 ml RPMI 1640 with 10 ml B27 supplement (with insulin) and 5 ml of penicillin-streptomycin stock solution. Aliquot, on ice, into 50 ml tubes and store at 4 °C.
- (Day 11, Differentiation Day 7) Remove RPMI differentiation media (without insulin) from each well and rinse with 1 ml DMEM/F12. Replace with 2 ml of the new RPMI differentiation media II (with insulin) to each well and return to the incubator. Note: Spontaneous beating should first be observed between days 7 and 10. If beating is not observed during this time, it typically indicates poor differentiation efficiency. The protocol can be continued until day 15 in an effort to observe beating, but if no beating is observed by day 15 it is best to start a new differentiation.
- (Day 12-24, Differentiation Day 8-20) Every day, remove the old differentiation media and replace with 2 ml of fresh RPMI differentiation media II per well to permit cell maturation and organization of the beating monolayer (Figure 1F, Video Figure 1). Note: Depending on the residual cell death, it may be necessary to rinse with 1 ml of DMEM/F12 through differentiation day 10.
3. (Day 24, Differentiation Day 20) Isolation of Cardiac Myocytes and Fibroblast-like Cells
- Dissociate Cells from the Monolayer
- Remove differentiation media and rinse once with 1 ml PBS.
- Dissociate the monolayers with 1 ml of the enzymatic dissociation solution (0.04% Trypsin/0.03% EDTA) to each well.
- Move plate into incubator for 10 min. Meanwhile, add 12 µl of ROCK inhibitor to 6 ml of trypsin neutralization solution.
- Gently add 1 ml of the trypsin neutralization solution containing the ROCK inhibitor to each well of the plate to neutralize the trypsin solution.
- Using a sterile transfer pipet, gently mix each well to break apart the cell clusters.
- Transfer all 12 ml from the 6-well plate to a 15 ml centrifuge tube. Note: Since two 6-well plates are used in this protocol, two 15-ml tubes will be needed.
- Transfer 3 ml of PBS to one well of the plate, and then sequentially transfer the same 3 ml to each subsequent well to collect any residual cells on the dish. Transfer the remaining 3 ml from the final well into the same 15 ml centrifuge tube containing the cells.
- Pellet at 300 x g for 5 min at 4 °C.
- Prepare Cells for Live Cell Sorting by FACS
- Prepare the staining buffer by adding 5 ml of Fetal Bovine Serum to 45 ml of PBS on ice with 50 µl of ROCK inhibitor.
- Remove the supernatant from the cell pellet (in 3.1.8) and resuspend in 1.2 ml of staining buffer.
- Transfer 200 µl of the cell suspension to a new, pre-chilled, 50 ml centrifuge tube on ice. This will be the negative staining control.
- Transfer the remaining 1 ml of cell suspension to a new, pre-chilled, 50 ml centrifuge tube on ice and add 2 µl SIRPα-PE/Cy7 (1:500 dilution) and 4 µl of CD90-FITC (1:250 dilution). Gently mix the cell suspension with a transfer pipet and return to the ice.
- Incubate both the negative control and sample, on a rocker shaker, on ice, at 4 °C for 1 hr.
- Prepare sample collection tubes by adding 3 ml of RPMI media (with insulin) to two 15 ml centrifuge tubes. Add 3 µl of ROCK inhibitor to each tube and store on ice.
- Pellet the stained cells at 300 x g for 5 min at 4 °C and rinse twice with at least 10 ml of ice-cold PBS per rinse.
- Add 1 µl of DAPI (1 µg/ml) to 5 ml of staining buffer. Gently resuspend the sample pellet with 1-3 ml of the DAPI-containing staining buffer using a transfer pipet.
- Add 500 µl of staining buffer (with no DAPI added) to the negative control.
- Gently filter both the negative control and sample through a 40 µm cell strainer to remove clumps of cells and transfer to polystyrene FACS tubes on ice. Immediately bring samples to the cell sorter.
- Similar to established live cell sorting methods18, use the negative control to set the gates, select for live cells (DAPI negative) and collect both the FITC+ (i.e., CD90+ fibroblasts) and PE/Cy7+ (i.e., SIRPα+ cardiomyocytes) populations independently at 20 psi (Figure 2). Note: After setting the gates, the negative control can be fixed in 4% PFA to determine differentiation efficiency by staining for cardiac troponin-T. Note: Some researchers prefer to use an isotype control rather than unstained control to set the gates in order to compensate for non-specific antibody binding. A so-called fluorescence minus one (FMO) control is another possibility. Due to the clear bimodal distribution of the FITC, DAPI and PE-Cy7 signals, we gated from the positive population, conservatively aiming far into the positive gate, which possibly excludes some true positives but helps to minimize any false positives.
- Cell Reaggregation in Preparation for Tissue Engineering
- After the cell sort, pellet both collection tubes and resuspend in 1 ml of DMEM containing 10% Neonatal Bovine Serum, 1% penicillin-streptomycin and 0.2% Amphotericin B (“NBS media”).
- Recombine the SIRPα+ and CD90+ cells in a 3:1 ratio and plate the combined cells in a non-tissue culture treated petri dish at a density of 2 million cells per 60 cm2 (10 cm dish). Add 10 ml of NBS media and 10 µl of ROCK inhibitor Y-27632.
- Place cell suspension in the tissue culture incubator for 48 hours to allow cell reaggregation into small clusters.
4. Human Cardiac Tissue Engineering
- Fabricate the Multi-tissue Bioreactor Note: To supplement the illustrations in Figure 3A, CAD files with detailed bioreactor design schematics are available upon request from the authors.
- Machine the PDMS master mold by drilling six evenly spaced holes of 0.5 mm diameter into a 9 x 33 x 3.25 mm cuboid of polytetrafluoroethylene.
- Using an endmill, machine a frame from polysulfone measuring 25 x 35 x 11 mm3. The purpose of the frame is to hold the PDMS posts (constructed from the cast made above) in alignment with the wells in the baseplate.
- Using a 1-mm endmill, machine 6 wells (6 x 1 x 1 mm3), 4 mm apart into a 20 x 40 x 5 mm3 piece of black polytetrafluoroethylene to form the baseplate.
- Mix the elastomeric base and curing agent for polydimethylsiloxane (PDMS) in a 10:1 w/w ratio and add to the custom polytetrafluoroethylene mold (step 4.1.1) to create two rows of six force-sensing posts, and incubate overnight and under vacuum at 80 °C. After curing, gently remove the PDMS from the master mold and carefully mark the tops of each post with a black permanent marker for enhanced contrast and automated real-time post deflection tracking. Note: Polytetrafluorethylene is fairly soft and can be damaged easily. Use care when cleaning the master mold prior to PDMS casting to ensure longevity of the system and consistent PDMS post geometry. A 0.5 mm wire can be used to clean the holes for the posts after each use, but take care to not scrape the inside of the holes. Note: An alternative is to degas the PDMS under vacuum for several hours at room temperature, then let the mixture cure at ambient pressure. This may lead to fewer residual gas bubbles forming in the PDMS during the curing process.
- Sterilize all Components in a Steam Autoclave. Note: The PDMS master mold (step 4.1.1) and polysulfone frame (step 4.1.2) are both reusable. The PDMS posts created from the cast (step 4.1.4) is also reusable but only for approximately 10 uses. However, more PDMS posts can be created as needed using the master mold.
- Collect Reaggregated Cardiac Cells
- Remove the reaggregated cells from the incubator and transfer all 10 ml of the reaggregation media to a 50 ml centrifuge tube.
- Rinse the plate with 3 ml of PBS and transfer the rinse to the same 50 ml centrifuge tube containing the reaggregation media.
- Add 3 ml of 0.04% Trypsin/0.03% EDTA to the 10 cm dish and return to the incubator for 5 min.
- After 5 min, examine the plate with an inverted compound microscope at 10X magnification to ensure complete cell dissociation from the dish. If some residual clusters are still attached, gently agitate the plate to detach the clumps. If the clusters remain attached, return to the incubator for another 2-3 min. Do not incubate longer than ten minutes or significant cell death may occur.
- Once all cells are detached, add 3 ml of trypsin neutralization solution. Gently mix the neutralization solution with the trypsin-cell solution and transfer to the 50 ml centrifuge tube containing the reaggregation media and rinse once with PBS.
- Rinse the entire dish with 5 ml of PBS and transfer to the same 50 ml tube containing the cells.
- Pellet the cells at 300 x g for 5 min at room temperature.
- Remove the supernatant and resuspend the pellet in 1 ml of NBS media, then transfer to a 1.5 ml microcentrifuge tube.
- Pellet at 300 x g for 5 min at room temperature and remove the supernatant. The cardiac cells (CD90+ stromal cells and SIRPα+ myocytes) are now ready for tissue construction.
- (Optional) Collect Supplemental Cells of Interest Note: In addition to the defined tissues containing only SIRPα+ cardiomyocytes and CD90+ fibroblast-like cells, it is possible to add additional cells of interest to interrogate their effect on tissue function. For example rat MSCs have been shown to enhance the function of rat engineered cardiac tissues1. The following optional step describes the collection of supplemental cells for the defined system.
- Collect the supplemental cell type of interest (e.g., mesenchymal stem cells) using 0.25% trypsin/0.1% EDTA.
- Pellet the cells at 300 x g for 5 min at room temperature then resuspend in 5 ml of appropriate cell culture media for the cell type of interest. For instance, for MSCs, use DMEM supplemented with 20% fetal bovine serum, 1% penicillin-streptomycin and 0.2% amphotericin-B to culture the cells.
- Perform a cell count using a hemocytometer, then pellet the cells again at 300 x g for 5 min at room temperature.
- Remove the supernatant, resuspend in 1 ml of cell culture media and transfer to a 1.5 ml microcentrifuge tube.
- Pellet the cells at 300 x g for 5 min at room temperature.
- Remove the supernatant. The supplemental cells are now ready for tissue construction. Note: if the supplemental cells are added at a concentration of 10% of the total cell number in the tissue, then for the defined tissues, this will require 50,000 supplemental cells per tissue as each tissue contains 500,000 cardiac cells (both CD90+ stromal cells and SIRPα+ myocytes).
- Create the Human Engineered Cardiac Tissues Note: Store all solutions on ice and the cells at room temperature. All volumes listed below are per tissue. Typically, approximately six tissues can be constructed from two 6-well plates of cardiac differentiations.
- Dilute 60.0 µl of the 5 mg/ml collagen stock to 3.125 mg/ml with 1.5 µl of 1 M NaOH, 9.6 µl of 10x PBS and 24.9 µl of sterile ultrapure deionized water. Critical step: Avoid the introduction of air bubbles to each solution during preparation as air bubbles will disrupt proper tissue formation.
- Add 12.0 µl of both 10x MEM and 0.2 N HEPES pH 9 to the dilute collagen mixture to create the collagen mix. Add both solutions down the side of the 15 ml centrifuge tube in order to avoid the introduction of air bubbles into the collagen mix.
- Add the basement membrane matrix to a final concentration of 0.9 mg/ml to the collagen mix and store on ice to create the final tissue mix. The final concentration of collagen should be 2 mg/ml.
- Add 500,000 of the reaggregated cells to the tissue mix (myocyte + fibroblast concentration is 20 million/ml) and fill to a final volume of 25 µl per tissue with either cell-free NBS media or 50,000 cells of the supplemental cell type of interest (e.g., hMSCs) and mix well to form an even cell suspension. Note: The number of supplemented cells will vary from for different applications. Here 10% supplementation is used.
- With care, pipet 25 µl of the cell suspension into each of the six wells in the bioreactor baseplate, without introducing air bubbles into the well.
- Push two rows of the PDMS force sensors onto either side of the polysulfone frame, forming 6 pairs of opposing posts, then invert the frame on top of the baseplate so that one pair of posts enters each well containing the cell suspension. Note: The polysulfone frame includes tabs to aid in alignment of the PDMS.
- Carefully place the bioreactor, baseplate down, into a 60 mm dish, then place the dish without its cover inside of a 10 cm dish, place the 10 cm cover on top, and move the entire bioreactor assembly into the tissue culture incubator, waiting two hours for the tissue to gel.
- After 2 hr, remove the bioreactor from the incubator and add 14 ml of NBS media to the entire assembly, enough to cover the baseplate. Return to the incubator, and change half of the media every day.
- Forty-eight hr later, carefully remove the baseplate by gently moving each side of the baseplate off the frame a few millimeters at a time, change the media, then return the bioreactor, tissues facing down, to the media.
- Continue changing half of the NBS culture media every day. Note: Spontaneous contractions of the tissue can be observed as early as 3 days after tissue construction, generating measureable twitch forces as early as day 5 to 7.
Representative Results
To obtain cardiac myocytes, a slightly modified version of the Boheler and Lian differentiation methods is used30,31. It is imperative that the differentiation starts during the log-phase of cell growth, but also that the starting population is sufficiently confluent to obtain a useable number of cells after sorting (approximately 75% is optimal). Typically, for H7 hESCs, plating at a density of 140,000 hESCs per well of a 6-well dish in essential 8 media and 5% CO2 incubator maintained at 37 °C yields sufficiently confluent cultures after 4 days to begin the differentiation, as illustrated in Figure 1A-D. Since insulin inhibits cardiac specification during the differentiation process32, insulin free media (differentiation media I, Table 1) is used for the first 10 days of differentiation. Once differentiation is initiated via the application of the GSK3 inhibitor CHIR99021, significant cell death will occur (Figure 1E). As a consequence it is essential to change the media every day. On day 2 of differentiation, the cells are removed from CHIR99021 and rested in differentiation media I (with no added small molecules) for 24 hr. After the 24 hr rest in differentiation media, the cells are treated with IWR-1 for 48 hr, after which they begin to self-organize into clusters (Figure 1F) that beat as early as seven days from starting the differentiation. Since cardiac specification has occurred after the application of IWR-1, the media is changed to media containing insulin (differentiation media II, Table 1). By 18 days of differentiation, the colonies of beating cells have connected and partially detached from the cell surface, forming robustly beating “webs” throughout the dish (Video Figure 1).
On day 20, the beating monolayer is gently dissociated via enzymatic methods to obtain single cells for live fluorescent associated cell sorting (FACS). Using the differentiation method described above, we conservatively harvest approximately 65% of the population as SIRPα+ and 10% as CD90+ (Figure 2). Generally 1-2 million SIRPα+ cells are obtained per 6-well plate.
After reaggregation for 48 hr in a 3:1 ratio of SIRPα+:CD90+ cells, the defined cell aggregates are mixed with a collagen-based hydrogel and pipetted into narrow wells in the baseplate of the multi-tissue bioreactor. Typically, before removal from the dish, small clusters of 5-10 cells will be seen beating on the surface of the dish. The defined tissue bioreactor consists of three components: a master mold to cast the PDMS posts; a frame to hold two PDMS racks of posts; and a black polytetrafluorethylene baseplate containing six wells for the tissue mix (Figure 3A). The entire tissue construction process is performed on ice and in aseptic conditions. After adding the liquid cell-matrix mixtures to the wells in the baseplate, the PDMS posts are attached to the polysulfone frame, and then inverted so that the posts align with the wells in the baseplate (Figure 3B). After 48 hr of incubation, the tissues should be sufficiently compacted that the baseplate can be removed. The baseplate is most easily detached by extracting the entire system from the media and gently removing any excess media from between the PDMS and baseplate. If all of the media is not removed, surface tension of the remaining fluid can pull the tissues off the posts. Once the media is removed, gently push the baseplate back against the PDMS posts to help loosen the tissue from the baseplate. Then gradually push the baseplate off by gently moving one side up a few millimeters, then the other side up the same amount. Repeat this process until the baseplate is removed, with all 6 hECTs attached to their respective PDMS end-posts (Figure 4A). Removing the baseplate should take no more than one minute. Replace the system into the cell culture media, inverted, so that all the tissues are covered by the cell culture media.
Approximately one day after removing the baseplate, weak localized contractions should be observable in the defined hECTs under bright field microscopy. After 7 days, the tissues have matured and compacted (Figure 4B-C) with aligned sarcomeres (Figure 4D). The tissues visibly deflect the PDMS posts with an average of 5-15 µN of force (Figure 4E). Twitch force measurements for the defined engineered cardiac tissues can be measured by real-time tracking of the post tip deflection with a high speed camera and custom data acquisition software, and applying the post deflection to beam bending theory after determining the Young’s modulus of the silicone posts using a force transducer, as explained in more detail previously.1,7 Maintaining sterility during data acquisition ensures the ability to measure changes in tissue function over time. We have maintained functional tissues for several weeks, as explained in more detail elsewhere1. Our preliminary findings indicate a substantial enhancement of hECT contractile force when tissues are supplemented with 10% human mesenchymal stem cells at both 1 Hz and 2 Hz pacing frequencies (Figure 4F).
 Figure 1: Representative images of H7 hESCs during the expansion and cardiac differentiation process. The expansion phase should last 4 days, with growth of the colonies expanding from 24 (A), to 48 (B), 72 (C) and 96 hr (D). After 96 hr of growth the differentiation is begun, resulting in substantial cell death during the first 48 hr of CHIR99021 treatment (E). After two days of IWR-1 treatment, reorganization occurs (F) to form clusters of cells that beat as early as day 7. Further organization into a robustly beating “web” occurs from day 7 to day 20, when the monolayers are dissociated for creating engineered cardiac tissues.
Figure 1: Representative images of H7 hESCs during the expansion and cardiac differentiation process. The expansion phase should last 4 days, with growth of the colonies expanding from 24 (A), to 48 (B), 72 (C) and 96 hr (D). After 96 hr of growth the differentiation is begun, resulting in substantial cell death during the first 48 hr of CHIR99021 treatment (E). After two days of IWR-1 treatment, reorganization occurs (F) to form clusters of cells that beat as early as day 7. Further organization into a robustly beating “web” occurs from day 7 to day 20, when the monolayers are dissociated for creating engineered cardiac tissues.
Video Figure 1: Representative cardiac monolayer after 17 days of differentiation.Please click here to view this video. Spontaneous beating within the monolayer is usually first observed between day 7 and day 10, and by 17 days of differentiation the monolayer has formed interconnected “webs” that beat robustly.
| Name | Composition | Differentiation days used |
| Differentiation Media I | RPMI 1640 B27 Supplement (minus insulin) Penicillin-Streptomycin | D0-D1 (with CHIR99021) D2 D3-D5 (with IWR-1) D5-D7 |
| Differentiation Media II | RPMI 1640 B27 Supplement (with insulin) Penicillin-Streptomycin | D7-D20 |
Table 1: Composition of Differentiation Medias.
 Figure 2: Representative live cell sorting results. Two samples were prepared: an unstained control and a stained control using 1:500 dilution of the SIRPα-PE/Cy7 antibody and 1:250 dilution of the CD90-FITC antibody. After staining, the cells were sorted at 20 psi in sorting buffer composed of PBS, 10% neonatal bovine serum, 10 µM ROCK inhibitor Y-27632, and 1 µg/ml DAPI. Both cell populations were gated in the forward and side scatter profiles to exclude debris (A, blue line) then single cells were selected by comparing the pulse width and height (pulse width analysis) in both the forward scatter (B, blue line) and side scatter channels (C, blue line). Next, live cells were selected by gating the DAPI- population (D, blue line). The final cell populations for collection were determined by examining the FITC and PE-Cy7 expression levels of the live populations. The unstained control (E, left) was used to establish the appropriate gate to select for the FITC+ (CD90) and SIRPα-PE/Cy7+ (cardiomyocyte) population present in the stained sample (E ,right). The FITC and SIRPα-PE/Cy7+poplations were collected in separate 15 ml centrifuge tubes.
Figure 2: Representative live cell sorting results. Two samples were prepared: an unstained control and a stained control using 1:500 dilution of the SIRPα-PE/Cy7 antibody and 1:250 dilution of the CD90-FITC antibody. After staining, the cells were sorted at 20 psi in sorting buffer composed of PBS, 10% neonatal bovine serum, 10 µM ROCK inhibitor Y-27632, and 1 µg/ml DAPI. Both cell populations were gated in the forward and side scatter profiles to exclude debris (A, blue line) then single cells were selected by comparing the pulse width and height (pulse width analysis) in both the forward scatter (B, blue line) and side scatter channels (C, blue line). Next, live cells were selected by gating the DAPI- population (D, blue line). The final cell populations for collection were determined by examining the FITC and PE-Cy7 expression levels of the live populations. The unstained control (E, left) was used to establish the appropriate gate to select for the FITC+ (CD90) and SIRPα-PE/Cy7+ (cardiomyocyte) population present in the stained sample (E ,right). The FITC and SIRPα-PE/Cy7+poplations were collected in separate 15 ml centrifuge tubes.
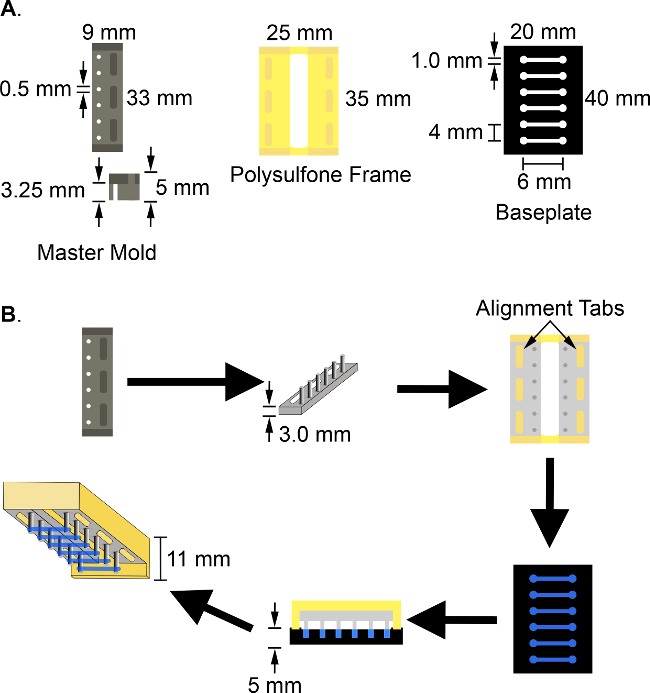 Figure 3: Schematic of the multi-tissue bioreactor and the cardiac tissue engineering process. Construction of the multi-tissue bioreactor requires three components (A): 1) a polytetrafluoroethylene master mold 9 x 33 x 5 mm3, with 6 evenly spaced holes 0.5 mm in diameter; 2) a 25 x 35 x 11 mm3 polysulfone frame to hold the PDMS posts during tissue construction and culture; and 3) a 20 x 40 x 5 mm3 black polytetrafluorethylene baseplate with 6 wells of 6 x 1 x 1 mm3 dimensions, spaced 4 mm apart along its length. To create human engineered cardiac tissues (B), the PDMS posts are fabricated by PDMS soft lithography using the polytetrafluorethylene master mold, two of which are then pressed onto the tabs of the polysulfone frame to form 6 pairs of force-sensing cantilever posts. The tissue solution is pipetted into the wells in the black polytetrafluorethylene baseplate. The frame and posts are then inverted and aligned on the polytetrafluorethylene baseplate so that one pair of posts enters each well containing the tissue mix. After 48 hr, the baseplate is removed and the defined tissues are cultured on the posts, remaining inverted in NBS media.
Figure 3: Schematic of the multi-tissue bioreactor and the cardiac tissue engineering process. Construction of the multi-tissue bioreactor requires three components (A): 1) a polytetrafluoroethylene master mold 9 x 33 x 5 mm3, with 6 evenly spaced holes 0.5 mm in diameter; 2) a 25 x 35 x 11 mm3 polysulfone frame to hold the PDMS posts during tissue construction and culture; and 3) a 20 x 40 x 5 mm3 black polytetrafluorethylene baseplate with 6 wells of 6 x 1 x 1 mm3 dimensions, spaced 4 mm apart along its length. To create human engineered cardiac tissues (B), the PDMS posts are fabricated by PDMS soft lithography using the polytetrafluorethylene master mold, two of which are then pressed onto the tabs of the polysulfone frame to form 6 pairs of force-sensing cantilever posts. The tissue solution is pipetted into the wells in the black polytetrafluorethylene baseplate. The frame and posts are then inverted and aligned on the polytetrafluorethylene baseplate so that one pair of posts enters each well containing the tissue mix. After 48 hr, the baseplate is removed and the defined tissues are cultured on the posts, remaining inverted in NBS media.
 Figure 4: Representative images and functional measurements of defined human engineered cardiac tissues. The multi-tissue bioreactor system holds six tissues in parallel (A, arrowheads). Defined tissues are self-assembled within seven days after tissue creation (top view, B and oblique side view, C). (D). Portion of a defined hECT stained for cardiac troponin-T (cTnT). (E) Representative twitch tracing shows raw force over time during 1 Hz pacing by electrical field stimulation. (F) Twitch tracing during stimulation at 1 Hz (0-2 sec) and 2 Hz (2-4 sec) pacing, with arrow marking change in frequency, for both unsupplemented defined hECTs (solid line) and tissues supplemented with 10% hMSCs (dotted line).
Figure 4: Representative images and functional measurements of defined human engineered cardiac tissues. The multi-tissue bioreactor system holds six tissues in parallel (A, arrowheads). Defined tissues are self-assembled within seven days after tissue creation (top view, B and oblique side view, C). (D). Portion of a defined hECT stained for cardiac troponin-T (cTnT). (E) Representative twitch tracing shows raw force over time during 1 Hz pacing by electrical field stimulation. (F) Twitch tracing during stimulation at 1 Hz (0-2 sec) and 2 Hz (2-4 sec) pacing, with arrow marking change in frequency, for both unsupplemented defined hECTs (solid line) and tissues supplemented with 10% hMSCs (dotted line).
Discussion
Construction of defined human engineered cardiac tissues (hECT) can provide a more consistent and reliable model of human cardiac myocyte function. Critically, all cellular and extracellular components in the system are known and can be manipulated as desired, thus removing the confounding influence of other unknown cell types resulting from the differentiation process. To balance rapid cell growth and high yield, it is preferable that the differentiation starts at 75% confluence of the hESCs, ideally four days after plating. Additionally, the use of ROCK inhibitor Y-27632 during both the cell dissociation and reaggregation after sorting greatly enhances cell viability. The presence of spontaneous beating of myocytes during the differentiation process is an important indicator of the efficiency and health of the differentiation. If spontaneous beating is not observed this may indicate poor differentiation efficiency, either due to ineffective reagents or a loss of pluripotency of the stem cells.
During tissue construction, the PDMS posts must align properly with the channels in the baseplate to create well-formed tissues that last long enough for measuring contractile function. To enhance the ease of device alignment, and strengthen tissue formation around the posts where they are susceptible to breaking, it is possible to add extra width (approximately one post diameter on each side of the post) to the ends of the channels, as demonstrated by the “dog bone”-shaped channels in Figure 3A. Without proper device alignment the tissue will either detach from the posts shortly after culture, or will remain in the well when the baseplate is removed. If a well-formed tissue falls off during baseplate removal, it is possible to gently reattach the tissue to the posts using fine forceps under a dissecting microscope, although it is easy to damage the delicate hECTs if adequate care is not taken.
A major strength of the described defined system is the ability to interrogate the contribution of direct cell-cell interaction mechanisms in cardiac cell therapies based on specific control of cell composition. The injection of cells in animals is imprecise and subject to low viability and retention33. Additionally, the effect of the injected cells on the target tissue is complicated by interactions with other physiologic systems, such as the immune system. As such, understanding the contribution of direct cell-cell interactions in cardiac cell therapies is challenging to address in model organisms. Therefore, an advantage to the described method is that cell-cell interactions are analyzed in a biomimetic three-dimensional environment, preserving key aspects of the cardiac niche, and in the presence of the specific cell types of interest — namely, the cardiac myocytes and fibroblasts. The utility of using the defined hECT system is demonstrated with the supplementation of 10% hMSCs derived from human marrow34,35 and used in clinical trials to the cell mixture during tissue creation. Those tissues that were supplemented with the hMSCs exhibited larger contractile force than the unsupplemented defined human tissues (Figure 4F). Since the tissue environment and composition has been controlled, the enhancement of tissue function with MSC supplementation reflects an inherent effect of hMSCs on cardiomyocyte function. Another strength to the system is that since the tissues are smaller than used previously,1,7 cell use is more efficient, an important consideration when using directed differentiations of pluripotent stem cells as the cell source.
Beyond studying mechanisms of cell therapy, the defined hECT system would enable the examination of cellular basis of cardiovascular cell-cell interactions. Perturbation of the biology of the cells prior to tissue construction, for example with shRNAs, would permit the investigation of molecular mechanisms governing cell-cell and cell-matrix interactions. An additional advantage to the tissue engineering system is the formation of aligned myofibrils capable of functional contractions. Thus, using the defined hECT system, the dynamics of myofibril formation, cardiomyocyte development and the organization of the tissue architecture can all be investigated via modification of the components of the extra-cellular matrix or molecular manipulation of the cells. The human and 3-D nature of the hECT system additionally increases the translatability of the findings.
While the described system offers a powerful method for understanding basic questions of cardiovascular biology, it is not without limitations. First, the engineered tissues exhibit an immature phenotype, consistent with published twitch force data from newborn myocardial samples7. While the phenotypic immaturity could be a confounding factor in evaluating cell interaction mechanisms, there is evidence that diseased cardiomyocytes revert to a fetal gene program7,36-38. If findings from the hECT system prove to be relevant in the context of a failing heart, this may be advantageous for testing cardiac therapies. The protocol also uses a hESC qualified basement membrane matrix during tissue construction. The composition of this matrix can vary from lot to lot, and while defined basement matrix alternatives are available they have yet to be evaluated in the context of hECTs.
Other limitations include the lack of a vascular system and no systems-level interaction effects. Given the size of the tissues, metabolic demands are met by diffusion alone so no vascular system is needed to ensure cell viability. However, endothelial cell signaling may be an important factor in specific cell therapies. If this is the case, a specific number of endothelial cells can simply be added to the defined cell mix during tissue construction. While systems-level interactions are important for complete understanding of cell mechanisms, the engineered tissue system offers a reductionist approach to simplify the problem in order to address the mechanism systematically. Complexity can be added to the defined tissue system as needed through the introduction of systems-related effects, such as leukocytes to incorporate an immune response, or by adding neighboring tissue types to interrogate paracrine signaling.
As an investigative tool, the defined human engineered cardiac tissue system offers the benefits of species-specific human cells, a biomimetic 3-D culture environment with controlled biocomplexity, straightforward and longitudinal monitoring of contractile function, and a defined cellular and matrix composition. Future directions for the hECT system include manipulating the therapeutic cell type of interest with siRNA or shRNA prior to introduction in the tissue in order to investigate specific molecular details of cell-cell interaction. Additionally, rather than using hESCs to form the cardiac cells, it is possible to use induced pluripotent stem cells (iPSCs) from patients with an inherited mutation in an effort to model related cardiac disease manifestations in the engineered tissues. Thus, our method of creating human engineered cardiac tissues with defined cell populations promises to open new avenues for studying cardiac cell therapies to help accelerate the development of novel treatments for patients with heart disease.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by NIH (1F30HL118923-01A1) to T.J.C., NIH/NHLBI PEN contract HHSN268201000045C to K.D.C., the research grant council of Hong Kong TRS T13-706/11(K.D.C), NIH (R01 HL113499) to B.D.G., the American Heart Association (12PRE12060254) to R.J., and Research Grant Council of HKSAR (TBRS, T13-706/11) to R.L. Additional funding was provided to T.J.C. by NIH DRB 5T32GM008553-18 and as a traineeship on NIDCR-Interdisciplinary Training in Systems and Developmental Biology and Birth Defects T32HD075735. The authors also wish to gratefully acknowledge Arthur Autz at The Zahn Center of The City College of New York for assistance with machining the bioreactor and Mamdouh Eldaly for technical assistance. We also thank Dr. Kenneth Boheler for advice on cardiac differentiation, and Dr. Joshua Hare for generously providing human mesenchymal stem cells.
References
- Serrao GW, et al. Myocyte-depleted engineered cardiac tissues support therapeutic potential of mesenchymal stem cells. Tissue Eng. Part A. 2012;18(13-14):1322–1333. doi: 10.1089/ten.tea.2011.0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, et al. Development of a drug screening platform based on engineered heart tissue. Circ. Res. 2010;107(1):35–44. doi: 10.1161/CIRCRESAHA.109.211458. [DOI] [PubMed] [Google Scholar]
- Fink C, Ergün S, Kralisch D, Remmers U, Weil J, Eschenhagen T. Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement. FASEB J. 2000;14(5):669–679. doi: 10.1096/fasebj.14.5.669. [DOI] [PubMed] [Google Scholar]
- Yildirim Y, et al. Development of a biological ventricular assist device: preliminary data from a small animal model. Circulation. 2007;116(11 Suppl):16–23. doi: 10.1161/CIRCULATIONAHA.106.679688. [DOI] [PubMed] [Google Scholar]
- Sekine H, et al. Cardiac Cell Sheet Transplantation Improves Damaged Heart Function via Superior Cell Survival in Comparison with Dissociated Cell Injection. Tissue Eng. Part A. 2011;17(23-24):2973–2980. doi: 10.1089/ten.tea.2010.0659. [DOI] [PubMed] [Google Scholar]
- Lesman A, et al. Transplantation of a tissue-engineered human vascularized cardiac muscle. Tissue Eng. Part A. 2010;16(1):115–125. doi: 10.1089/ten.TEA.2009.0130. [DOI] [PubMed] [Google Scholar]
- Turnbull IC, et al. Advancing functional engineered cardiac tissues toward a preclinical model of human myocardium. FASEB J. 2014;28(2):644–654. doi: 10.1096/fj.13-228007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavandiran N, Dubois N, et al. Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proc. Natl. Acad. Sci. U. S. A. 2013;110(49):4698–4707. doi: 10.1073/pnas.1311120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf S, et al. Human engineered heart tissue as a versatile tool in basic research and preclinical toxicology. PloS One. 2011;6(10):e26397. doi: 10.1371/journal.pone.0026397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch NL, et al. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ. Res. 2011;109(1):47–59. doi: 10.1161/CIRCRESAHA.110.237206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes SS, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nature Methods. 2013;10(8):781–787. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, et al. Three-dimensional filamentous human diseased cardiac tissue model. Biomaterials. 2014;35(5):1367–1377. doi: 10.1016/j.biomaterials.2013.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012;125(13):3015–3024. doi: 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes Soares C, Midlej V, de Oliveira MEW, Benchimol M, Costa ML, Mermelstein C. 2D and 3D-organized cardiac cells shows differences in cellular morphology, adhesion junctions, presence of myofibrils and protein expression. PloS One. 2012;7(5):e38147. doi: 10.1371/journal.pone.0038147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Keller G, Gold JD, Wu JC. Production of De Novo Cardiomyocytes: Human Pluripotent Stem Cell Differentiation and Direct Reprogramming. Cell Stem Cell. 2012;10(1):16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery CL, Zhang J, Ng ES, Elliott DA, Elefanty AG, Kamp TJ. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ. Res. 2012;111(3):344–358. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. Non-cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell-derived cardiomyocytes during differentiation. Stem Cells Dev. 2010;19(6):783–795. doi: 10.1089/scd.2009.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois NC, et al. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nature Biotechnol. 2011;29(11):1011–1018. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudon-David F, Bouzeghrane F, Couture P, Thibault G. Thy-1 expression by cardiac fibroblasts: lack of association with myofibroblast contractile markers. J. Mol. Cell. Cardiol. 2007;42(5):991–1000. doi: 10.1016/j.yjmcc.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Gago-Lopez N, et al. THY-1 receptor expression differentiates cardiosphere-derived cells with divergent cardiogenic differentiation potential. Stem Cell Reports. 2014;2(5):576–591. doi: 10.1016/j.stemcr.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare JM, Traverse JH, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J. Am. Coll. Cardiol. 2009;54(24):2277–2286. doi: 10.1016/j.jacc.2009.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378(9806):1847–1857. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wollert KC, et al. Intracoronary autologous bone-marrow cell transfer after myocardial infarction: the BOOST randomised controlled clinical trial. Lancet. 2004;364(9429):141–148. doi: 10.1016/S0140-6736(04)16626-9. [DOI] [PubMed] [Google Scholar]
- Hirsch A, et al. Intracoronary infusion of autologous mononuclear bone marrow cells or peripheral mononuclear blood cells after primary percutaneous coronary intervention: rationale and design of the HEBE trial--a prospective, multicenter, randomized trial. Am. Heart. J. 2006;152(3):434–441. doi: 10.1016/j.ahj.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Jeevanantham V, Butler M, Saad A, Abdel-Latif A, Zuba-Surma EK, Dawn B. Adult bone marrow cell therapy improves survival and induces long-term improvement in cardiac parameters: a systematic review and meta-analysis. Circulation. 2012;126(5):551–568. doi: 10.1161/CIRCULATIONAHA.111.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer GP, et al. Intracoronary bone marrow cell transfer after myocardial infarction: 5-year follow-up from the randomized-controlled BOOST trial. Eur. Heart J. 2009;30(24):2978–2984. doi: 10.1093/eurheartj/ehp374. [DOI] [PubMed] [Google Scholar]
- Meyer GP, et al. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months' follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation. 2006;113(10):1287–1294. doi: 10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- Hirsch A, et al. Intracoronary infusion of mononuclear cells from bone marrow or peripheral blood compared with standard therapy in patients after acute myocardial infarction treated by primary percutaneous coronary intervention: results of the randomized controlled HEBE trial. Eur. Heart J. 2011;32(14):1736–1747. doi: 10.1093/eurheartj/ehq449. [DOI] [PubMed] [Google Scholar]
- Simari RD, et al. Bone marrow mononuclear cell therapy for acute myocardial infarction: a perspective from the cardiovascular cell therapy research network. Circ. Res. 2014;114(10):1564–1568. doi: 10.1161/CIRCRESAHA.114.303720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, et al. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J. Vis. Exp. 2014. p. e52010. [DOI] [PMC free article] [PubMed]
- Lian X, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. U. S. A. 2012;109(27):1848–1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, et al. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PloS One. 2011;6(4):e18293. doi: 10.1371/journal.pone.0018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean TJ, Lin P, Caplan AI, Dennis JE. MSCs: Delivery Routes and Engraftment, Cell-Targeting Strategies, and Immune Modulation. Stem Cells Int. 2013. p. 732742. [DOI] [PMC free article] [PubMed]
- Trachtenberg B, et al. Rationale and design of the Transendocardial Injection of Autologous Human Cells (bone marrow or mesenchymal) in Chronic Ischemic Left Ventricular Dysfunction and Heart Failure Secondary to Myocardial Infarction (TAC-HFT) trial: A randomized, double-blind, placebo-controlled study of safety and efficacy. Am. Heart J. 2011;161(3):487–493. doi: 10.1016/j.ahj.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Williams AR, et al. Intramyocardial stem cell injection in patients with ischemic cardiomyopathy: functional recovery and reverse remodeling. Circ. Res. 2011;108(7):792–796. doi: 10.1161/CIRCRESAHA.111.242610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104(24):2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail. Rev. 2007;12(3-4):331–343. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010;1188:191–198. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]