

Abstract
The fission yeast Schizosaccharomyces pombe has been an invaluable model system in studying the regulation of the mitotic cell cycle progression, the mechanics of cell division and cell polarity. Furthermore, classical experiments on its sexual reproduction have yielded results pivotal to current understanding of DNA recombination and meiosis. More recent analysis of fission yeast mating has raised interesting questions on extrinsic stimuli response mechanisms, polarized cell growth and cell-cell fusion. To study these topics in detail we have developed a simple protocol for microscopy of the entire sexual lifecycle. The method described here is easily adjusted to study specific mating stages. Briefly, after being grown to exponential phase in a nitrogen-rich medium, cell cultures are shifted to a nitrogen-deprived medium for periods of time suited to the stage of the sexual lifecycle that will be explored. Cells are then mounted on custom, easily built agarose pad chambers for imaging. This approach allows cells to be monitored from the onset of mating to the final formation of spores.
Keywords: Molecular Biology, Issue 109, Fission yeast, Schizosaccharomyces pombe, live-cell imaging, fluorescence microscopy, nitrogen starvation, pheromone, G protein-coupled receptor, sex, mating, shmoo, cell-cell fusion
Introduction
Though genetic exchange between two cells is the central event in sexual reproduction, it relies on a chain of events that promote cell differentiation, allow for partner choice, carry out cell-cell fusion and maintain genomic stability. Thus the sexual lifecycle presents itself as a model system to study a number of biological questions regarding developmental switches, response to extrinsic stimuli, plasma membrane fusion, chromosome segregation, etc. Exploring the fission yeast sexual cycle to study these phenomena brings the benefits of the model system's powerful genetics, well-established high-throughput approaches and sophisticated microscopy. Sex in fission yeast is a heterotypic event between a P-cell and an M-cell of distinct mating types. The two cell types differentially express a number of genes1,2 including those for production of the secreted P- and M-pheromones, pheromone-receptors Map3 and Mam2 as well as pheromone-proteases Sxa1 and Sxa2. Homothallic strains, such as the commonly used h90 strain, carry the genetic information for both mating types in a single genome and cells undergo a complex pattern of mating type switching throughout the mitotic lifecycle (reviewed in Ref.3). Multiple isolates of heterothallic fission yeast that rarely or never switch mating type are also commonly used4, most prominently the h+N (P-type) and h-S (M-type) strains.
In fission yeast, entry into the sexual lifecycle is under strict nutritional regulation. Only nitrogen-starved fission yeast cells arrest mitotic reproduction and produce diffusible pheromones to signal presence of a mating partner and promote further steps of the sexual cycle (reviewed in Ref.5). Nitrogen deprivation de-represses the key transcriptional regulator of mating Ste11 that acts as a developmental switch and promotes expression of mating specific genes including the pheromone receptor and the pheromone production genes6,7. Pheromone-receptor engagement activates the receptor-coupled protein G-alpha and downstream MAPK signaling which further enhances Ste11 transcriptional activity8-10, thus increasing pheromone production in a positive feedback between mating partners. Pheromone levels are crucial to induce different cell polarization states by regulating the master organizer of cell polarity, the Rho-family GTPase Cdc4211. Upon exposure to low pheromone concentrations, active Cdc42 is visualized in dynamic patches exploring the cell periphery, and no cell growth is observed at this stage. Increased pheromone levels promote the stabilization of Cdc42 activity to a single zone and growth of a polarized projection, termed the shmoo, which brings partner cells in contact. Subsequently, the two haploid mating partners fuse to form a diploid zygote. Recent work reveals the existence of a novel actin structure essential for fusion that is assembled by the mating-induced formin Fus112. This fusion focus concentrates type-V myosin dependent processes and positions the cell wall degradation machinery, thus allowing remodeling of the cell wall to permit plasma membrane contact without cell lysis12. Upon cell-cell fusion, the nuclei come in contact and undergo karyogamy. A prominent dynein-dependent back-and-forth movement of the nucleus inside the zygote (the horse-tail movement) then promotes the pairing of chromosome homologs13,14, which is followed by meiosis. Finally, the four products of meiosis are packaged into individual spores during sporulation.
Because of its complexity and the numerous steps involved, detailed monitoring of mating has been challenging. Two notable difficulties are that the entire process takes well over fifteen hours and that cells are difficult to synchronize. These difficulties are circumvented by single-cell microscopy approaches. Here a general protocol to investigate the sexual lifecycle in fission yeast is presented. With minor adjustments, this protocol permits the study of all the different steps of the process, namely the induction of mating gene product, cell polarization and pairing between sister-cells after mating type switching and between non-sister partners, cell-cell fusion, and post-fusion horse-tail movement, meiosis and sporulation. This method allows to 1) easily visualize fluorescently tagged proteins over time pre-, during and post-fusion; 2) discriminate the behavior of cells of opposite mating type; and 3) measure and quantify parameters such as shmooing, mating, fusion or sporulation efficiency.
Protocol
Microscopy analysis of fission yeast sexual reproduction
1. Media Preparation
- Prepare Minimum Sporulation medium (MSL-N)15 by mixing the following components: Glucose: 10 g/L, KH2PO4: 1 g/L, NaCl: 0.1 g/L, MgSO4·7H2O: 0.2 g/L. Add Trace elements (10,000x): 100 µl/L, Vitamins (1,000x): 1 ml/L, and 0.1 M CaCl2 ml/L. Filter-sterilize using a 0.22 μm pore size filter and store at room temperature (RT).
- Use the following Vitamins (1,000x) stock: Pantothenate: 1 g/L, Nicotinic Acid: 10 g/L, Inositol: 10 g/L, Biotin: 10 mg/L. Filter-sterilize using a 0.22 μm pore size filter and store at 4 °C.
- Use the following Trace elements (10,000x) stock: Boric Acid: 5 g/L, MnSO4: 4 g/L, ZnSO4·7H2O: 4 g/L, FeCl2·6H2O: 2 g/L, MoO3: 0.4 g/L, KI: 1 g/L, CuSO4·5H2O: 0.4 g/L, Citric Acid: 10 g/L. Filter-sterilize using a 0.22 μm pore size filter and store at 4 °C.
Prepare MSL-N Agarose 2% (used to make agarose pad chambers) by combining 10 ml of MSL-N with 0.2 g of agarose. Melt for ~2 min at high power in a microwave oven until agarose dissolves and aliquot 0.5 ml in microcentrifuge tubes. Store at RT.
Prepare Minimum Sporulation medium with nitrogen (MSL+N) from MSL-N by adding (NH4)2SO4: 1 g/L, Leucine: 0.225 g/L, Adenine: 0.225 g/L, Uracil: 0.225 g/L. Filter-sterilize using a 0.22 µm pore size filter. Store at RT. Note: Leucine, adenine and uracil are added to this medium to allow the growth of auxotrophic strains. Additional amino-acid supplement should be included if the strain used carries a distinct auxotrophy (for instance his3Δ). Please also note that work with fully prototroph strains is recommended, as only such strains will allow high mating efficiency.
Prepare 200 ml VALAP for chamber sealing. In a beaker add equal weights of Lanolin, Vaseline (or other petroleum jelly), and Paraffin. Heat mixture at low temperature and stir occasionally until thoroughly blended. Aliquot the mix into several small petri dishes. Store at RT.
2. Culturing Fission Yeast Strains for Mating Experiments (Figure 1).
- Day 1, Evening: Inoculate freshly streaked strains from solid media into culture tubes containing 3 ml of MSL+N. Use flat-bottom tubes of about 2.5 cm diameter. Use other culture tubes or flasks with adapted culture volume. Incubate overnight (O/N) with shaking at 25 °C, 200 rpm. If working with heterothallic strains, inoculate separately.
- Dilute cell suspensions in media the following morning to ensure that cultures have optical density measured at 600 nm (O.D.600) of 0.4-0.8 in the evening. Note: O.D.600 measurements as a proxy of cell concentration for fission yeast are linear in the range of approximately 0.1-1.0. For our spectrophotometer O.D.600 = 0.1 corresponds to 1.4 x 106 cells per ml. Thus, if initial O.D.600 reads are outside this range samples need to be diluted/concentrated for reliable measurements.
Day 2, Evening: Dilute cells in 20 ml of MSL+N media to O.D.600 = 0.025 in 100 ml flasks. Incubate strains O/N at 30 °C, 200 rpm. Note: Wild-type cell cultures should reach O.D.600 ~0.8 the following morning (15 hr later). If working with strains with longer generation time adjust dilution accordingly.
- Day 3, Morning: Measure O.D.600 to verify that cultures have cell density of O.D.600 ~0.8. If working with heterothallic strains, mix equal numbers of partner cells. Pellet cells at 1,000 x g and transfer to a 1.5 ml tube. Wash cells three times in 1 ml of MSL-N medium. Re-suspend cells in 3 ml of MSL-N medium and dilute cells to O.D.600 = 1.5.
- To monitor mating between sister cells directly mount cells for imaging (see Protocol Section 2.4). Use homothallic h90 strains, in which mating type switching will take place during the mitotic divisions occurring after nitrogen deprivation. Immediate mounting of cells on the agarose pad ensures that, after cell division, sister-cells remain next to each other.
- To monitor cell polarization (exploratory dynamics and shmooing) incubate 1-3 ml of cell culture at 30 °C, 200 rpm for 3-4 hr prior to mounting cells for imaging. Within these 3-4 hr, the last mitotic division will have occurred. A few cells will have initiated polarization, but most will just be starting their exploratory polarization dynamics.
- To monitor cell-cell fusion incubate 1-3 ml of cell culture at 30 °C, 200 rpm for 4-6 hr prior to mounting cells for imaging.
- To monitorpost-fusion events incubate 1-3 ml of cell cultures at 30 °C, 200 rpm for 8 hr prior to mounting cells for imaging.
- To monitor response to a specific pheromone concentration (only for heterothallic strains) incubate 3 ml of cell culture at 30 °C, 200 rpm for 3-4 hr prior to mounting cells for imaging.
- Remove the MSL-N-containing microcentrifuge tube from the 95 °C heat block (see Protocol section 2.4.1. below). Add P-factor or M-factor at the desired concentration directly in the melted MSL-N agarose. Mix well by vortexing before immediate pad preparation.
- After cell mounting, incubate the pad for 15-30 min at 25 °C prior to imaging. P-factor and M-factor pheromones were used from a stock solution of 1 mg/ml in methanol. Note: If working with mutant strains incubation times may vary. Clumping of cells may occur after prolonged incubation in liquid media and may be particularly strong in some mutant strains. Clumped cells cannot be spotted onto agarose pads in a single layer and thus are difficult to autofocus and image. In case of extensive cell clumping mount cells on agarose pads immediately after washes and re-suspension in nitrogen-lacking media (as in the Protocol Section 2.3.1.) and incubate them at 30 °C prior to imaging.
- Mounting Cells for Imaging:
- Prepare MSL-N agarose pads16 by melting an aliquot in a 95 °C heat-block for 10-15 min and adding 200 μl between two glass slides separated by spacers (Figure 1B). Use spacers with a thickness of ~0.5 mm. To make spacers, use strips cut out of cardboard, such as that delivered with restriction enzymes.
- Pellet 100 μl of cells at 1,000 x g for 1 min, remove the supernatant and re-suspend cells in residual 2-4 μl medium. Do NOT re-suspend in fresh medium as it delays mating.
- After 2-3 min carefully remove spacers and the top glass slide. If several strains are to be mounted, prepare the cell concentrates before preparing the agarose pad.
- Add 1 μl of cells to the pad and wait until the drop starts drying (~1 min) before covering with cover slip and sealing with VALAP.
- Let the pad sit for at least 30 min before imaging at 25 °C or RT.
- To obtain a mixture of cells in various stages of mating, make a pad immediately after washes in MSL-N (see Section 2.3.) and incubate at 18 °C O/N (~15 hr). This approach is useful for imaging cells at distinct mating stages with high temporal resolution or samples with weak signal.
3. Live-cell Imaging of Mating Yeast Cells
- Adjust Image Acquisition Settings. Note: Image acquisition settings presented here were optimized for the DeltaVision platform composed of a customized Olympus IX-71 inverted microscope with Plan Apo 60X/1.42 NA or U-Plan Apo 100X/1.4 NA objectives, a CoolSNAP HQ2 camera and an Insight SSI 7 color combined unit illuminator. The hardware is controlled by softWoRx v4.1.2 that also enables digital autofocusing.
- Ensure that the autofocusing intervals do not exceed 15 min since the Z-drift over longer time can surpass the capacity of the software autofocusing system. If using a hardware-based autofocusing system, use larger intervals.
- Adjust imaging interval. Take images every 10 min for ~15 hr as a starting point when working with a fluorescently tagged protein for the first time. This interval provides a good overview of the entire mating process without extensive bleaching.
- Image with 5 min intervals or shorter to more precisely time events such as fusion. Shorter intervals require strong fluorophores enabling low exposure times and little bleaching. For imaging with intervals below 1 min avoid O/N microscopy and instead prepare a mixture of all mating stages as detailed in Protocol Section 2.4.6.
- Adjust image exposure time. Test exposure times between 50 and 300 msec. Aim to strike a balance between the signal-to-noise ratio and photobleaching as to achieve a large number of time-points with a discernable signal (typically over 100).
- Adjust Z-sectioning. Sections every 0.3 µm covering 5 µm provide a good imaging depth. Note that Z-sectioning further increases photo-damage, which can be minimized by using the OAI (optical axis integration - a single sweep acquisition of the total sample depth). Good auto-focus system reduces the need for many Z-sections and is highly recommended.
- Tips to Study Mating Type-specific Behaviors:
- For heterothallic strains, use h- and h+ cells expressing distinct fluorophores, so that the two cell types can be distinguished. Use any genetically encoded fluorophore expressed as a cytosolic version or fusion to an endogenous protein, though we have had good success with sfGFP, a fast-folding variant of GFP17. Endogenous proteins are usually tagged at their endogenous genomic locus by homologous recombination14.
- For homothallic strains, express a genetically encoded cytosolic fluorescent protein under the control of mating type specific promoters such as those of the pheromone receptor genes map3 and mam2 to drive the expression in P and M cells, respectively. The promoters of map3 and mam2 are inactive during vegetative growth and are induced upon nitrogen starvation18,19. Constructs driving the expression of cytosolic GFP or mCherry under the control of these promoters are typically integrated at a genomic locus (ura4, leu1) that does not interfere with the normal progression of the sexual life cycle12.
Use mating type specific cytosolic fluorophores (as in Protocol Section 3.2.2) to determine precise timing of cell fusion visualized as the transfer of fluorescent signal from one partner cell to the other.
4. Quantification of Mating and Fusion Efficiencies
To quantify mating and fusion efficiencies11,12, spot cells directly after MSL-N wash onto MSL-N agarose pads, as in step 2.3.1 above, and leave for 24 hr at 25 °C prior to imaging (Figure 1A). Using this protocol a homothallic wild-type strain mating efficiency reaches ~60% and fusion efficiency 99%. To test whether any reduction in these values observed in mutant strains reflect a terminal phenotype or a delay, repeat the experiment with a 36 hr incubation time.
Calculate mating efficiency as
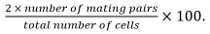 Mating pairs include zygotes, asci, and unfused cell pairs identified from their position and growth towards each others.
Mating pairs include zygotes, asci, and unfused cell pairs identified from their position and growth towards each others.Calculate fusion efficiency as
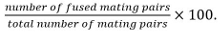 Note: Fused mating pairs are identified by their ability to form spores if it is known that the mutant under study does not influence sporulation. If sporulation cannot be used as read-out, fusion is determined by observing the spreading of a cytosolic marker expressed in only one mating partner into both partners.
Note: Fused mating pairs are identified by their ability to form spores if it is known that the mutant under study does not influence sporulation. If sporulation cannot be used as read-out, fusion is determined by observing the spreading of a cytosolic marker expressed in only one mating partner into both partners.
Representative Results
Fission Yeast Growth and Mating Dynamics Upon Removal of a Nitrogen Source As nitrogen starvation is a prerequisite for initiation of sexual reproduction in fission yeast, wild-type homothallic h90 strain was monitored upon shift from nitrogen-rich to nitrogen-deprived medium (Figure 2), following the protocol outlined in Figure 1. Briefly, cells were grown O/N to exponential phase (O.D.600 = 0.5) in MSL+N medium, collected, washed and re-suspended in MSL-N liquid medium to final O.D.600 = 1.5. Every 2 hr aliquots of cells were calcofluor-stained and imaged (Figure 2A-D).
Calcofluor staining (Figure 2A) revealed that in nitrogen-rich medium ~21% of cells were septating (n >300, Figure 2B) and that average cell length at division was 15.2 ± 1.4 µm (n = 50, Figure 2C). Non-dividing cells showed a broad distribution of cell lengths (8-14 µm; n >200, Figure 2D). However, two hours after the shift to MSL-N medium there was a drastic change in the length distribution of non-dividing cells, with over 60% of cells being shorter than 9 µm (n >200, Figure 2D). Cell septation was also detected at the reduced length of 9.8 ± 0.8 µm (n = 50, Figure 2A, 2C). A further reduction in length of both non-dividing and dividing cells was also observed, as time in MSL-N increased. Indeed, after 8 hr the septation index of the culture decreased below 2% (n >300) and over 85% of cells arrested their cell cycle at lengths shorter than 7 µm (n >200, Figure 2D).
Cells started to form mating pairs four hours after the shift to liquid MSL-N medium (<1%, Figure 2A). Subsequently the number of mating pairs rapidly increased and 8 hr after the shift 10% of cells engaged a mating partner (n >200, Figure 2B).
To monitor mating dynamics, 6 hr after starvation induction, an aliquot of cells was also mounted onto MSL-N agarose pad for imaging. Cells underwent shmooing, fusion and sporulation (Figure 2E, 2F) in the subsequent 21 hr without any apparent synchrony (Movie S1). The mating efficiency depended on the relative position and density of cells in a given field of view, with over 60% of cells engaged a partner when positioned densely in a monolayer (Figure 2G and Movie S1).
The wild-type heterothallic h+ and h- fission yeast strains were also subjected to the same protocol with an additional step of mixing h+ and h- cells in a one-to-one ratio at the time of nitrogen removal. Similar starvation and mating dynamics were observed, but the mating efficiency was slightly lower (Figure 2G, Movie S2).
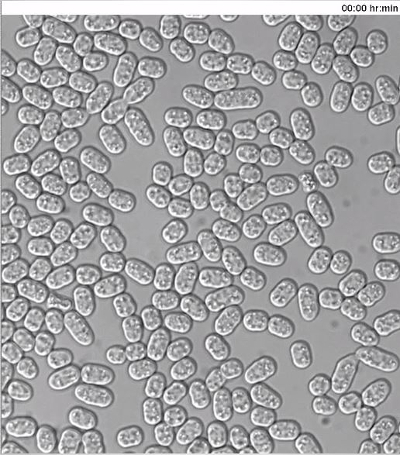
Monitoring Dynamics of Fluorescently Tagged Proteins in Mating Fission Yeast Cells To monitor the dynamics of type-V myosin Myo52 (Ref20) in mating cells, exponentially grown h- cells expressing Myo52-3GFP from the native locus were mixed with h+ cells similarly expressing Myo52-tdTomato, as well as cytosolic GFP from the P-cell specific map3 promoter. The cell suspension was washed, diluted in MSL-N medium and incubated for 6 hr at 30 ºC, 200 rpm prior to mounting cells onto MSL-N agarose pads for O/N imaging at 10 min intervals.
As previously reported11,12 Myo52 initially formed dynamic zones throughout the cell cortex (Figure 3A, arrowheads and Movie S3). Myo52 signal was then stabilized (Figure 3A, arrows and Movie S3) in a single focus just before the fusion event, which was visualized by the transfer of the cytosolic GFP signal from the h+ into the h- cell (Figure 3A and Movie S3). The Myo52-tdTomato signal could also be observed at the fusion neck during its expansion but no discernable signal was evident as the zygote proceeded to sporulation (Figure 3A and Movie S3).
Monitoring the Behavior of Heterothallic Fission Yeast Cells Exposed to External Pheromones Heterothallic h- cells lacking the Sxa2 protease that degrades P-factor for desensitization readily respond to synthetic P-factor. An h-sxa2Δ strain, expressing Scd2-GFP as a marker for active Cdc4211, was grown in MSL+N medium, accordingly to the described protocol. Cells were washed in MSL-N medium and incubated for 4 hr at 30 ºC. MSL-N agarose pads containing methanol (data not shown), 0.1 μg/ml (low pheromone, Figure 3B) or 1 μg/ml (high pheromone, Figure 3C) of P-factor were prepared, cut and positioned on a new slide to generate mini-pads containing different pheromone amounts. 0.5 μl of cell suspension was mounted on each mini-pads for O/N imaging at 5 min intervals.
In concordance with published results11, low P-factor levels promoted the formation of dynamic Scd2 zones without growth (Figure 3B, Movie S4), while high levels of P-factor stabilized a single Scd2 zone, thus inducing shmoo elongation from one cell pole (Figure 3C, Movie S5).
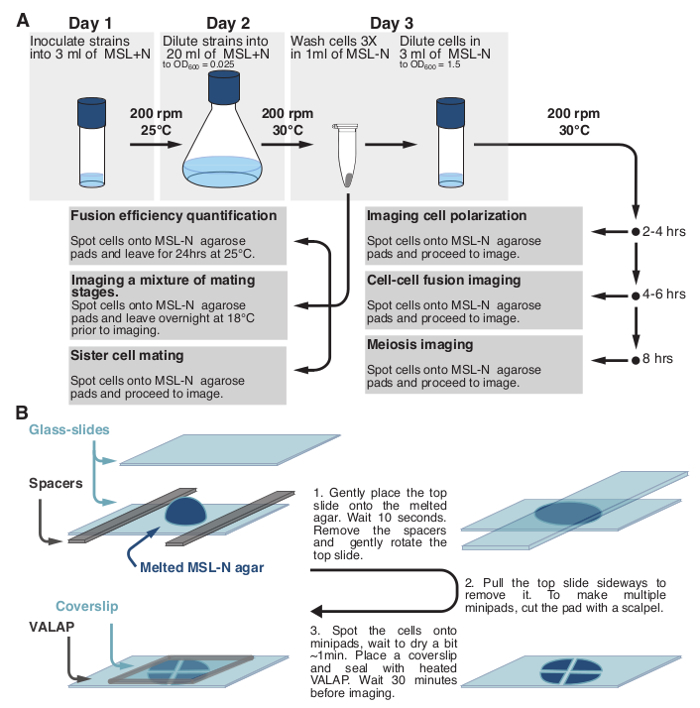 Figure 1: Schematic Representation of the Protocol. (A) Schematic description of the protocols used to monitor fission yeast sexual lifecycle and (B) to prepare fission yeast samples for long-term imaging. Please click here to view a larger version of this figure.
Figure 1: Schematic Representation of the Protocol. (A) Schematic description of the protocols used to monitor fission yeast sexual lifecycle and (B) to prepare fission yeast samples for long-term imaging. Please click here to view a larger version of this figure.
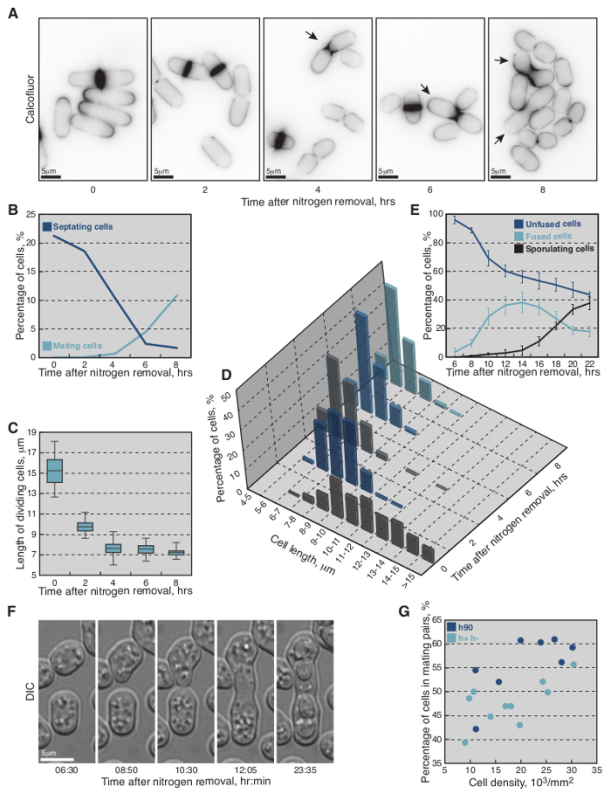 Figure 2: Growth and Mating Dynamics of Fission Yeast Cells Shifted from MSL+N to MSL-N Media. (A) Epifluorescence micrographs of calcofluor-stained cells shifted to MSL-N media for the indicated periods of time. (B) Percent of septating (dark-blue line, n >300 per timepoint) and mating (light-blue line, n >300 per timepoint) cells shifted to MSL-N media for the indicated periods of time. (C) Average length of septating cells shifted to MSL-N media for the indicated periods of time (n = 50 per timepoint except 8 hr timepoint where n = 20). (D) Length distribution of non-dividing cells shifted to MSL-N medium for the indicated periods of time (n >200 per timepoint). (E) Average fraction of unfused (dark-blue line), fused (light-blue line) and sporulating (black line) cells in a population of h90 wild-type yeast shifted to MSL-N media for the indicated periods of time and mounted onto an agar pad at timepoint 6 hr (n >900 cells per timepoint from three different timelapses). The cells were considered fused when no refractive cell wall between partners was visible in the DIC micrographs and formation of spore cell wall was used to score for sporulating cells. (F) DIC micrographs of mating cells shifted to MSL-N media for the indicated periods of time. (G) Mating efficiency as a function of cell density on agarose pads for h90 (dark-blue dots) and h+/h- (light-blue dots) cells shifted to MSL-N media for 24 hr. The most suitable density of cells for long-term imaging is achieved at approximately 25,000 cells/mm2. Please click here to view a larger version of this figure.
Figure 2: Growth and Mating Dynamics of Fission Yeast Cells Shifted from MSL+N to MSL-N Media. (A) Epifluorescence micrographs of calcofluor-stained cells shifted to MSL-N media for the indicated periods of time. (B) Percent of septating (dark-blue line, n >300 per timepoint) and mating (light-blue line, n >300 per timepoint) cells shifted to MSL-N media for the indicated periods of time. (C) Average length of septating cells shifted to MSL-N media for the indicated periods of time (n = 50 per timepoint except 8 hr timepoint where n = 20). (D) Length distribution of non-dividing cells shifted to MSL-N medium for the indicated periods of time (n >200 per timepoint). (E) Average fraction of unfused (dark-blue line), fused (light-blue line) and sporulating (black line) cells in a population of h90 wild-type yeast shifted to MSL-N media for the indicated periods of time and mounted onto an agar pad at timepoint 6 hr (n >900 cells per timepoint from three different timelapses). The cells were considered fused when no refractive cell wall between partners was visible in the DIC micrographs and formation of spore cell wall was used to score for sporulating cells. (F) DIC micrographs of mating cells shifted to MSL-N media for the indicated periods of time. (G) Mating efficiency as a function of cell density on agarose pads for h90 (dark-blue dots) and h+/h- (light-blue dots) cells shifted to MSL-N media for 24 hr. The most suitable density of cells for long-term imaging is achieved at approximately 25,000 cells/mm2. Please click here to view a larger version of this figure.
 Figure 3: Dynamic Localization of Fluorescent Proteins during Fission Yeast Mating. (A) Deconvolved single z-plane epifluorescence micrographs of cells with the indicated genotypes shifted from MSL+N to MSL-N media for 6 hr and mounted onto MSL-N agarose pad for the indicated time. Arrowheads indicate dynamic Myo52-3GFP zones and the arrow points out Myo52 localization at the fusion focus. (B), (C) Deconvolved single z-plane epifluorescence micrographs of cells with indicated genotypes shifted from MSL+N to MSL-N media for 4 hr and mounted on MSL-N agarose mini-pads containing 0.1 μg/ml (B) or 1 μg/ml (C) of P-factor for the indicated time. Arrowheads indicate dynamic (B) or stable (C) Scd2-GFP zones. Please click here to view a larger version of this figure.
Figure 3: Dynamic Localization of Fluorescent Proteins during Fission Yeast Mating. (A) Deconvolved single z-plane epifluorescence micrographs of cells with the indicated genotypes shifted from MSL+N to MSL-N media for 6 hr and mounted onto MSL-N agarose pad for the indicated time. Arrowheads indicate dynamic Myo52-3GFP zones and the arrow points out Myo52 localization at the fusion focus. (B), (C) Deconvolved single z-plane epifluorescence micrographs of cells with indicated genotypes shifted from MSL+N to MSL-N media for 4 hr and mounted on MSL-N agarose mini-pads containing 0.1 μg/ml (B) or 1 μg/ml (C) of P-factor for the indicated time. Arrowheads indicate dynamic (B) or stable (C) Scd2-GFP zones. Please click here to view a larger version of this figure.
 Supplemental Movie 1:
DIC timelapse of wild-type h90 fission yeast cells that were nitrogen-starved for 6 hr prior to imaging.
(Right click to download).
Supplemental Movie 1:
DIC timelapse of wild-type h90 fission yeast cells that were nitrogen-starved for 6 hr prior to imaging.
(Right click to download).
 Supplemental Movie 2:
DIC timelapse of wild-type h+ and h- fission yeast cells that were mixed and nitrogen-starved for 6 hr prior to imaging.
(Right click to download).
Supplemental Movie 2:
DIC timelapse of wild-type h+ and h- fission yeast cells that were mixed and nitrogen-starved for 6 hr prior to imaging.
(Right click to download).
 Supplemental Movie 3: Deconvolved single z-plane epifluorescence and DIC timelapse of h- cells expressing Myo52-3GFP from the native locus mixed with h+ cells expressing Myo52-tdTomato from the native locus and cytosolic GFP from the P-cell specific map3 promoter. Cells were grown in MSL-N medium for 6 hr at 30 ºC prior to imaging. Transfer of cytosolic GFP into the h- cell defines the fusion time. (Right click to download).
Supplemental Movie 3: Deconvolved single z-plane epifluorescence and DIC timelapse of h- cells expressing Myo52-3GFP from the native locus mixed with h+ cells expressing Myo52-tdTomato from the native locus and cytosolic GFP from the P-cell specific map3 promoter. Cells were grown in MSL-N medium for 6 hr at 30 ºC prior to imaging. Transfer of cytosolic GFP into the h- cell defines the fusion time. (Right click to download).
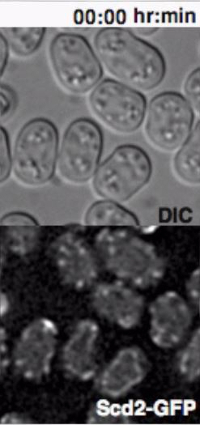 Supplemental Movie 4: Deconvolved single z-plane epifluorescence and DIC timelapse of h-sxa2 cells expressing Scd2-GFP from its native promoter treated with 0.1 μg/ml P-factor. Cells were grown in MSL-N medium for 4 hr at 30 ºC before imaging. (Right click to download).
Supplemental Movie 4: Deconvolved single z-plane epifluorescence and DIC timelapse of h-sxa2 cells expressing Scd2-GFP from its native promoter treated with 0.1 μg/ml P-factor. Cells were grown in MSL-N medium for 4 hr at 30 ºC before imaging. (Right click to download).
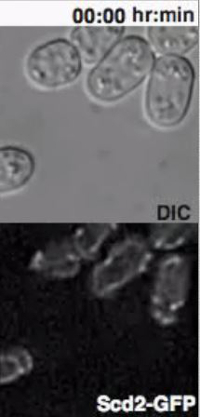 Supplemental Movie 5: Deconvolved single z-plane epifluorescence and DIC timelapse of h-sxa2 cells expressing Scd2-GFP from its native promoter treated with 1 μg/ml P-factor. Cells were grown in MSL-N medium for 4 hr at 30 ºC before imaging. (Right click to download).
Supplemental Movie 5: Deconvolved single z-plane epifluorescence and DIC timelapse of h-sxa2 cells expressing Scd2-GFP from its native promoter treated with 1 μg/ml P-factor. Cells were grown in MSL-N medium for 4 hr at 30 ºC before imaging. (Right click to download).
| YSM995 | h- wt |
| YSM1371 | h+ wt |
| YSM 1396 | h90 wt |
| YSM2534 | h- myo52-3GFP::kanMX |
| YSM2730 | h+ myo52-tdTomato::natMX Pmap3:GFP::ura4+@ura4locus |
| YSM2731 | h- scd2-GFP::natMX sxa2∆::kanMX |
Table S1: Strains Used in This Study.
Discussion
Environmental conditions, and nutrient availability in particular, strongly affect the fission yeast physiology. Nitrogen starvation is necessary for commitment to the sexual reproduction and initially leads to striking changes in the mitotic cell cycle progression (Ref.21 and Figure 2). Upon nitrogen removal from exponentially growing population, cell size at division rapidly decreases (Figure 2C) and the majority of cells arrest mitotic progression shorter than the length of newly born exponentially grown cells (Ref.21 and Figure 2D). As cells of opposite mating types arrest mitotic progression they engage mating partners and proceed to form zygotes and sporulate (Figure 2B, 2E, 2F). In the case of a two-dimensional monolayer of wildtype cells, over sixty percent of h90 cells will engage a partner, and almost all will undergo cell fusion (Figure 2G). The mating efficiency is slightly lower in the mixture of heterothallic strains and depends on the density of cells and their relative position (Figure 2G). One of the most critical aspects of the protocol to obtain high mating efficiencies is to ensure that cells are kept within the indicated densities throughout. Monitoring of cell size and mating efficiency parameters as shown in Figure 2 then provides a simple control that cells are entering sexual reproduction efficiently.
We note that, since mating medium lack any nitrogen source (even low amounts of amino-acids and nitrogen bases are excluded), high mating efficiencies such as those presented in Figure 2E and 2G are only obtained with fully prototrophic strains. Auxotrophic strains can be used, but require additional care to ensure that cells are at all times kept within cell densities stated in the protocol. Even with care, only lower mating efficiencies will be observed. Mating efficiency is also influenced by temperature, with lower efficiencies observed at elevated temperature, which may limit the use of temperature-sensitive mutant alleles during the mating process.
The method presented here is primarily used for long-term imaging of fluorescent reporters. Because imaging is performed over long time period, successful imaging of fission yeast mating is highly dependent on the microscopy setup available. For this, the stability of the microscope set-up, its sensitivity and access to an autofocus system are critical. Either software-based or built-in hardware autofocus systems are possible. In addition, to increase throughput, a motorized stage enabling multipoint acquisition is another important quality.
Imaging quality is limited by the properties of the fluorophore of interest. The distinct incubation times in liquid MSL-N medium detailed in Protocol Section 2.3 are aimed at optimizing fluorescence on pheromone-induced tagged proteins and minimizing unnecessary bleaching by imaging only mating stages of interest. While abundant or highly focalized fluorescently tagged proteins allow acquisition of hundreds of time-points over the course of the entire sexual lifecycle (Movie S3), other proteins are challenging to image over such long times due to photo-bleaching. Image quality also depends on the chosen fluorophore, typically a GFP derivative. We repeatedly observed that sfGFP17 (superfolder GFP), which folds with rapid kinetics, provided superior signal during the mating process, possibly because strong autophagy induces rapid protein turnover. Importantly, we did not monitor the availability of oxygen necessary for fluorescent protein maturation17,22. Thus, it is recommended to be cautious in using fluorescence measurements to quantify the levels of proteins expressed after cells are mounted onto slides, or to use alternative oxygen permeable microfluidics devices to conduct such experiments. In addition, agarose pads spotted with cells in the evening and kept at 18 ºC O/N have been very useful to image weak reporters as such pads contain a mixture of cells at all stages of sexual reproduction.
Mating in fission yeast does not exhibit synchrony and cells can be readily seen initiating shmooing alongside other cells already sporulating (Movies S1, S2). Such population dynamics renders classical, bulk biochemical techniques difficult to use, making single cell microscopy described here a method of choice. All microscopy-based approaches can in principle be applied to cells prepared with the protocol described here. Importantly, these approaches can monitor processes that exhibit mating type-specific dynamics such as cell-cell fusion described by Dudin and colleagues12. To monitor asymmetry, mating type reporters have been particularly useful (Figure 3A). Differentiating between the two mating types is easily achieved when working with heterothallic strains by employing distinct fluorophores to tag proteins in the h- and h+ cells. However, this is not feasible for homothallic strains. An approach developed to differentiate the mating type of h90 cells is to express cytosolic fluorescent proteins under the control of mating type specific promoters. In particular, the promoters of the pheromone receptor genes map3 and mam2 were used to drive the expression of GFP or mCherry proteins in P and M cells respectively. Furthermore, these markers allowed precise timing of the cell-cell fusion, visualized as the transfer of fluorescent signal from one partner cell to the other.
Mating pheromones are crucial determinants for progressing through distinct stages of sexual reproduction in yeast11,23. Different levels of synthetic pheromone lead to distinct cell polarization states in fission yeast (Figure 3B, 3C and Ref.11). The protocol included above allows evaluating how distinct exogenous pheromone levels affect cell physiology. Because the pheromone protease Sxa2 rapidly degrades P-factor, a heterothallic mutant strain where the protease was deleted was used to obtain reproducible results. However, in mixtures of cells of opposite mating types, not only the levels but also the spatial distribution of pheromones emitted by a mating partner are likely to be important. Analyzing the effects of pheromone spatial distribution on cell response will require development of more sophisticated setups such as microfluidics devices employed on budding yeast24,25.
In summary, we present a basic method for long-term microscopy of the sexual lifecycle of fission yeast. Minor adjustments to this protocol allow research focus on cellular responses at distinct stages of the sexual lifecycle. Combining this approach with single cell biochemistry tools and microfluidics devices will further promote fission yeast sexual reproduction as a powerful model system.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
AV was supported by an EMBO long-term postdoctoral fellowship. Research in the Martin lab is funded by an ERC Starting grant (GeometryCellCycle) and a Swiss National Science Foundation grant (31003A_155944) to SGM.
References
- Mata J, Bahler J. Global roles of Ste11p, cell type, and pheromone in the control of gene expression during early sexual differentiation in fission yeast. Proc Natl Acad Sci U S A. 2006;103(42):15517–15522. doi: 10.1073/pnas.0603403103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue-Franzen Y, Kjaerulff S, Holmberg C, Wright A, Nielsen O. Genomewide identification of pheromone-targeted transcription in fission yeast. BMC Genomics. 2006;7:303. doi: 10.1186/1471-2164-7-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar AJ. Lessons learned from studies of fission yeast mating-type switching and silencing. Annu Rev Genet. 2007;41:213–236. doi: 10.1146/annurev.genet.39.073103.094316. [DOI] [PubMed] [Google Scholar]
- Beach DH, Klar AJ. Rearrangements of the transposable mating-type cassettes of fission yeast. EMBO J. 1984;3(3):603–610. doi: 10.1002/j.1460-2075.1984.tb01855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini L, Dudin O, Martin SG. Mate and fuse: how yeast cells do it. Open Biol. 2013;3(3):130008. doi: 10.1098/rsob.130008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki N, Yamamoto M. Reduction in the intracellular cAMP level triggers initiation of sexual development in fission yeast. Mol Gen Genet. 1992;233(1-2):17–24. doi: 10.1007/BF00587556. [DOI] [PubMed] [Google Scholar]
- Sugimoto A, Iino Y, Maeda T, Watanabe Y, Yamamoto M. Schizosaccharomyces pombe ste11+ encodes a transcription factor with an HMG motif that is a critical regulator of sexual development. Genes Dev. 1991;5(11):1990–1999. doi: 10.1101/gad.5.11.1990. [DOI] [PubMed] [Google Scholar]
- Kjaerulff S, Lautrup-Larsen I, Truelsen S, Pedersen M, Nielsen O. Constitutive activation of the fission yeast pheromone-responsive pathway induces ectopic meiosis and reveals ste11 as a mitogen-activated protein kinase target. Mol Cell Biol. 2005;25(5):2045–2059. doi: 10.1128/MCB.25.5.2045-2059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara T, Nakafuku M, Yamamoto M, Kaziro Y. Isolation and characterization of a gene encoding a G-protein alpha subunit from Schizosaccharomyces pombe: involvement in mating and sporulation pathways. Proc Natl Acad Sci U S A. 1991;88(13):5877–5881. doi: 10.1073/pnas.88.13.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Shimanuki M, Yanagida M. Fission yeast genes that confer resistance to staurosporine encode an AP-1-like transcription factor and a protein kinase related to the mammalian ERK1/MAP2 and budding yeast FUS3 and KSS1 kinases. Genes Dev. 1991;5(1):60–73. doi: 10.1101/gad.5.1.60. [DOI] [PubMed] [Google Scholar]
- Bendezu FO, Martin SG. Cdc42 explores the cell periphery for mate selection in fission yeast. Curr Biol. 2013;23(1):42–47. doi: 10.1016/j.cub.2012.10.042. [DOI] [PubMed] [Google Scholar]
- Dudin O, et al. A formin-nucleated actin aster concentrates cell wall hydrolases for cell fusion in fission yeast. J Cell Biol. 2015;208(7):897–911. doi: 10.1083/jcb.201411124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikashige Y, et al. Telomere-led premeiotic chromosome movement in fission yeast. Science. 1994;264(5156):270–273. doi: 10.1126/science.8146661. [DOI] [PubMed] [Google Scholar]
- Bahler J, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14(10):943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Egel R, Willer M, Kjaerulff S, Davey J, Nielsen O. Assessment of pheromone production and response in fission yeast by a halo test of induced sporulation. Yeast. 1994;10(10):1347–1354. doi: 10.1002/yea.320101012. [DOI] [PubMed] [Google Scholar]
- Tran PT, Marsh L, Doye V, Inoue S, Chang F. A mechanism for nuclear positioning in fission yeast based on microtubule pushing. J Cell Biol. 2001;153(2):397–411. doi: 10.1083/jcb.153.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Shimoda C. The Schizosaccharomyces pombe mam2 gene encodes a putative pheromone receptor which has a significant homology with the Saccharomyces cerevisiae Ste2 protein. EMBO J. 1991;10(12):3743–3751. doi: 10.1002/j.1460-2075.1991.tb04943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Davey J, Imai Y, Yamamoto M. Schizosaccharomyces pombe map3+ encodes the putative M-factor receptor. Mol Cell Biol. 1993;13(1):80–88. doi: 10.1128/mcb.13.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi F, Arai R, Mabuchi I. Identification of two type V myosins in fission yeast, one of which functions in polarized cell growth and moves rapidly in the cell. Mol Biol Cell. 2001;12(5):1367–1380. doi: 10.1091/mbc.12.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajiki K, Pluskal T, Shimanuki M, Yanagida M. Metabolomic analysis of fission yeast at the onset of nitrogen starvation. Metabolites. 2013;3(4):1118–1129. doi: 10.3390/metabo3041118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Nagai T, Mizuno H. Mechanisms of protein fluorophore formation and engineering. Curr Opin Chem Biol. 2003;7(5):557–562. doi: 10.1016/s1367-5931(03)00097-8. [DOI] [PubMed] [Google Scholar]
- Dyer JM, et al. Tracking shallow chemical gradients by actin-driven wandering of the polarization site. Curr Biol. 2013;23(1):32–41. doi: 10.1016/j.cub.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beta C, Bodenschatz E. Microfluidic tools for quantitative studies of eukaryotic chemotaxis. Eur J Cell Biol. 2011;90(10):811–816. doi: 10.1016/j.ejcb.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Lee SS, et al. Quantitative and dynamic assay of single cell chemotaxis. Integr Biol (Camb) 2012;4(4):381–390. doi: 10.1039/c2ib00144f. [DOI] [PubMed] [Google Scholar]