

Abstract
In this work, we validate and analyze the results of previously published cross docking experiments and classify failed dockings based on the conformational changes observed in the receptors. We show that a majority of failed experiments (i.e. 25 out of 33, involving four different receptors: cAPK, CDK2, Ricin and HIVp) are due to conformational changes in side chains near the active site. For these cases, we identify the side chains to be made flexible during docking calculation by superimposing receptors and analyzing steric overlap between various ligands and receptor side chains. We demonstrate that allowing these side chains to assume rotameric conformations enables the successful cross docking of 19 complexes (ligand all atom RMSD < 2.0 Å) using our docking software FLIPDock. The number of side receptor side chains interacting with a ligand can vary according to the ligand's size and shape. Hence, when starting from a complex with a particular ligand one might have to extend the region of potential interacting side chains beyond the ones interacting with the known ligand. We discuss distance-based methods for selecting additional side chains in the neighborhood of the known active site. We show that while using the molecular surface to grow the neighborhood is more efficient than Euclidian-distance selection, the number of side chains selected by these methods often remains too large and additional methods for reducing their count are needed. Despite these difficulties, using geometric constraints obtained from the network of bonded and non-bonded interactions to rank residues and allowing the top ranked side chains to be flexible during docking makes 22 out of 25 complexes successful.
Keywords: Induce fit, Docking, Side chain, Flexibility, cAPK, CDK2, Ricin, HIV protease
Introduction
The rapidly growing number of available three-dimensional structures for biologically and pharmacologically important macromolecules greatly facilitates many areas of research in structure-based molecular design. Macromolecules are known to undergo conformational changes when binding various ligands. This pliability can range from the rearrangement of amino acid side chains to backbone flexibility of loops near the active site, large scale motions such as domains moving relative to each other or even partial unfolding/re-folding of the structure. Although molecular docking is becoming an established method for drug lead discovery and optimization, most modern automated docking procedures still hold the receptor rigid because the representation of its flexibility in a brute force method (i.e. allowing each atom to move) is computationally expensive. The incorporation of the receptor flexibility in the automated docking process still remains one of the main challenges for the computational prediction of ligand–receptor interactions and protein–protein interactions [1].
FLIPDock (Flexible Ligand–Protein Docking) [2], our program for docking flexible ligands in to flexible receptors, was designed to address this shortcoming. We first developed the Flexibility Tree (FT) data structure to selectively encode the conformational sub-spaces [3] of macromolecules in a hierarchical and multi-resolution manner. A FT is built by recursively partitioning a molecule or molecular system into molecular fragments moving relative to each other. Motion descriptors (hinge, shear, twist, screw, etc.) can be assigned to any such fragment in order to describe this fragment's motion. Additional motion descriptors (rotameric side chains, normal modes, essential dynamics, etc.) can be used to describe conformational changes occurring within a fragment. The conformational subspace encoded by a FT can be designed to span a range of conformations known to be important for the biological activity of a protein. A variety of motions can be combined, ranging from domain moving as rigid bodies or backbone atoms undergoing normal mode-based deformations, to side chains assuming rotameric conformations. In addition, these conformational subspaces are parameterized by a small number of variables which can be optimized during the docking process, thus effectively modeling the conformational changes in a flexible receptor during a docking simulation.
FLIPDock uses two FTs to represent the conformational spaces of a receptor and a ligand molecule, respectively. Randomizing FT motion variables yields a putative docking, i.e. a random conformation of both molecules (within the conformational spaces encoded by the FTs) and a random position and orientation of the ligand molecule relative to the receptor. Hence, the docking problem corresponds to the optimization of the FT motion variables for a given objective function. FLIPDock was designed to support multiple search engines and multiple scoring functions. In this work we used a Genetic Algorithm (GA) to search the solution space and scoring function based on the AutoDock 3.05 force field [4].
FLIPDock's ability to deal with numerous flexible receptor side chains is in and of itself valuable as it has been observed that most induced fit motion, especially in the catalytic residues, is accounted for by sidechain flexibility [5, 6]. One recent study on ligand induced changes in eight binding sites suggests that the structure of active site is generally preserved, small sidechain motions are sufficient to accommodate 60 different ligands [7]. Several methods for incorporating sidechain flexibility in protein–ligand docking have been reported. Nested torsions were used to represent flexible amino acids [8]. In this approach each single bond in the sidechain can rotate freely and the corresponding torsion angle can be represented by a “gene” when using GA based optimizations techniques. However, the number of genes that GA optimization can handle limits this approach to a small number of flexible side chains in practice. An alternative is to employ a rotamer library that contains a set of energetically favored conformations [9–11]. This approach makes it possible to include multiple flexible sidechains into GA optimization since only one “gene” is need for each side chain, i.e. the rotamer index in the library.
In previous studies [9–11], the choices of the flexible sidechains near the active site were based on intuition or experimental observations. Furthermore, these methods do not search for the “feasible” rotamer combination during the docking process. Instead, an ensemble of low energy conformers of the receptor were generated and used as rigid receptors in the docking programs. In a previous study, we predicted eight flexible side chains from a single HIV protease complex with no a priori knowledge [2]. We then used our FLIPDock software to cross dock 20 HIV protease inhibitors into alternative receptor conformations. The success rate of such cross docking was improved from 73%, assuming the rigid receptors, to 93% by allowing eight side chains to be flexible in the docking process, as described in [2]. The geometric constraint based method successfully identified the two Arg8 side chains to be the most flexible ones near the active site of HIV-1 protease, supported by experimental observation [12] and computational tests [13].
This manuscript is organized as follows: We first identified a list of difficult protein–ligand docking cases using cross docking experiments. We then identify cases where side chain flexibility is crucial for successful docking. Allowing these side chains to be flexible in FLIPDock dramatically increases the success rate of automated docking calculations. We discuss distance-based methods for progressively including more side chains around a known active site and a geometric-constraints-based method for ranking these residues in order to assist users in the selection of receptor side-chains to be made flexible.
Materials and methods
Compiling the challenging docking cases
In order to build a set of challenging docking cases we started with the data set that Huang and Zou [14] used to validate their ensemble docking program. We repeated the cross docking experiment on this data set using our docking program FLIPDock using rigid receptors. We found 33 cases for which both the modified version of DOCK used in [14] and FLIPDock failed to reproduce the expected ligand pose, i.e., RMSD (root mean square deviations) <2.5 Å. In order to identify potential reasons for docking failure we performed an overlap analysis and further classified these 33 complexes into three groups based on the type of overlaps (i.e. with receptor side-chains, with receptor backbone atoms, or no severe clash). We found that 25 of these 33 complexes had severe clashes with receptor side chain atoms and used these 25 complexes in our flexible side chain docking experiments.
The data set used by Huang and Zou [14] contains 10 ensembles of structures, comprising 105 crystal structures and 87 ligands where an ensemble of structures is a collection of conformations of a given receptor with different ligands bound. In [14] the HIV protease complexes were split manually into two ensembles based on ligand types (HIVpa: non-peptide inhibitors such as cyclic urea, cyclic sulfamide and cyclic cyanoguanidines; HIVpb: substrate-based inhibitors, with structural water present in the active site). In our cross docking we combined these two ensembles since they involve the same receptor.
Cross docking study and the overlap analysis mentioned above requires building ligand–receptor complexes for each receptor conformation with every non-native ligand, within an ensemble. This was done by superimpositions of the receptor conformations using Cα atoms. The PDB codes for structure used as a reference for superimposition are: 1P38 for P38 ensemble; 1HXW for HIVp; 1RA3 for DHFR, 3LCK for LCK, 1IFT for Ricin; 1TPP for Trypsin; 1AHC for aMMC; 1HCL for CDK2 and 1BX6 for cAPK. The transformation used to superimpose each receptor to the reference structure was also applied to this receptor's native ligand, thus defining an “expected pose” for every ligand relative to every receptor in the ensemble.
When docking the ligands into the rigid receptors using FLIPDock, we identified, out of 10 FLIPDock runs, the solution with the best RMSD from the expected pose. The reason we did not chose the predictions with the best score is that in this work we focus on the effects of various induced fit rather than the performance of FLIPDock's scoring function. A docking complex was considered as “challenging” when: (1) none of the 10 FLIPDock dockings (using a rigid receptor) resulted in a predicted pose of the ligand close enough to the expected pose (i.e. an RMSD < 2.5 Å); and (2) the same complex was known as a difficult case by [14, S.-Y. Huang and X. Zou, pers. Commun.].
The steric overlap analysis was performed as follows. Using the expected poses, steric overlaps were identified in the 33 “challenging” complexes, where both FLIPDock and modified version of Dock failed to produce a good result. Note that the expected ligand pose was transformed from the experimentally observed position by superimposing receptor atoms. Therefore some bias is introduced from the selection of superimposition atoms (Cα or backbone atoms) and the choice of reference conformation. When the distance between a ligand atom and a receptor is slightly less than the sum of atomic radii, it could be introduced by this bias. Here we defined a steric overlap as a distance of 1.0 Å or less between a ligand atom and a receptor atom to capture severe overlaps. The 33 complexes were further classified into three groups based on the type of overlaps detected. The first group contains the complexes that have steric overlap between atoms in the ligand and only the side chain atoms. The second group contains the cases where there are no severe bad contact between the ligand and the receptor. Finally, the third group contains the complexes with steric overlap between the ligand and at least one receptor backbone atoms. In this work, we focus on the first group to study the induced fit problem at the receptor side chain level. For the first group, the receptor side chains that overlap with the ligand were identified and we will refer to these side chains as the “critical side chains” as they are required to be made flexible in a docking calculation.
Docking with sidechain flexibility
In this work we use a rotamer library to represent the set of conformations a receptor side-chain is able to adopt. FLIPDock uses genetic algorithm to optimize the ligand position, orientation, internal torsions of the ligand, as well as the rotamer indices for each moving sidechain. The parameters for the genetic algorithm used for this work were set as follows: population size = 200, replacement rate = 80%, mutation rate = 30%, crossover probability = 50%. The stopping criteria were: deviation of scores in the last generation less than 0.0001, number of energy evaluation more than 1,000,000, or 100 generations, whichever is met first. A docking is considered successful if the RMSD of all ligand atoms is less than 2.0 Å (2.5 Å is used when compiling the challenging docking cases [14]).
Results and discussions
Seeking challenging docking cases
Tables 1–3 list the cases for which both FLIPDock and the modified version of Dock used in [14] failed to produce a successful docking. Table 1 contains the cases where the only steric overlaps occur between ligand atoms and side chains atoms of the receptor. These are the docking cases we used to test FLIPDock's ability to successfully dock ligands when multiple receptor side chains are made flexible. Table 2 lists the cases where ligand atoms overlap with at least one receptor backbone atom. These cases were discarded because modeling backbone flexibility is beyond the scope of this paper. Table 3 lists the cases where the ligand has no severe overlap with the receptor (atomic distance <1.0 Å), yet no solution near the expected pose was found. It is possible that the 1.0 Å distance cutoff is too restrictive therefore some overlaps were not captured. A number of other reasons can also lead to the failure in these cases, including: a lack of accuracy in the scoring functions and/or an ineffective search. These problems are also beyond the scope of this paper hence were dismissed as well.
Table 1.
Challenging docking cases: after superimposition, the receptor side chains have severe clashes with the ligand atoms
| Protein | Ligand | Receptor | Sidechain |
|---|---|---|---|
| CAPK | 1STC | 1BKX | 1BKX:A:PHE327 |
| 1BX6 | 1BKX | 1BKX:A:PHE54 | |
| 1STC | 1YDT | 1YDT:E:PHE327 | |
| CDK2 | 1FVV | 1JSV | 1JSV:A:LYS33 |
| 1DI8 | 1JSV | 1JSV:A:LYS33 | |
| 1E9H | 1JSV | 1JSV:A:LYS33 | |
| 1FVV | 1H1P | 1H1P:A:LYS89 | |
| 1FVV | 1E1X | 1E1X:A:LYS33 | |
| 1FVT | 1JSV | 1JSV:A:LYS33 | |
| 1FVT | 1E1X | 1E1X:A:LYS33 | |
| 1FVT | 1E9H | 1E9H:A:LYS89 | |
| 1JSV | 1H1P | 1H1P:A:LYS89 | |
| 1DM2 | 1JSV | 1JSV:A:LYS33 | |
| 1DI8 | 1E1X | 1E1X:A:LYS33 | |
| Ricin | 1FMP | 1OBT | 1OBT:_:TYR80 |
| 1IFU | 1OBT | 1OBT:_:TYR80 | |
| 1OBT | 1IFU | 1IFU:_:TYR80 | |
| 1OBT | 1IFS | 1IFS:_:TYR80 | |
| 1IFS | 1OBT | 1OBT:_:TYR80 | |
| 1APG | 1OBT | 1OBT:_:TYR80 | |
| 1OBT | 1FMP | 1FMP:_:TYR80 | |
| HIVp | 1C70 | 2UPJ | 2UPJ:B:PRO81 |
| 1HTF | 1G35 | 1G35:B:ARG8 | |
| 1HTF | 1G2K | 1G2K:B:ARG8 | |
| 1HTF | 1AAQ | 1AAQ:B:ARG8 |
Table 3.
Challenging docking cases: no severe clashes between the ligand and the receptor were found after superimposition
| Protein | Ligand | Receptor |
|---|---|---|
| CDK2 | 1H1P | 1H1Q |
| 1AQ1 | 1JSV | |
| 1G5S | 1JSV | |
| Ricin | 1OBT | 1APG |
| P38 | 1BL7 | 1M7Q |
| HIVp | 1HTF | 7UPJ |
Table 2.
Challenging docking cases: the ligands have severe clashes with backbone atoms in the receptor after superimposition
| Protein | Ligand | Receptor | Backbone |
|---|---|---|---|
| P38 | 1m7q | 1DI9 | 1DI9:A:MET109 |
| 1m7q | 1A9U | 1A9U:_:MET109 |
It is noteworthy that the superimposition of the receptors within an ensemble depends on the choice of the reference structure, as well as the choice of the atoms used for the superimposition. These choices affect the resulting expected poses of non-native ligands. Hence, when using these expected poses for measuring success, the RMSD criterion should not be made too stringent.
Docking with flexible critical side chains
While FLIPDock supports explicit torsion angles to represent side chain flexibility, in this work we use a rotamer library as it greatly reduces the computational cost of docking calculation by reducing a few torsion angles to a single variable to be optimized (i.e. the rotamer index). In addition, it was shown that restricting receptor side chains to rotameric positions is often a good enough representation of flexibility, and substantially increases the success rate in cross docking experiments [2]. The critical side chains identified by the overlap analysis were made flexible in the 25 FLIPDock experiments. To be consistent with the cross docking reported in [14], the ligands are kept rigid. In Table 4, we list the best RMSDs for ligand-atom of 10 runs. In our 25 cases, only one or two side chains are critical for each receptor: Phe54 and Phe327 for cAPK, Lys33 and Lys89 for CDK2, Try80 for Ricin, Arg8 and Pro81 for HIVp. This is consistent with a recent statistical analysis [5] that shows in most cases (∼85% of the 980 pockets) three or less residues change conformation in the binding pocket. In this study, making the critical side chains flexible in FLIPDock is sufficient to dock 19 out of the 25 ligands (76%, Table 4) to alternative receptor conformations. In the six failed cases, the centroid atom in 1BX6 ligand shifted 4 Å away from the expected position, leading to a RMSD >5 Å. The 1FMP, 1OBT and 1APG ligands were also predicted to adopt slightly different binding modes leading to RMSD values above 2.0 Å. For these failed cases, although the critical side chains can adopt rotameric conformations, the surrounding rigid side chains prevent them from adopting alternative conformations that could accommodate the rigid ligand. Allowing more side chains near the active site to be flexible is expected to improve the prediction for these failed cases.
Table 4.
The docking results when the critical sidechains were made flexible in FLIPDock
| Ligand | Receptor | RMSD | Ligand | Receptor | RMSD |
|---|---|---|---|---|---|
| 1STC | 1BKX | 0.97 | 1DI8 | 1E1X | 1.03 |
| 1BX6 | 1BKX | 5.04 | 1FMP | 1OBT | 2.42 |
| 1STC | 1YDT | 0.66 | 1IFU | 1OBT | 1.25 |
| 1FVV | 1JSV | 0.75 | 1OBT | 1IFU | 2.96 |
| 1DI8 | 1JSV | 1.75 | 1OBT | 1IFS | 3.12 |
| 1E9H | 1JSV | 1.08 | 1IFS | 1OBT | 0.92 |
| 1FVV | 1H1P | 1.07 | 1APG | 1OBT | 2.96 |
| 1FVV | 1E1X | 1.62 | 1OBT | 1FMP | 3.22 |
| 1FVT | 1JSV | 0.51 | 1C70 | 2UPJ | 1.71 |
| 1FVT | 1E1X | 0.67 | 1HTF | 1G35 | 0.29 |
| 1FVT | 1E9H | 0.97 | 1HTF | 1G2K | 0.16 |
| 1JSV | 1H1P | 1.18 | 1HTF | 1AAQ | 0.44 |
| 1DM2 | 1JSV | 1.30 |
The numbers are ligand all-atom RMSD values in Å
Attempt to predict the critical side chains
In this paper we focus on the side chains rearrangement induced by various ligands. In such a context, the number and identity of the side chains that should be made flexible in a docking simulation to accommodate ligands with different sizes and binding mechanisms is a natural question. Including too few would miss the ones that could be crucial for alternative ligands while including too many would be computationally infeasible for docking. In addition, if the active site is defined as the set of residues interacting with the ligand, the size of such an active site varies with respect to the size of different binding ligands. Therefore, techniques for defining the neighborhood of an active site are important.
We discuss here two methods for defining the neighborhood of an active site. Both methods are based on using a distance cutoff to select receptor atoms beyond the ones directly in contact with the ligand, but they differ in how the distance is measured. The first method uses a distance in Euclidian space. All receptor atoms within a cutoff distance of any ligand atom are selected and every residue with at least one atom selected becomes part of the extended active site. The cutoff distance is defined as the sum of the van der Waals radii of a pair of ligand-receptor atoms plus a user defined distance. In the second method, we use the molecular surface to guide the expansion of the active site. In this approach we start from the molecular surface patches corresponding to the receptor atoms directly in contact with a ligand atom. We then grow this initial patch by selecting surface points within a cutoff distance (on the surface) from a point in the initial patch. The grown patch is then used to select the atoms contributing to this part of the surface leading to a set of residues forming the extended active site. As an approximation of the distance on the surface we use the distances between the surface vertices in the triangulated molecular surface. In Fig. 1 the triangles defined by red and green vertices correspond to the initial patch for the native ligand of the Ricin-AMP complex (PDB code: 1OBT). The green vertices define the boundary of the patch. The blue vertices are the vertices that are within 5.0 Å of a green vertex. This distance is measured as the sum of the length of the edges forming the shortest path to a green vertex on the triangulated surface.
Fig. 1.
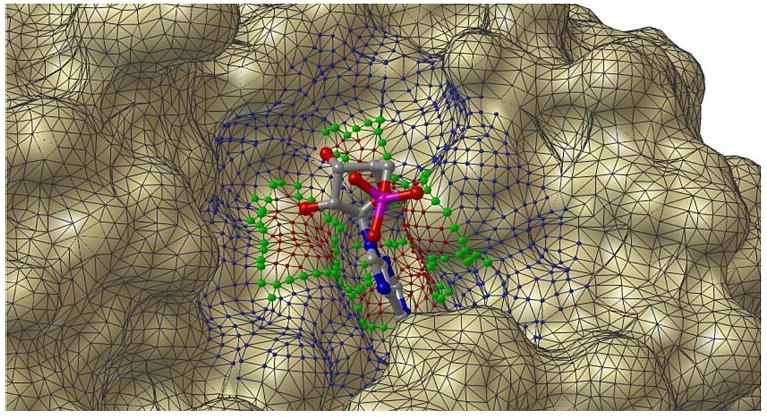
Surface expansion method. The initial surface patch that contact the AMP ligand is shown with red spheres on the vertices and green spheres vertices defining the boundary of the patch. The patch is expanded along the surface. The blue spheres show vertices of the surface that are within 5.0 Å of the green vertices. The distance is measured on the surface as the sum of the edge length for the shortest path between a green and a blue vertex. All the atoms that contribute to the surface patch and the expansion are included in the grown active site. This figure is prepared by PMV [15], surface calculated from MSMS [16]
We find that defining active site neighborhood using the molecular surface distance method is in general more efficient than the Euclidian distance based method as it includes the known critical side chains earlier (i.e. for a smaller cut-off distance) while less non-critical side chains are selected than using the Euclidian distance method. However, it remains a challenging problem to predict residues that need to be made flexible when starting from an arbitrary protein–ligand complex. When the shape and size of the various ligands binding to a receptor differ significantly, the complex that contains the largest or the most space-spanning ligand is the best structure for defining the extended active site as it involves more side chains participating in the interactions. Surface-based active site expansion can serve as the first step of active site analysis. However, since current search techniques cannot handle all side chains as flexible, additional methods for assisting the user in picking which side chains to be made flexible during docking calculations are desirable. In [2] we have described a method based on computed geometric constraints for each atom in a protein. The constraints are obtained from all bonded and non-bonded interactions. We observed that the two critical residues in HIV-1 protease correspond to the least constrained side chains in the active site. This method yielded mixed results for the ensembles considered in this work.
Docking with multiple side chains flexible
The side chains to be made flexible were identified by first defining a list of residues forming the extended active site. Side chains to be made flexible where then selected from this list by choosing the ones with the smallest geometric constraints. We choose the complexes with the most space spanning ligands in the dataset as they provide the best active site definition (1YDT for cAPK, 1FVV for CDK2, 1HBV for HIVp, and 1OBT for Ricin). These complexes were also used to compute the constraints. Table 5 lists the side chains that were chosen to be flexible in the cross docking experiment. The side chains identified earlier as critical are highlighted in bold in this table. The geometric constraint computed for these side chains are also given in this table. While the critical side chains often rank high in the list of active site residues order by increasing constraints, we were unable to find a universal cutoff allowing us to automatically include all known critical side chains.
Table 5.
Side chains in the active sites ranked by constraints
| cAPK | Constraints |
| 1YDT:E:THR183 | 200.0 |
| 1YDT:E:LEU74 | 208.5 |
| 1YDT:E:VAL57 | 210.8 |
| 1YDT:E:LEU173 | 214.1 |
| 1YDT:E:PHE54 | 234.4 |
| 1YDT:E:PHE327 | 237.7 |
| CDK2 | Constraints |
| 1FVV:A:LYS89 | 170.4 |
| 1FVV:A:LYS33 | 194.2 |
| 1FVV:A:LEU134 | 212.0 |
| 1FVV:A:PHE80 | 250.4 |
| HIVp | Constraints |
| 1HBV:B:ARG8 | 169.9 |
| 1HBV:A:ILE47 | 189.8 |
| 1HBV:A:VAL82 | 190.9 |
| 1HBV:A:PRO81 | 196.1 |
| 1HBV:B:ILE84 | 208.9 |
| 1HBV:B:ASP25 | 211.6 |
| Ricin | Constraints |
| 1OBT:_:ILE172 | 204.3 |
| 1OBT:_:TYR123 | 228.2 |
| 1OBT:_:TYR80 | 228.3 |
Critical sidechains are marked in bold and italic
Table 6 provides the best RMSDs out of 10 runs for all ligand atoms. In 22 out of the 25 cases (88%), the rigid ligands can be docked into non-native receptor conformations. Of the three failed cases, the azepane (hexahydroazepine) group in 1BX6 ligand flipped almost 180°, leading to a different binding mode; the AMP in 1OBT and the 1DI8 ligand were also predicted to adopt slightly different binding modes leading to RMSD values above 2.0 Å.
Table 6.
Docking results with multiple sidechains flexible in FLIPDock
| Ligand | Receptor | RMSD | Ligand | Receptor | RMSD |
|---|---|---|---|---|---|
| 1STC | 1BKX | 0.85 | 1DI8 | 1E1X | 1.11 |
| 1BX6 | 1BKX | 6.20 | 1FMP | 1OBT | 1.62 |
| 1STC | 1YDT | 0.56 | 1IFU | 1OBT | 1.46 |
| 1FVV | 1JSV | 0.91 | 1OBT | 1IFU | 1.51 |
| 1DI8 | 1JSV | 3.45 | 1OBT | 1IFS | 1.71 |
| 1E9H | 1JSV | 1.25 | 1IFS | 1OBT | 1.28 |
| 1FVV | 1H1P | 0.97 | 1APG | 1OBT | 1.29 |
| 1FVV | 1E1X | 1.57 | 1OBT | 1FMP | 3.21 |
| 1FVT | 1JSV | 0.54 | 1C70 | 2UPJ | 1.58 |
| 1FVT | 1E1X | 0.60 | 1HTF | 1G35 | 0.27 |
| 1FVT | 1E9H | 0.93 | 1HTF | 1G2K | 0.21 |
| 1JSV | 1H1P | 1.31 | 1HTF | 1AAQ | 0.39 |
| 1DM2 | 1JSV | 1.37 |
It is interesting to notice that the success rate increases from 76% (making only the critical side chains flexible) to 88% when more sidechains are flexible, indicating that our chosen definition of “critical side chains” (distance between a ligand and side chain atoms <1.0 Å) can only catch very severe clashes. Other bad contacts that could prohibit success docking can be removed when we allow more side chains than the critical ones (Table 5) to be flexible in docking.
Note that a priori knowledge was utilized to decide which complex to use to define active site neighborhood and the critical side chains were known before docking. In a more realistic scenario, such information is not always available. Allowing multiple side chains to change conformations can be helpful in such a docking test. However, when multiple side chains are included, it would be more demanding for the searching engine and the scoring function to eliminate false positive and false negative predictions.
Conclusions
In this work, we compiled a set of challenging docking cases that are confirmed by FLIPDock and a modified version of the DOCK program. Although it is not compiled from the most comprehensive sources, it is a good start and can serve as a baseline for evaluating various flexible receptor docking protocols.
FLIPDock was designed to incorporate backbone motions as well as side chain flexibility. We have demonstrated that using rotamer based representation is sufficient to simulate most conformational changes in receptor side chains upon binding of different ligands. FLIPDock can successfully dock 22 ligands out of the 25 challenging cases (88%) when allowing multiple side-chains to be flexible during optimization.
Our side chain flexibility ranking is based on geometric cutoff values of various binding/non-binding interactions [2]. It serves as a quantitative indicator for side chain flexibility. The ranking of critical sidechains in the receptors under study is consistent with the flexibility scale derived from statistical analysis of side chain conformational variances [5]: Lys and Arg are ranked high while Phe is much lower in ranking. However the comparison of the flexibility ranking and the observed critical side chains also suggests that there is no direct correlation between side-chain flexibility and its role in biological function: not all the highly flexible side chains are critical for ligand binding; the functional sidechains may not necessarily be highly flexible. Under such circumstances, a potential solution can be to allow a relatively large number of sidechains to be flexible in a docking. FLIPDock can handle multiple side chains in docking simulations [2]. We have experimented with problems involving ten and more flexible sidechains. Although the optimal solution can be found, the internal energy among the moving sidechains usually dominates the FLIPDock scores leading to the optimization of the side chain packing rather than the optimization of the ligand receptor interactions. Further calibration will be necessary to address this problem.
FLIPDock package (http://www.flipdock.scripps.edu) is freely available for non-profit research. The source code for surface distance expansion will be available in MGLTools (http://www.mgltools.scripps.edu).
Acknowledgments
The financial support from NIH Grant BISTI, GM65609, 2003–2007 and NIH RR08605 are appreciated. This work was partially supported by the National Center for Supercomputing Applications under TG-ASC070027N and utilized the TeraGrid Tungsten machine. This is manuscript 19007-MB from the Scripps Research Institute.
Contributor Information
Yong Zhao, Email: yongzhao@scripps.edu.
Michel F. Sanner, Email: sanner@scripps.edu.
References
- 1.Ahmed A, Kazemi S, Gohlke H. Protein flexibility and mobility in structure-based drug design 2007 [Google Scholar]
- 2.Zhao Y, Sanner MF. Proteins. 2007;68:726. doi: 10.1002/prot.21423. [DOI] [PubMed] [Google Scholar]
- 3.Zhao Y, Stoffler D, Sanner M. Bioinformatics. 2006;22:2768. doi: 10.1093/bioinformatics/btl481. [DOI] [PubMed] [Google Scholar]
- 4.Morris G, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson A. J Comput Chem. 1999;19:1639. [Google Scholar]
- 5.Najmanovich R, Kuttner J, Sobolev V, Edelman M. Proteins. 2000;39:261. doi: 10.1002/(sici)1097-0134(20000515)39:3<261::aid-prot90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 6.Gutteridge A, Thornton J. J Mol Biol. 2005;346:21. doi: 10.1016/j.jmb.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Fradera X, de la Cruz X, Silva CHTP, Gelpi JL, Luque FJ, Orozco M. Bioinformatics. 2002;18:939. doi: 10.1093/bioinformatics/18.7.939. [DOI] [PubMed] [Google Scholar]
- 8.Huey R, Morris GM, Olson AJ, Goodsell DS. J Comput Chem. 2007;28:1145. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 9.Leach AR. J Mol Biol. 1994;235:345. doi: 10.1016/s0022-2836(05)80038-5. [DOI] [PubMed] [Google Scholar]
- 10.Kallblad P, Dean PM. J Mol Biol. 2003;326:1651. doi: 10.1016/s0022-2836(03)00083-4. [DOI] [PubMed] [Google Scholar]
- 11.Frimurer TM, Peters GH, Iversen LF, Andersen HS, Moller NPH, Olsen OH. Biophys J. 2003;84:2273. doi: 10.1016/S0006-3495(03)75033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ala PJ, DeLoskey RJ, Huston EE, Jadhav PK, Lam PY, Eyermann CJ, Hodge CN, Schadt MC, Lewandowski FA, Weber PC, McCabe DD, Duke JL, Chang CH. J Biol Chem. 1998;273:12325. doi: 10.1074/jbc.273.20.12325. [DOI] [PubMed] [Google Scholar]
- 13.Österberg F, Morris G, Sanner M, Olson A, Goodsell D. Proteins. 2002;46:34. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 14.Huang SY, Zou X. Proteins. 2007;66:399. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 15.Sanner MF. Structure. 2005;13:447. doi: 10.1016/j.str.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Sanner MF, Olson AJ, Spehner JC. Biopolymers. 1996;38:305. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]