

Abstract
Protein methyltransferases (PMTs) play essential roles in many biological processes through methylation of histones and diverse nonhistone substrates. Dysregulation of these enzymes has been implicated in many diseases including cancers. While PMT-associated biology can be probed via genetic perturbation, this approach targets full-length PMTs rather than their methyltransferase activities and often lacks temporal, spatial and dose controls (timing, location and amount of dosed compounds). In contrast, small-molecule inhibitors of PMTs can be designed to specifically target the methyltransferase domains in a temporal, spatial and dose-dependent manner. This utility has motivated the development of hundreds of PMT inhibitors, but meanwhile can make it challenging to select the most suitable PMT inhibitors to interrogate PMT-associated biology. This perspective aims to provide timely guidance to evaluate these PMT inhibitors in their relevant biological contexts.
Keywords: PKMT, PRMT, chemical probe, MOA, target engagement and off-target effects
Introduction
Protein methyltransferases (PMTs) have received much attention for their emerging roles on epigenetics through methylating histones and diverse nonhistone substrates [1]. PMTs catalyze methylation reactions by transferring the methyl group from the cofactor S-5′-adenosyl-L-methionine (SAM) to lysine or arginine side chains of their protein substrates [1]. The human genome encodes 9 protein arginine methyltransferases (PRMTs) and >50 protein lysine methyltransferases (PKMTs) [1]. PRMTs belong to the Class-I family of methyltransferases, featured with seven β-strand topology [2]. In comparison, the vast majority of PKMTs belong to the Class-V family of methyltransferases, which contain a characteristic pseudo-knot SET domain (Suppressor of variegation 3–9, Enhancer of zeste and Trithorax) [1]. DOT1L (KMT4), CaM-KMT, METTL21A, METTL21C, METTL21D (or VCP-KMT) and METTL10 are several well-characterized human non-SET PKMTs and are structural homologues of PRMTs [3–8]. PMT-mediated posttranslational modifications are expected to either modulate the functions of target proteins directly or via their interactome and thus downstream effector proteins [9].
PMT-involved epigenetic modulation has been implicated in many essential biological processes [2]. Its dysregulation has been linked to many diseases including developmental disorders and cancer [10–12]. PMTs are multi-functional proteins that contain a methyltransferase domain for catalysis as well as other motifs for recognizing DNA, RNA and other binding partners [13]. PMT biology can be perturbed by target-specific shRNA/siRNA silencing or genomic editing approaches. However, these approaches target full-length PMTs rather than their catalytic domains alone. In contrast, small-molecule inhibitors of PMTs can be designed to specifically target their methyltransferase activities and thus allow their roles to be dissected from other functions of the full-length protein [1, 10–12]. PMT inhibitors also have the common merits of chemical tools for temporal, spatial and dose controls (accurate timing, location and amount of dosed compounds) and can potentially be applied as therapeutic agents in PMT-relevant diseases [1, 11, 12]. These advantages have led to development of many PMT inhibitors in the past [10–12]. However, many emerging PMT inhibitors have not reached the criteria of “chemical probes”, which require high cellular potency, well-defined selectivity, and a clear mechanism of action (MOA) [12, 14]. The lack of such characterization makes it challenging to select suitable PMT inhibitors among many available candidates. It is still possible to use even well-characterized PMT chemical probes incorrectly and thus misinterpret the biological outcomes associated with these compounds. This perspective aims to provide general guidance to evaluate PMT inhibitors and thus leverage their strength to interrogate the functions of PMTs in a precise and reliable manner.
Application of PMT inhibitors within relevant contexts
Combined efforts in the past decade have led to identification of hundreds of inhibitors against human PMTs [10, 11]. In the course of developing PMT inhibitors of high quality, early lead candidates are subject to stepwise optimization with the initial focus on in vitro potency and selectivity, followed by target engagement and efficiency under cellular settings, and eventually pharmacological properties for in vivo application. Some PMT inhibitors have been rigorously examined at each stage, while others can still be in a rudimentary stage with only their potency examined with an in vitro biochemical assay [10, 11]. The literature associated with PMT inhibitors often documents in details how these compounds were characterized. These data can indicate how the PMT inhibitors should be correctly used as chemical tools to interrogate PMT-associated biology and thus need to be carefully reviewed for their application in relevant contexts. Some of curial parameters for assessment of PMT inhibitors include effective doses, whose values are expected to be at least 10-fold higher than the IC50/EC50 to achieve > 95% target engagement; relevant contexts for which these compounds can effectively engage such inhibition against PMTs (e.g. in vitro biochemical settings, inside living cells or in animals); the methods for in vivo administration (e.g. oral, intraperitoneal or intravenous). For in vitro biochemical experiments, the potency of PMT inhibitors under specific settings can be altered significantly by PMT constructs (catalytic domains versus fully-length proteins), the concentrations of substrates and the SAM cofactor, as well as the presence of other PMT-binding partners (see MOA of PMT inhibitors for more details). It is also worth noting that IC50 and EC50 values of PMT inhibitors can be highly context-dependent and different across cell lines (see MOA of PMT inhibitors for more details). Even for the best-characterized PMT inhibitors, their target engagement and in vivo efficiency must be rigorously confirmed under unprecedented biological settings such as the CNS (central nervous system), as not all PMT inhibitors may be able to cross the blood brain barrier, and cell types and tissues that highly express xenobiotic transporters and thus prevent the accumulation of PMT inhibitors through efflux mechanism.
Inhibitors of PMTs that methylate H3K9
Seven human PMTs including G9a (KMT1C/EHMT2) and GLP (KMT1D/EHMT1) have been shown to methylate H3K9 [10]. H3K9 methylation is a common mark of gene suppression. BIX01294 (Figure 1) was first identified from a high throughput screening as a dual inhibitor of G9a and GLP [15]. However, this HTS hit shows low potency against G9a and GLP, with in vitro IC50 of 1~10 μM, and likely interacts with other cellular targets besides the two enzymes [15]. UNC0321 (Figure 1), a BIX01294 derivative, was developed later as a more potent and specific inhibitor of G9a and GLP with a Morrison Ki value of 63 pM (100-fold more potent than BIX01294) [16, 17]. Despite the improved potency of UNC0321, its utility is limited to in vitro biochemical assays because of its poor cell membrane permeability. In contrast, UNC0638 shows not only excellent potency and specificity but also the desired cellular uptake [18]. UNC0638 (Figure 1) has demonstrated its use as a dual specific chemical probe of G9a and GLP under multiple cellular settings [12]. However, this compound is less suitable for in vivo experiments because of its poor pharmacological kinetics. This issue was solved later by developing UNC0642 and UNC1479 (Figure 1) as chemical probes of G9a and GLP for animal studies (Figure 1) [19]. UNC0642 and UNC1479 show comparable cellular potency and target selectivity against G9a and GLP. In addition, the latter is more suitable to explore G9a/GLP’s roles in the CNS due to its 2-fold better brain penetration (brain/plasma ratios of 0.33 versus 0.68 in male Swiss albino mice) [19]. Collectively, despite high structural similarity among these Ga9/GLP inhibitors, only UNC0642 and UNC1479 demonstrated a broad use as chemical probes of G9a and GLP in vitro, in cellular contexts and in vivo; the use of UNC0321 and UNC0638 should be restricted to in vitro settings.
Figure 1.

Stepwise evolution of representative G9a/GLP inhibitors.
Inhibitors of PMTs that methylate H3K27
EZH1 and EZH2 (KMT6) act on histone H3K27 and this methylation often marks gene suppression [12]. Somatic EZH2 mutations are often observed in follicular and diffuse large B-cell lymphomas and are expected to play key oncogenic roles. EZH2 and its mutants are therefore of interest as novel anti-cancer targets [20]. Structurally similar GSK343, EPZ005687, and EI1 (Figure 2) are among the first class of potent and selective inhibitors of human EZH2 (KMT6) [20, 21]. The use of these compounds as chemical probes has been demonstrated under cellular settings but not yet in animals [20, 21]. In contrast, GSK126, EPZ-6438, CPI-360 and CPI-1692 (Figure 2) were characterized as chemical probes of EZH2 with their suitable use demonstrated both in cells and in animals [22–24]. GSK126, EPZ-6438, CPI-360 and CPI-1692 can be administrated intraperitoneally or intravenously [22–24]. In comparison with GSK343, EPZ005687, and EI1, which selectively target EZH2 versus EZH1, UNC1999 and a compound containing a distinguished tetramethylpiperidinyl benzamide scaffold (TMPB) were characterized as dual specific inhibitors against both EZH1 and EZH2 (Figure 2) [25, 26]. Compound 1 was documented as a cellular chemical probe, while UNC1999 showed excellent oral bioavailablity in mice and can be used for in vivo mouse experiments [25, 26]. However, given that GSK126, EPZ-6438 and UNC1999 are substrates of the xenobiotic efflux transporters (e.g. P-gp/ABCB1 or BCRP/ABCG2) [27], these compounds are not suitable for targeting CNS and similar tissues with high expression of these efflux transporters. It is also worth noting that many of these inhibitors also target mutant-type EZH2 such as its Y641 variants [20].
Figure 2.
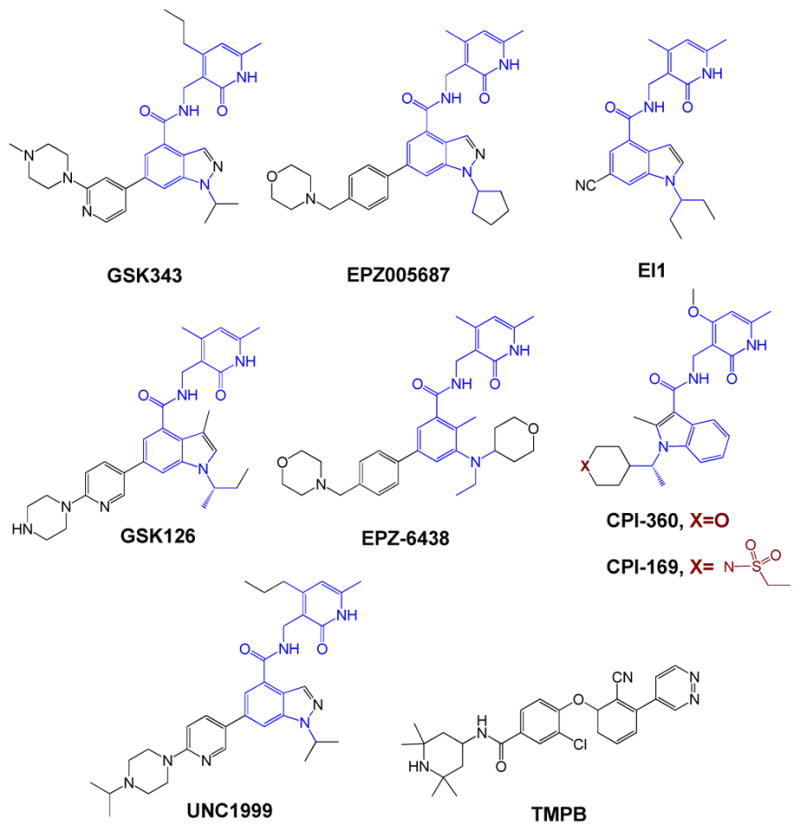
Structures of emerging inhibitors against EZH1/2.
Inhibitors of PMTs that methylate H3K36
SETD2 (KMT3A) is the sole PMT that catalyze trimethylation of H3K36, a histone mark implicated in transcription elongation and RNA splicing [28, 29]. Pr-SNF and Bn-SNF (Figure 3) were characterized as inhibitors of human SETD2 [30]. The potency and selectivity of the two compounds were examined across a panel of human PMTs with in vitro biochemical assays [30]. However, Pr-SNF and Bn-SNF have not been shown to be effective as cell-based or in vivo chemical probes [30], given the poor membrane permeability of the family of compounds containing similar polar amine and carboxylic moieties [31]. SETD2’s functions have been implicated in transcription elongation, RNA splicing, DNA repairing and tumor suppression [13, 28, 30, 32, 33]. However, Pr-SNF and Bn-SNF have yet to fulfill the criteria to be used to perturb and examine these biological processes.
Figure 3.
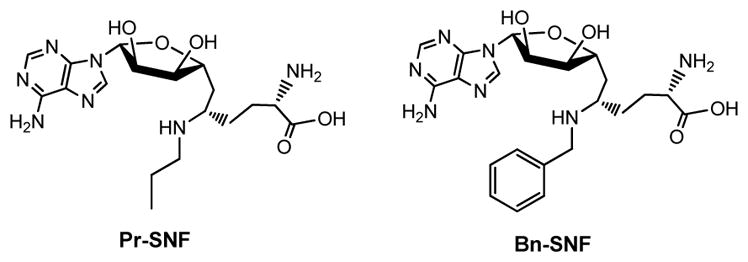
Structures of Pr-SNF and Bn-SNF as SETD2 inhibitors.
Inhibitors of PMTs across species
Previous industrial and academic efforts have been mainly focused on developing small-molecule inhibitors of human PMTs. In contrast, limited progress has been made to develop inhibitors against PMTs of other species. While PMTs can be highly homologous across eukaryotic organisms, caution should be taken upon assuming that the potency and selectivity profile of a compound for human targets can be recapitulated against their counterpart targets in remotely related species. There are many examples that altering a single amino acid in target proteins can render potent inhibitors into completely inactive compounds (e.g. the M184V resistance mutation of HIV reverse transcriptase for lamivudine; the T315I resistance mutation of Bcr-Abl for imatinib) [34, 35]. An inhibitor that is characterized against human PMTs thus needs to be evaluated carefully before applying it to perturb the PMT homologues across species.
Known MOA of PMT inhibitors
Along the reaction path of PMT-involved methylation, PMTs facilitate the catalysis by first recruiting substrates and the cofactor SAM to two nearby binding sites, followed by deprotonation of Lys/Arg nucleophile and finally the transmethylation reaction [1]. For many PMTs, efficient catalysis also requires the participation of remote residues (e.g. dimerization interface of PRMT3) or the presence of other PMT-binding proteins (e.g. WDR5 and RBBP5 for MLL1-mediated H3K4 methylation associated with transcription activity; EED and SUZ12 for EZH2-mediated H3K27 methylation associated with transcription suppression) [36, 37]. Methyltransferase activities of PMTs can thus be inhibited by perturbing SAM/substrate binding or catalytically essential protein-protein interactions. One of key steps in developing high quality small-molecule inhibitors of PMTs is to characterize MOA. Defining the MOA of PMT inhibitors not only is informative to optimize the potency and selectivity of lead compounds but also essential to correctly correlate biological outcomes with PMT inhibition under complicated cellular settings (see discussion below). Current PMT inhibitors can be classified into at least three distinct MOA categories based on whether they target SAM-binding sites, substrate-binding sites or allosteric sites of PMTs.
SAM-competitive PMT inhibitors
A common strategy for developing PMT inhibitors is to engage small molecules to selectively fit SAM-binding pockets of certain PMTs and compete with the cofactor SAM (SAM-competitive inhibitors, Figure 4). EPZ004777 (an inhibitor of DOT1L-mediated H3K79 methylation), Pr-SNF/Bn-SNF (inhibitors of SETD2-mediated H3K36 methylation), GSK126 (an inhibitor of EZH2-mediated H3K27 methylation) and UNC1999 (a dual inhibitor of EZH1/2-mediated H3K27 methylation) are several examples of well-characterized SAM-competitive PMT inhibitors [22, 25, 30, 38, 39]. Inhibition profiles of these compounds are characterized by a linear increase of their apparent IC50 as a function of increased concentration of the SAM cofactor. The structural data of DOT1L and SETD2 in complex with EPZ004777 and Pr-SNF/Bn-SNF further support this MOA, with the inhibitors occupying the SAM-binding pockets of the PMTs and thus preventing SAM’s access for catalysis [30, 38, 40]. While Pr-SNF/Bn-SNF were characterized as SAM-competitive inhibitors, their apparent IC50 also depends upon the presence of peptide substrate. These compounds achieve the highest affinity through forming an inhibitor-SETD2-substrate ternary complex (partially uncompetitive for peptide substrate) (Figure 4) [30]. In comparison, EPZ004777, GSK126 and UNC1999 are canonical SAM-competitive inhibitors with their apparent IC50 independent of the concentration of peptide substrates (noncompetitive for peptide substrate: the presence of the inhibitor does not alter the apparent Km value of the peptide substrate) [22, 25, 38, 39].
Figure 4.

Schematic description of MOA of representative PMT inhibitors. In general, the current PMT inhibitors can be classified into three categories: SAM-competitive, substrate-competitive and allosteric. In addition, these compounds can also display substrate-uncompetitive or SAM-uncompetitive character.
MOA of Pr-SNF/Bn-SNF and EPZ004777 revealed by the X-ray crystal structures of the inhibitor-PMT complexes could also rationalize the selectivity of these inhibitors against their targets versus even closely related PMT homologues [30, 38, 40]. In both scenarios, SETD2 and DOT1L are subject to dramatic conformational changes upon binding their inhibitors. The existence of such structurally distinct inhibitor-bound conformers is expected to associate with potency and selectivity of Pr-SNF/Bn-SNF and EPZ004777 against their respective PMT targets [30, 38, 40]. Similar to kinases [41], PMTs may adopt a set of conformers that are highly dynamic but can be captured upon interacting with structurally matched inhibitors. Exploring and targeting structurally distinct conformers of PMTs may pave a new path for developing SAM-competitive inhibitors of high selectivity. It remains to be examined whether the specificity of SAM-competitive inhibitors GSK126 and UNC1999 also stems from their preferential interactions with certain structurally-distinct conformers of EZH1/2.
Substrate-competitive PMT inhibitors
Another common strategy for developing PMT inhibitors is to engage small molecules to occupy substrate-binding pockets of PMTs (substrate-competitive inhibitors). Individual PMTs are expected to adapt distinct peptide-binding pockets to recognize diverse substrates with limited sequence consensus. In comparison with SAM-competitive inhibitors, substrate-competitive inhibitors are expected to achieve high target specificity by targeting distinct substrate-binding pockets. UNC0638/UNC0642 (G9a/GLP inhibitors), (R)-PFI-2 (an inhibitor SETD7/KMT7, a promiscuous PMT acting on diverse nonhistone substrates), LLY-507 (an inhibitor of SMYD2/KMT3C against K370 methylation of p53) and EPZ015666 (an inhibitor of PRMT5 against SmD3 methylation) are several examples of well-characterized substrate-competitive PMT inhibitors with cell-based activities (Figure 4) [19, 42–44]. Inhibition profiles of these compounds are characterized by a linear increase of their apparent IC50 as a function of the increased concentration of substrates. The substrate-competitive MOA is also supported by the X-ray crystal structures of the PMTs in complex with these inhibitors at the anticipated substrate-binding pockets [19, 42–44]. Herein substrate-competitive inhibitors can be further classified by their dependence on the cofactor SAM for target engagement (Figure 4). UNC0638 and UNC0642 are canonical substrate-competitive inhibitors with their apparent IC50 independent of the presence of SAM [19]. In contrast, (R)-PFI-2 and EPZ015666 are substrate-competitive, SAM-uncompetitive inhibitors that achieve their higher affinity via the formation of the inhibitor-PMT-SAM ternary complexes (Kd values of the inhibitors decrease in the presence of increased concentration of SAM; the Lineweaver–Burk plot of varied concentrations of the inhibitors is characterized by parallel lines to the original enzyme-SAM plot with higher y-intercepts) [42, 44]. AZ-505, for which though with no report of its cellular activity, was also characterized as a substrate-competitive, SAM-uncompetitive inhibitor of SMYD2 [45]. It remains to be investigated whether the SMYD2 inhibitor LLY-507 also follows the substrate-competitive, SAM-uncompetitive MOA for target inhibition.
Allosteric PMT inhibitors
Besides the SAM/substrate-competitive inhibitors, a distinct set of PMT inhibitors were developed to target the allosteric sites of PMTs that are less obvious but essential for enzyme catalysis [46, 47]. It has been well documented that the methyltransferase activities of certain PMTs depend upon the participation of the residues remote from their catalytic sites. Such examples include the dimerization arm of PRMT3 or the formation of a higher-order complex with their partner proteins (e.g. MLL1 and EZH2) [37, 46, 47]. Dimerization of PRMT3 occurs through extensive interactions between the outer surface of the SAM-binding domain of one PRMT3 subunit and the three-helix arm extending from β-barrel domain of the neighboring PRMT3 subunit [46]. A cavity at the base of the dimerization arm, which is more than 15 Å away from PRMT3’s catalytic site, was determined to be the target site of SGC707 (Figure 4), a PRMT3-specific inhibitor [46]. This observation is consistent with SGC707’s noncompetitive character with respect to both substrate and the SAM cofactor (IC50 independent upon the presence of substrate or SAM; the presence of the inhibitor does not change the Km values of substrate or SAM) [46]. SGC707’s binding to PRMT3 is expected to cause undesired conformational constraints for catalysis [46]. As another example, the H3K4 methyltransferase activity of MLL1 depends on the formation of a core complex with at least three other partner proteins WDR5, ASH2L and RbBP5. The MLL1-WDR5 interaction is crucial for the catalysis of the MLL1 complex (Figure 4). MM-401 was developed as an allosteric inhibitor of MLL1 by targeting WDR5’s central channel and thus preventing MLL1’s occupancy at the same site [47]. MM-401-mediated inhibition of MLL1’s methyltransferase activity is expected to be independent of the concentrations of MLL1’s substrates and the SAM cofactor, but sensitive to the concentration of MLL1. Similar strategies have been successfully adapted to develop MI-2/3/463/503, OICR-9429 and SAH-EZH2 as disruptors of protein-protein interaction for MLL1-WRD5, MLL1-Menin and EZH2-EED complexes, respectively [48–51].
Target engagement of PMT inhibitors
Given the distinct MOA of PMT inhibitors, different strategies should be employed to maximally engage these compounds with their targets under complicated biological settings. For canonical SAM-competitive PMT inhibitors, the efficiency of target engagement in a cellular context negatively correlates with the presence of the SAM cofactor, whose intracellular concentration may vary by several fold across cell or tissue types (e.g. liver, testes versus kidney) [52]. However, as long as the amount of these PMT inhibitors is sufficient to compete with SAM, the methyltransferase activities of the targeted PMTs shall be inhibited regardless of the presence of peptide substrates. In contrast, the efficiency of canonical substrate-competitive PMT inhibitors for target engagement in a cellular context negatively correlates with the affinity of PMTs to their substrates (Km, substrate) and the concentrations of the substrates. Because PMTs can methylate diverse histone and nonhistone targets with a dynamic range of Km, substrate and intracellular concentrations, EC50 values of substrate-competitive PMT inhibitors will likely vary from one substrate to another even when cellular conditions and inhibitor concentrations are identical. It is thus feasible to use modest doses of substrate-competitive PMT inhibitors to selectively perturb the methyltransferase activities of PMTs on a subset of substrates with large Km, substrate and low intracellular concentrations, but spare those substrates with small Km, substrate or high intracellular concentrations from such inhibition.
The target engagement can be more complicated for noncanonical SAM/substrate-competitive PMT inhibitors. For substrate-competitive, SAM-uncompetitive inhibitors such as (R)-PFI-2 and EPZ015666, the optimal setting is to engage PMT inhibition in the presence of a large amount of the SAM cofactor and a minimal amount of peptide substrates. In contrast, for SAM-competitive, substrate-uncompetitive inhibitors such as Pr-NSF and Bn-NSF, the optimal setting to engage PMT inhibition is the presence of a minimal amount of the SAM cofactor and a large amount of peptide substrates.
Such scenarios can be further complicated under cellular contexts in the presence of PMT oligomerization or PMT complexes containing multiple subunits. For instance, EZH2 needs to form a core complex with EED and SUZ12 to catalyze H3K27 methylation in vivo [37]. The methyltransferase activity of the core EED-SUZ12-EZH2 complex can be further enhanced allosterically through the interaction between its EED subunit and H3K27me3 peptide, a product of EZH2-catalyzed methylation [37]. The allosteric activation of EZH2 is expected to facilitate the self-propagation of H3K27me3 mark to adjacent chromatin. Interestingly, the SAM-competitive EZH2 inhibitor GSK126 showed around 10-fold increase in the affinity to the EED-SUZ12-EZH2 complex after recruiting H3K27me3 peptide as the allosteric activator. This observation indicates that, despite the potential competition from high concentration of the SAM cofactor under a cellular setting, the efficiency of GSK126 against EZH2 can be increased by the presence of allosterically activated H3K27me3-EED-SUZ12-EZH2 complex [53]. Mutually, the presence of GSK126 is expected to stimulate the EED-SUZ12-EZH2 core complex to recruit H3K27me3 peptide. Therefore, the treatment with GSK126 can not only perturb the methyltransferease activity of EZH2 but also alter the components of the EZH2 complex under cellular contexts. However, this effect is expected to be dynamic and will diminish gradually as GSK126 blocks the formation of H3K27me3 for a prolonged period. It is thus of interest to document the potency of PMT inhibitors under diverse cellular scenarios. Given that DOT1L-mediated methylation on H3K79 can be enhanced upon the formation of the DOT1L-AF10 complex [54], it remains to be explored whether the DOT1L inhibitor EPZ004777 and the DOT1L-binding protein AF10 can mutually alter their affinities to DOT1L through the formation of an EPZ004777-DOT1L-AF10 ternary complex. SGC707 inhibits PRMT3 by targeting the dimerization interface of PRMT3 and is expected to be potent against the PRMT3 dimer complex inside cells [46]. It would be also interesting to examine whether such MOA can be applied to other PRMT3 complexes under cellular settings.
Evaluating target engagement and off-target effects of PMT inhibitors
Given that multiple factors affect potency of PMT inhibitors inside cells, their target engagement and potential off-target effects should be rigorously evaluated. To directly evaluate target engagement of PMT inhibitors in a cellular context, these compound are often appended with a functional anchor (e.g. a terminal alkyne, azide or biotin) [55]. Such derivatives can be designed to maintain target affinity comparable to that of their parent compounds, while gaining the ability to pull down the engaged PMT targets from cell lysates after immobilizing the inhibitor-PMT complex via the functional anchor (e.g. alkyne-azide cycloaddition with a biotin moiety, followed by biotin-streptavidin pulldown) [56]. The target engagement can be correlated to the pulldown efficiency of target proteins. Recently, a cellular thermal shift assay (CTSA) has gained attention for its merit to monitor drug target engagement in living cells and tissues [57, 58]. Herein the binding of small molecules is expected to increase the thermal stability of target proteins. CTSA can thus be applicable to evaluate target engagement of PMT inhibitors.
Target engagement of PMT inhibitors can also be evaluated indirectly by their efficiency in blocking methylation of designated substrates (methylation marks) in relevant cellular contexts. For a SAM-competitive or allosteric PMT inhibitor, a robust practice is to confirm a dose-dependent decrease of a methylation mark such as H3K9me2 for G9a/GLP and H3K27me3 for EZH1/2 [18, 22, 25]. Because of the SAM-competitive or allosteric MOA, the lack of PMT-associated methylation often indicates that the current dose is sufficient to saturate the SAM-binding or allosteric site of the targeted PMT and thus there is no free PMT available for catalysis. In contrast, for a substrate-competitive PMT inhibitor, target engagement should be evaluated with the most robust methylation mark (the substrates with the highest concentration and the lowest Km, substrate), given the dynamic range of PMT substrates available to compete with the inhibitor. Otherwise, a specific dose of a PMT inhibitor may only inhibit the subset of methylation events involved with the PMT substrates with low concentrations and low affinity, while sparing others from such inhibition.
It is also worth noting that the engagement of inhibitors with their PMT targets can also have other outcomes, besides the inhibition of methyltransferase activities, such as altering the conformation of PMTs or changing the affinities of PMTs to their binding partners. For instance, substrate-competitive PMT inhibitors are expected to perturb the binding of PMTs to their substrates. Such perturbation thus not only renders the inhibition of the PMT-mediated methylation but also alters the integrity of the PMT-substrate complex. As other examples, the binding of GSK126 to the EED-SUZ12-EZH2 complex promotes the interaction of its EED subunit with H3K27me3 peptide [53]; the binding of MM-401 to the MLL1-WDR5-ASH2L-RbBP5 complex disrupts the interaction of WDR5 with MLL1 [47]. Therefore, the treatment of these PMT inhibitors alters the components of the PMT complexes and exhibits the effects beyond the inhibition of substrate methylation.
Besides target engagement, off-target effects of PMT inhibitors should be evaluated prior to their use as chemical tools. The target specificity of PMT inhibitors is often evaluated in vitro by their cross-inhibition profiles against a panel of PMTs, as well as kinases, GPCRs and channels/transporters [18, 25]. Similar to the approaches used to evaluate target engagement, off-target effects of PMT inhibitors can be roughly evaluated through a pulldown approach with the inhibitor derivatives appended with a terminal alkyne/azide/biotin or CTSA for the capability of the inhibitors to stabilize off-target proteins of interest. Because the off-target effects of PMT inhibitors are dose-dependent, such experiment should be carried out at the concentrations that are relevant to those used for PMT inhibitors to engage their own targets. However, there are several challenges of examining off-target effects in a definitive manner with the methods described above. For instance, off-target candidates (e.g. kinases, GPCRs and channels/transporters) for cross-inhibition analysis mainly originate from the human genome and thus only allow the set of data to be applied to human cell lines. A compound that is characterized as a target-specific inhibitor against a human PMT may lose such specificity in other species. In addition, only a small number of off-target candidate proteins can be evaluated in vitro in comparison with potential > 20K human off-target candidates in a cellular context. The robust evaluation of off-target effects of PMT inhibitors with the derivative-based pulldown or CTSA is also limited by our inability to profile the full proteome with mass spectrometry or individual antibodies.
Given the limitation of these methods for definitive evaluation of off-target effects, multiple complimentary strategies can be adapted to minimize the off-target impact of PMT inhibitors in specific biological contexts. A routine check of the off-target impact is to correlate in vitro IC50 with cellular EC50 of PMT inhibitors. In general, in vitro IC50 values of PMT inhibitors against methylation marks are expected to be smaller than the EC50 values for their cellular treatment given the potential cell membrane barrier and metabolic degradation for small-molecule compounds. Additional caution is warranted if this is not the case. Potential off-target effects can also be evaluated with structurally similar but inactive derivatives of PMT inhibitors as negative controls [18, 25]. The assumption here is that potential off-target effects but not PMT-associated perturbation will be shared by the structurally similar pairs of active and inactive compounds. Alternatively, it is an excellent practice to examine the biological effects of several distinct PMT inhibitors in parallel (e.g. TMPB paired with other EZH1/2 inhibitors in Figure 2). The logic behind this experiment is that off-target effects are unlikely to be shared among a set of PMT inhibitors with different structures and distinct MOA for target engagement.
Potential off-target effects of PMT inhibitors can also be evaluated in combination with genetic rescue experiments. There are several common scenarios for this approach: (1) overexpression of target PMTs is expected to decrease potency of the inhibitors make the inhibitors less potent (increased EC50); (2) knocking in inhibitor-resistant or catalytically-active PMT variants is expected to make the cells more resistant to inhibitor treatment; (3) knocking down PMTs with shRNA/siRNA or knocking out PMTs with CRISPR/Cas9 is expected to mimic the phenotypes of inhibitor treatment; (4) the combination of genetic and chemical perturbation is expected to mimic the maximal outcomes of the single treatment alone. However, it should be emphasized that the discrepancy between genetic and chemical perturbation is not always caused by the off-target effects of the latter. As delineated above, a PMT inhibitor can act through distinct MOA in a highly context-dependent manner and its efficiency can be readily altered by multiple parameters including the presence of SAM, substrates and PMT-binding partners. The phenotypes of eliminating a full-length PMT, which often function through substrate methylation and protein-protein interaction, is not expected to be identical to the outcomes associated with PMT inhibitors, which only perturb a specific subset of such functions. Indeed, the discrepancies between the two approaches may lead to new insights into the complexity of PMT-associated biology besides substrate methylation.
Conclusion and future perspective
Developing PMT inhibitors has been significantly accelerated in the past decade, along with the rapid emergence of PMT-associated biology [2, 10–12]. However, there is still a great need for high-quality PMT inhibitors against >60 human PMTs as well as 7~70 PMTs across other eukaryotic model organisms. Given our interest in exploring PMT-involved epigenetic biology under diverse normal and disease settings, we expect to witness the increased access to PMT inhibitors and likely extend their use towards broader biological settings. When selecting suitable inhibitors among many available candidates, it is important to rigorously evaluate the strength and weakness of these compounds in the relevant contexts for correct use. Target engagement and potential off-target effects should be examined before applying even well-characterized PMT inhibitors in unprecedented biological settings. In addition, it is essential to know the MOA of each PMT inhibitor to reasonably interpret the outcome of a treatment experiment. PMT inhibitors will soon become convenient tools, which can complement genetic perturbation to study PMT biology. The highlighted principles in this perspective endeavor to provide general guidance to leverage PMT inhibitors as convenient chemical tools. Here the author apologizes that the limited space allows only a small set of PMT inhibitors to be discussed here.
Executive Summary.
Introduction
Protein methyltransferases (PMTs) have been implicated in epigenetic-associated diseases.
Development of PMT inhibitors is of critical need but also presents the challenge of their utility.
Considering PMT inhibitors within relevant contexts
PMT inhibitors must be carefully evaluated and used in relevant contexts.
Optimization of PMT inhibitors is a multi-step process as exemplified with G9a/GLP inhibitors and EZH1/2 inhibitors.
The potency and specificity of inhibitors against human PMTs may not be transferred to PMT homologues of other species.
Knowing mechanism of action (MOA) of PMT inhibitors
PMT inhibitors can be further classified on the basis of their MOA.
Efficiency of SAM-competitive inhibitors will be negatively affected by the presence of intracellular SAM.
Efficiency of substrate-competitive inhibitors is dynamically correlated with concentrations and Km, substrate values of cellular substrates.
PMT inhibitors can show SAM/substrate-uncompetitive or allosteric MOA.
Evaluating target engagement and off-target effects of PMT inhibitors
Target engagement and off-target effects of PMT inhibitors can be evaluated in vitro by their inhibition profiles against candidate proteins, and in cellular contexts through target pulldown methods or cellular thermal shift assay.
Target engagement of PMT inhibitors can be evaluated indirectly by their abilities to erase methylation marks.
Off-target effects of PMT inhibitors can be evaluated by their EC50, inactive analogues or a set of structurally distinct inhibitors.
Inhibition of PMTs can have the effects beyond substrate methylation, which are complimentary but may not identical to genetic perturbation.
Conclusion and future perspective
PMT inhibitors can be used as convenient chemical tools to interrogate PMT-associated biology.
We anticipate critical need and broader utility of PMT inhibitors in the future.
Acknowledgments
Financial and competing interest disclosure
The work carried out by the Luo laboratory and discussed in this article received support from NIGMS (1R01GM096056), the NIH Director’s New Innovator Award Program (1DP2-OD007335), NIH/NCI Cancer Center Support Grant 5P30 CA008748-44, Starr Cancer Consortium, Mr. William H. Goodwin and Mrs. Alice Goodwin Commonwealth Foundation for Cancer Research, The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center, Tri-I Therapeutics Discovery Grant, and Sohn Conference Foundation. The author has no financial interest in or financial conflict with the subject matter discussed in the manuscript apart from those disclosed. The author thanks Michael Langberg and Ryan Blawski for proof-reading the manuscript. No writing assistance was involved in the production of this manuscript.
References
- 1*.Luo M. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem Biol. 2012;7(3):443–463. doi: 10.1021/cb200519y. A general review of PMTs from chemistry perspective. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Yang Y, Bedford Mt. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13(1):37–50. doi: 10.1038/nrc3409. A broad review of PRMT-related biology. [DOI] [PubMed] [Google Scholar]
- 3.Shimazu T, Barjau J, Sohtome Y, Sodeoka M, Shinkai Y. Selenium-based S-adenosylmethionine analog reveals the mammalian seven-beta-strand methyltransferase METTL10 to be an EF1A1 lysine methyltransferase. Plos One. 2014;9(8):e105394. doi: 10.1371/journal.pone.0105394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kernstock S, Davydova E, Jakobsson M, et al. Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat Commun. 2012;3:1038. doi: 10.1038/ncomms2041. [DOI] [PubMed] [Google Scholar]
- 5.Jakobsson Me, Moen A, Bousset L, et al. Identification and characterization of a novel human methyltransferase modulating Hsp70 protein function through lysine methylation. J Biol Chem. 2013;288(39):27752–27763. doi: 10.1074/jbc.M113.483248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Min J, Feng Q, Li Z, Zhang Y, Xu Rm. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112(5):711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Hsu Yh, Mo C, et al. METTL21C is a potential pleiotropic gene for osteoporosis and sarcopenia acting through the modulation of the NF-kappaB signaling pathway. J Bone Miner Res. 2014;29(7):1531–1540. doi: 10.1002/jbmr.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magen S, Magnani R, Haziza S, et al. Human calmodulin methyltransferase: expression, activity on calmodulin, and Hsp90 dependence. PLoS One. 2012;7(12):e52425. doi: 10.1371/journal.pone.0052425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang R, Luo M. A journey toward Bioorthogonal Profiling of Protein Methylation inside living cells. Curr Opin Chem Biol. 2013;17(5):729–737. doi: 10.1016/j.cbpa.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Liu Y, Liu K, Qin S, Xu C, Min J. Epigenetic targets and drug discovery: part 1: histone methylation. Pharmacol Ther. 2014;143(3):275–294. doi: 10.1016/j.pharmthera.2014.03.007. A comprehensive review of PMT inhibitors. [DOI] [PubMed] [Google Scholar]
- 11**.Kaniskan Hu, Konze Kd, Jin J. Selective inhibitors of protein methyltransferases. J Med Chem. 2014;58(4):1596–1629. doi: 10.1021/jm501234a. A comprehensive review of PMT inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12**.Kaniskan Hu, Jin J. Chemical probes of histone lysine methyltransferases. ACS Chem Biol. 2014;10(1):40–50. doi: 10.1021/cb500785t. A comprehensive review of PMT chemical probes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner Ej, Carpenter Pb. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13(2):115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Frye Sv. The art of the chemical probe. Nat Chem Biol. 2010;6(3):159–161. doi: 10.1038/nchembio.296. Definition of chemical probes. [DOI] [PubMed] [Google Scholar]
- 15.Kubicek S, O’sullivan Rj, August Em, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25(3):473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 16.Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53(15):5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, et al. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J Med Chem. 2011;54(17):6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vedadi M, Barsyte-Lovejoy D, Liu F, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7(9):648–648. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu F, Barsyte-Lovejoy D, Li F, et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem. 2013;56(21):8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knutson Sk, Wigle Tj, Warholic Nm, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 21.Verma Sk, Tian X, Lafrance Lv, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012;3(12):1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mccabe Mt, Ott Hm, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 23.Knutson Sk, Warholic Nm, Wigle Tj, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradley Wd, Arora S, Busby J, et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21(11):1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Konze Kd, Ma A, Li F, et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nasveschuk Cg, Gagnon A, Garapaty-Rao S, et al. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med Chem Lett. 2014;5(4):378–383. doi: 10.1021/ml400494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang P, De Gooijer Mc, Buil Lc, Beijnen Jh, Li G, Van Tellingen O. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-Inhibitors. Int J Cancer. 2015 doi: 10.1002/ijc.29566. [DOI] [PubMed] [Google Scholar]
- 28.De Almeida Sf, Grosso Ar, Koch F, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol. 2011;18(9):977–983. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 29.Edmunds Jw, Mahadevan Lc, Clayton Al. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. Embo J. 2008;27(2):406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30**.Zheng W, Ibanez G, Wu H, et al. Sinefungin Derivatives as Inhibitors and Structure Probes of Protein Lysine Methyltransferase SETD2. J Am Chem Soc. 2012;134(43):18004–18014. doi: 10.1021/ja307060p. Characterization of Pr-SNF and Bn-SNF as SAM-competitive and substrate-uncompetitive inhibitors of SETD2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Q, Jiang Fy, Schultz Pg, Gray Ns. Design of allele-specific protein methyltransferase inhibitors. J Am Chem Soc. 2001;123(47):11608–11613. doi: 10.1021/ja011423j. [DOI] [PubMed] [Google Scholar]
- 32.Zhu X, He F, Zeng H, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46(3):287–293. doi: 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F, Mao G, Tong D, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153(3):590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Modugno M. New resistance mechanisms for small molecule kinase inhibitors of Abl kinase. Drug Discov Today Technol. 2014;11:5–10. doi: 10.1016/j.ddtec.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Gupta R, Hill A, Sawyer Aw, Pillay D. Emergence of drug resistance in HIV type 1-infected patients after receipt of first-line highly active antiretroviral therapy: a systematic review of clinical trials. Clin Infect Dis. 2008;47(5):712–722. doi: 10.1086/590943. [DOI] [PubMed] [Google Scholar]
- 36.Patel A, Dharmarajan V, Vought Ve, Cosgrove Ms. On the Mechanism of Multiple Lysine Methylation by the Human Mixed Lineage Leukemia Protein-1 (MLL1) Core Complex. J Biol Chem. 2009;284(36):24242–24256. doi: 10.1074/jbc.M109.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margueron R, Justin N, Ohno K, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu W, Chory Ej, Wernimont Ak, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3 doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 39.Daigle Sr, Olhava Ej, Therkelsen Ca, et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basavapathruni A, Jin L, Daigle Sr, et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80(6):971–980. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- 41.Lee Gm, Craik Cs. Trapping moving targets with small molecules. Science. 2009;324(5924):213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Barsyte-Lovejoy D, Li F, Oudhoff Mj, et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc Natl Acad Sci USA. 2014;111(35):12853–12858. doi: 10.1073/pnas.1407358111. Characterization of (R)-PFI-2 as a substrate-competitive and SAM-uncompetitive inhibitor of SETD7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen H, Allali-Hassani A, Antonysamy S, et al. LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J Biol Chem. 2015;290(22):13641–13653. doi: 10.1074/jbc.M114.626861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44**.Chan-Penebre E, Kuplast Kg, Majer Cr, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11(6):432–437. doi: 10.1038/nchembio.1810. Characterization of EPZ015666 as a substrate-competitive and SAM-uncompetitive inhibitor of PRMT5. [DOI] [PubMed] [Google Scholar]
- 45**.Ferguson Ad, Larsen Na, Howard T, et al. Structural Basis of Substrate Methylation and Inhibition of SMYD2. Structure. 2011;19(9):1262–1273. doi: 10.1016/j.str.2011.06.011. Characterization of AZ-505 as a substrate-competitive and SAM-uncompetitive inhibitor of SMYD2. [DOI] [PubMed] [Google Scholar]
- 46.Kaniskan Hu, Szewczyk Mm, Yu Z, et al. A Potent, Selective and Cell-Active Allosteric Inhibitor of Protein Arginine Methyltransferase 3 (PRMT3) Angew Chem Int Ed. 2015;54(17):5166–5170. doi: 10.1002/anie.201412154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao F, Townsend Ec, Karatas H, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53(2):247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grembecka J, He S, Shi A, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borkin D, He S, Miao H, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 2015;27(4):589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grebien F, Vedadi M, Getlik M, et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol. 2015;11(8):571–578. doi: 10.1038/nchembio.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim W, Bird Gh, Neff T, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9(10):643–650. doi: 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caudill Ma, Wang Jc, Melnyk S, et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J Nutr. 2001;131(11):2811–2818. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- 53**.Van Aller Gs, Pappalardi Mb, Ott Hm, et al. Long residence time inhibition of EZH2 in activated polycomb repressive complex 2. ACS Chem Biol. 2013;9(3):622–629. doi: 10.1021/cb4008748. Characterization of the formation of H3K27me3-EED-SUZ12-EZH2-GSK126 complex. [DOI] [PubMed] [Google Scholar]
- 54.Deshpande Aj, Deshpande A, Sinha Au, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. 2014;26(6):896–908. doi: 10.1016/j.ccell.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konze Kd, Pattenden Sg, Liu F, et al. A chemical tool for in vitro and in vivo precipitation of lysine methyltransferase G9a. ChemMedChem. 2014;9(3):549–553. doi: 10.1002/cmdc.201300450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prescher Ja, Bertozzi Cr. Chemistry in living systems. Nat Chem Biol. 2005;1(1):13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 57.Savitski Mm, Reinhard Fb, Franken H, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346(6205):1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 58.Jafari R, Almqvist H, Axelsson H, et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9(9):2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]