

Abstract
Background
Podocytes are major components of the filtration barrier and a renal source of vascular endothelial growth factor (VEGF). Chronic renovascular disease (RVD) progressively deteriorates renal function, accompanied by podocyte damage and a progressive reduction in VEGF. We showed that the endothelin (ET) pathway contributes to this pathological process and ET-A (but not ET-B) receptor antagonism protects the kidney in RVD. We hypothesize that ETA-induced renoprotection is largely driven by protection of podocyte integrity and function.
Methods
To mimic renal environment of chronic RVD, human podocytes were incubated under chronic hypoxia for 96 hours and divided in untreated or treated with an ET-A or ET-B receptor antagonist. Cells were quantified after 96 hours. Cell homogenates and media were obtained after 1, 24 and 96 hours to quantify production of VEGF, anti-VEGF soluble receptor s-Flt1, and the expression of apoptotic mediators. A separate set of similar experiments was performed after addition of a VEGF-neutralizing antibody (VEGF-NA).
Results
Hypoxia decreased podocyte number, which was exacerbated by ET-B but improved after ET-A antagonism. Production of VEGF was preserved by ET-A antagonism whereas s-Flt1 increased in hypoxic cells after ET-B antagonism only, accompanied by a greater expression of pro-apoptotic mediators. On the other hand, treatment with VEGF-NA diminished ET-A-induced protection of podocytes.
Conclusion
ET-A antagonism preserves podocyte viability and integrity under chronic hypoxia, whereas ET-B antagonism exacerbates podocyte dysfunction and death. Enhanced bioavailability of VEGF after ET-A antagonism could be a pivotal mechanism of podocyte protection that significantly contributes to ET-A receptor blockade-induced renal recovery in chronic RVD.
Keywords: podocytes, VEGF, hypoxia, endothelin antagonists, apoptosis
Introduction
Chronic kidney disease (CKD) is a progressive disease affecting almost 14% of the general population1. CKD constitutes an independent cardiovascular risk factor and these patients have higher rates of hospitalization, greater mortality, shorter life expectancy, and healthcare costs that increase up to 4 times as CKD progresses2. A frequent etiology of CKD is chronic renovascular disease (RVD), which affects between 9–11% of the general population and can deteriorate renal function, lead to CKD, and progress to end-stage renal disease3–5. Our previous studies in a swine model of chronic RVD (induced by unilateral renal artery stenosis) showed that the stenotic kidney develops a progressive deterioration of hemodynamics, filtration, and tubular function, accompanied by a significant vascular rarefaction and the development of albuminuria, glomerulosclerosis, and tubule-interstitial fibrosis6–9.
There is a significant systemic and renal up-regulation of the endothelin (ET) system in this model of RVD. The ET system includes three major isoforms: ET-1, ET-2, and ET-3, which exert their actions through specific ET-A and ET-B receptors. ET-1 is the major isoform of the ET family and plays important physiological roles in controlling renal function and blood pressure. However, it has been shown to be highly active in pathological situations and a major contributor to renal injury in atherosclerosis10, 11, diabetes12, 13, and RVD8, 14. Indeed, we showed that up-regulation of the ET-1/ET-A pathway can lead to progressive renal dysfunction, fibrosis, and microvascular damage of the stenotic kidney in RVD, and such deleterious changes were largely attenuated by specific ET-A but not ET-B receptor antagonism8, 14. However, the potential mechanisms of this distinct ET-A antagonism-induced renoprotection are not fully elucidated.
Podocytes are terminally specialized epithelial cells that are key components of the glomerular filtration barrier. Damage and even loss of podocytes have been described in hypertensive-induced renal injury15, 16, pre-eclampsia17, and diabetes18. A major factor for podocyte health and function is vascular endothelial growth factor (VEGF). Podocytes are both sources and targets of VEGF in autocrine and paracrine fashion, express VEGF receptors19 and also express ET-A and B receptors20. Our recent studies showed that the excretion of nephrin from the stenotic kidney (a marker suggestive of podocyte injury) is exacerbated in the swine model of RVD but is reduced by ET-A antagonism9. Furthermore, the stenotic kidney has a significant decrease in the expression and availability of VEGF21, 22 that is largely restored after ET-A antagonism8, 9, 14. Therefore, the current study was designed to test the hypothesis that renoprotection by ET-A antagonism may be driven by a distinct protection on podocytes steered by recovering VEGF bioavailability. To test our hypothesis and extend our previous in vivo findings8, 9 by elucidating potential mechanisms of ET-1/ET-A-mediated podocyte injury and protection, human podocytes that were chronically exposed to hypoxia (to mimic the stenotic kidney environment in RVD23) were treated with specific ET-A or ET-B antagonists. In addition, to determine whether protection of podocytes by ET-A antagonism is mediated by VEGF, a separate similar set of experiments was carried out after VEGF blockade.
Results
Podocyte phenotype
Since cells were used after 7–12 passages, we first determine whether cells still have podocyte characteristics. Homogenates from normoxic cells collected at the 0-hour time-point were used to determine the expression of Wilms’ tumor (WT)-124 by western blot (duplicates), showing preserved expression of this podocyte marker (Figure 1).
Figure 1. Wt-1 expression is preserved in podocytes after 7–12 passages, indicating preserved podocyte characteristics.

Cell homogenates from normoxic podocytes were used and expression determined in duplicates by western blotting.
Podocyte counts
Cell counting after 96 hours showed that hypoxia induced a significant reduction in the number of podocytes. Cell number was improved by ET-A but not ET-B antagonism. The average number of cells per mL in each group (mean± SD) at the 96-hour time-point was as follows: Normoxic: 9.3 × 106±2.7 × 106; Hypoxic: 4.6 × 106±1.9 × 106; Hypoxic+ET-A: 5.3 × 106±1.9 × 106; and Hypoxic+ET-B: 2.9 × 106±3.3 × 105.
Hypoxia increases ET-1 and reduces podocyte activity
Hypoxia increased the concentration of ET-1 in media compared to normoxic cells (0.29±0.00 vs. 0.31±0.00 pg/μg, respectively, p<0.05 vs. normoxic). ET-A antagonism did not increase ET-1 (0.27±0.02 pg/μg) whereas ET-B antagonism resulted in a further increase in ET-1 concentration (0.35±0.00 pg/μg, p<0.05 vs. normoxic and hypoxic). Quantification of VEGF in cell media showed that ET-A antagonism improved VEGF secretion (suggesting preserved podocyte activity) whereas hypoxic and ET-B antagonists-treated podocytes showed a similar reduction in VEGF (compared to normoxic cells after 96 hours, Figure 2A). Furthermore, concentration of s-Flt1 was similar in the media of normoxic, hypoxic and ET-A antagonist treated podocytes but progressivley up-regulated in media from ET-B antagonists-treated ones (Figure 2B).
Figure 2. Chronic hypoxia reduces podocyte activity.

Production of VEGF (A) and s-Flt1 (B) after 1, 24, and 96 hours of hypoxia. Only ET-A antagonism significantly improved VEGF availability (expressed in pg/protein of interest/μg of total protein), whereas ET-B antagonism progressively increased production of s-Flt1. * p<0.05 vs. Normoxic; † p<0.05 vs. Hypoxic; ‡ p<0.05 vs. Hypoxic+ET-A
ET-A blockade restore the expression of podocin, nephrin, and angiogenic factors
Hypoxic and ET-B treated podocytes showed decreased expression of VEGF, which was preserved by ET-A antagonism. In addition, ET-A antagonism improved expression of podocin (ANOVA p<0.01) and nephrin, which are both major podocyte slit diaphragm-associated proteins19, 25, implying an overall reduction in hypoxia-induced podocyte damage that was not fully achieved after ET-B antagonism (Figure 3A–B).
Figure 3. ET-A blockade restore the expression of podocin, nephrin, and VEGF.

ET-A antagonism distinctly preserved the expression of VEGF and the slit-associated proteins podocin and nephrin (determined by western blot at the 96 hours time-point and quantified related to β-actins, A and B, respectively), suggesting a reduction in podocyte damage. * p<0.05 vs. Normoxic; † p<0.05 vs. Hypoxic; ‡ p<0.05 vs. Hypoxic+ET-A.
ET-A blockade improved expression of apoptotic and survival factors
A proteome profiler apoptotic array showed the following factors with a higher expression in hypoxic podocytes compared to normoxic controls: BAX, p53, SMAC, heat-shock protein (HSP) 60, cytochrome (Cyt)-C and apoptosis induced factor (AIF). The differences in the expression of these factors was then confirmed by western blot and showed a significant attenuation by ET-A antagonism (ANOVA p<0.05 for all). ET-A blockade also improved the expression of pro-survival p-akt (ANOVA p=0.02) and Bcl-2 (ANOVA p=0.04), suggesting a decrease in apoptotic activity. Interestingly, ET-B antagonism resulted in a similar or higher expression of apoptotic and survival factors as observed in hypoxic cells, indicating a non-protective effect on hypoxic podocytes (Figure 4A). These changes in expression of apoptotic and survival factors (and cell counts) were accompanied by modifications in podocyte morphology (nuclear condensation/fragmentation) suggestive of apoptosis (Figure 4B).
Figure 4. ET-A blockade improved expression of apoptotic and survival factors.
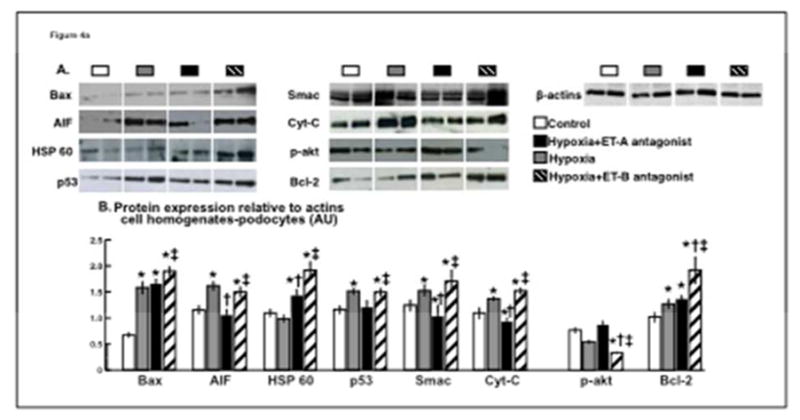

4a) Podocyte expression (determined by western blot at the 96 hours time-point, A) and quantification (B) of pro-apoptotic BAX, AIF, HSP 60, p53 and Smac were all increased by hypoxia and ET-B antagonism but restored by ET-A blockade, accompanied by improved expression of pro-survival p-akt. 4b) Representative pictures (96 hours) from normoxic (A), hypoxic (B), and hypoxic podocytes treated with ET-A (C) or ET-B (D) antagonist. Cell counts and morphological changes suggestive of apoptosis (e.g.: nuclear condensation and fragmentation) were improved after ET-A but not ET-B antagonism. These results suggest a decrease in pro-apoptotic activity in hypoxic podocytes after ET-A antagonism.
* p<0.05 vs. Normoxic; † p<0.05 vs. Hypoxic; ‡ p<0.05 vs. Hypoxic+ET-A
VEGF-NA abolished the beneficial effects of ET-A blockade on podocyte survival
To determine whether VEGF plays a role in ET-A induced protection of podocytes, a separate similar set of experiments were performed after incubation of podocytes to VEGF-NA. Neutralization of VEGF abolished the protective effects of ET-A antagonism in cell counts, resulting in a significant reduction in the number of podocytes (Figure 5A, all groups). The average number of cells per mL in each group (mean± SD) after VEGF-NA at the 96-hour time-point was as follows: Normoxic+VEGF-NA: 5.6 × 106±3.2 × 106; Hypoxic+VEGF-NA: 1.4 × 106±5.0 × 105; Hypoxic+ET-A+VEGF-NA: 1.7 × 106±4.5 × 105; and Hypoxic+ET-B+VEGF-NA: 1.2 × 106±5.8 × 105. The reduction in the number of podocytes was accompanied by increased expression of pro-apoptotic AIF, SMAC, and p53. These data suggest that protection by ET-A antagonism may be largely mediated by availability of VEGF in podocytes (Figure 5B).
Figure 5. VEGF-NA decreased the beneficial effects of ET-A blockade on podocyte counts and pro-apoptotic signaling.
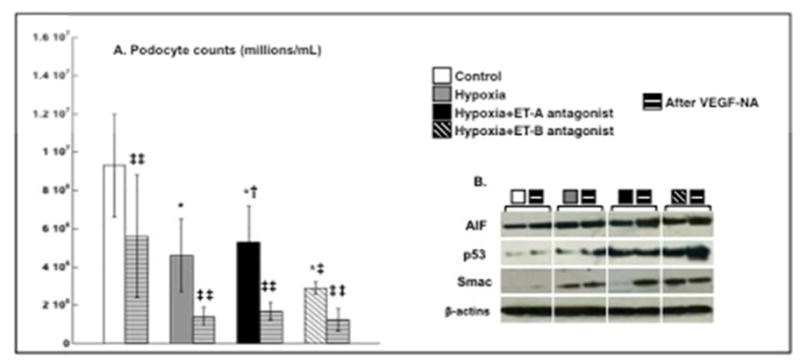
Incubation for 96 hours with VEGF-NA further decreased cell count (A) and diminished the protective actions of ET-A on cell survival and apoptotic signaling (B), suggesting the ET-A antagonism-induced protection is largely mediated by VEGF.
* p<0.05 vs. Normoxic; † p<0.05 vs. Hypoxic; ‡ p<0.05 vs. Hypoxic+ET-A; ‡‡ p<0.05 vs. untreated.
Discussion
Podocytes are increasingly recognized as a major target of ET-A receptor blockers. Preservation of podocyte integrity and function has been suggested as a central mechanism behind the improvements in proteinuria in CKD patients treated with ET-A antagonists, regardless of the etiology26. The current study extends our previous findings on the therapeutic efficacy of ET antagonists on the stenotic kidney in chronic RVD. Using a hypoxic in vitro setting, we observed that podocytes exposed to chronic hypoxia (as occurs in the stenotic kidney23) are progressively damaged, a process that may be partly driven by a sustained decrease in VEGF and accelerated apoptosis. ET-A antagonism significantly improved podocyte survival, preserved VEGF bioavailability, and decreased apoptotic signaling, effects that were not observed after ET-B blockade or VEGF-NA. Therefore, our study suggests that preservation of VEGF is a prominent mechanism by which ET-A antagonism protects the integrity and survival of podocytes in the ischemic kidney. These findings contribute to the understanding of ET-A induced renoprotection that is consistently observed in several pre-clinical and clinical studies, as in turn also support the notion of VEGF as a potential therapeutic target to protect the kidney.
Podocytes are the major component of the glomerular filtration barrier. Quantification of podocytes in urine has been proposed as a marker of renal injury27 since damage and loss of podocytes may reflect the severity of glomerular disease. Podocytes are terminally differentiated epithelial cells and it is still not fully clear whether their regeneration or proliferation occur once they are severely damage20. However, emerging evidence suggest that several therapeutic interventions that recover renal function are mainly driven by podocyte protection26, 28, indicating that podocytes may be a therapeutic target for repair and recovery. Among these interventions is the use of antagonists of the ET receptors. Podocytes have a fully functional ET system and express both ET-A and -B receptors29. Previous studies have shown that podocytes can generate and secrete ET-1 in vitro as exposure of podocytes to ET-1 induce a disruption of their cytoskeleton30, 31. Recent preclinical and clinical studies support the notion that ET-A antagonism reduces proteinuria in CKD possibly by improving podocyte health32. Our previous studies in a swine model of RVD demonstrated that the ET-1/ET-A pathway is up-regulated and plays a prominent pathophysiological role in deteriorating RBF and GFR as well as in promoting inflammation and fibrosis8, 9, 14. Furthermore, blunted GFR and glomerulosclerosis in this model are associated with increased excretion of nephrin and attenuated expression of podocin, suggesting substantial podocyte damage. However, the mechanisms of ET-A mediated podocyte damage and protection are unclear.
Our results extend our previous in vivo studies8, 9, 14 by elucidating potential underlying mechanisms of podocyte protection by ET-A antagonism. We first observe that blockade of the ET-A, but not ET-B receptors improved the release of VEGF by podocytes under hypoxia to a greater extent. Indeed, untreated and ET-B antagonist treated hypoxic podocytes showed a smaller VEGF production (compared to normoxic) after 96 hours. Although hypoxia may serve as a powerful stimulus for release of VEGF, this may be the case in the early phase but not when hypoxia in renal cells is sustained, which concurs with studies from our and other laboratories on in vivo and in vitro settings33, 34. This may be partly driven by alteration of the hypoxia-induced factor (HIF)-1α/VEGF axis combined with possible loss or exhaustion of VEGF sources22, 35. VEGF is a pivotal cytokine for maintenance and generation of the vascular networks by mediating endothelial cell differentiation, survival, proliferation, and migration towards formation of new vessels and vascular repair36. Importantly, podocytes are one of the major sources of renal VEGF and present fully functional VEGF receptors19, 37, 38. VEGF is also necessary for podocyte survival and function via interaction with nephrin through VEGFR2, which together with podocin are crucial slit-diaphragm associated proteins for podocyte integrity. Loss of VEGF in the advanced stage of renal disease is thought to be driven by the loss of podocytes39, 40, underscoring the importance of VEGF for renal health. Our study shows that VEGF bioavailability and expression is improved after ET-A but not ET-B antagonism, accompanied by restored expression of podocin and nephrin. Furthermore, the generation of s-Flt1, which acts as a VEGF “trap” thus reducing the availability of VEGF and its effects41, was dramatically increased after ET-B antagonism, indicating that blockade of ET-B receptors do not lead to protection of podocytes and may further compromise their survival. The s-Flt1 receptor has important physiological functions in guiding VEGF effects in vascular sprouting42, for example, but also has been implicated in the pathophysiology of pre-eclampsia43 and CKD44 when is elevated. Therefore, our data may extend our recent studies by offering a potential mechanistic explanation for the deleterious effects of single ET-B antagonism in the stenotic kidney of the swine model of RVD8.
In line with these observations, cell counts were only improved after ET-A antagonism, underscoring a potentially important role of VEGF on podocyte viability. Hypoxia is a powerful stimulus for renal apoptosis45, 46 and we showed that the ET-1/ET-A pathway contributes to apoptosis in the stenotic kidney14. Our study shows that hypoxic podocytes have increased expression of pro-apoptotic factors such as AIF, BAX, p-53, and SMAC, suggesting that hypoxia may stimulate both caspase-independent and -dependent pathways47–49. We also observed that hypoxia increased Cyt-C, a factor necessary for activation of apoptosis especially when coupled with BAX50, which was also elevated. Hypoxia also blunted the expression of pro-survival p-akt although anti-apoptotic BCL-2 was up-regulated, possibly reflecting a potential (but likely insufficient) mechanism to counteract the enhanced pro-apoptotic activity. The increased expression of pro-apoptotic mediators (and reduction in cell counts) was accompanied by changes in podocyte morphology suggestive of apoptotic cell death (e.g.: nuclear condensation and fragmentation51, 52). Notably, most of these changes were restored only by ET-A antagonism whereas the expression of all the pro-apoptotic factors was further increased by ET-B blockade.
Our study strongly suggests that blockade of the ET-A receptors may protect podocytes by preserving VEGF activity after performing a series of experiments after VEGF-NA. The use of VEGF inhibitors is among the first line of therapy in some types of cancer. However, VEGF inhibition carries significant renal damage, proteinuria, and hypertension, underscoring the prominent role of VEGF in the kidneys53. It is possible that some of these deleterious effects are driven by accelerated loss of podocyte since we observed that VEGF-NA further decreased cell counts and increased the expression of some of the pro-apoptotic factors in hypoxic podocytes. More importantly, these effects were also observed after ET-A receptor blockade, suggesting that the beneficial effects of ET-A antagonism on podocytes are largely dependent on VEGF bioavailability. These findings offer a mechanistic explanation and a pathophysiological link between renal dysfunction, damage, and loss of VEGF that were recovered after chronic ET-A (but not ET-B) antagonism in the swine model of chronic RVD8, 9.
Our study has some limitations. Although the in vitro setting offers an opportunity to elucidate mechanisms, we are aware that the degree of hypoxia was relatively severe and of short duration, which may not reflect the level of renal hypoxia or its full development and pathophysiological consequences in the stenotic kidney. It is also possible that other factors not investigated in this study may play a role in causing podocyte damage in addition to loss of VEGF, such as transforming growth factor (TGF)-β54, which has a powerful anti-angiogenic and pro-fibrotic effect55 and, as we have shown, was up-regulated in the stenotic kidney but attenuated by ET-A blockade8. Nevertheless, our study represents a mechanistic extension of our in vivo studies that showed the therapeutic efficacy of clinically available ET-A antagonists for renoprotection in RVD8, 9. Furthermore, being that podocytes are a central component of the glomerular filtration barrier and the vital role VEGF plays in podocyte health, a mechanistic understanding of their protection by ET-A antagonists may open new avenues for future research to protect the kidney that may go beyond RVD and contribute to the treatment of other forms of chronic renal disease.
Methods
All experiments and measurements were performed in triplicates
Podocyte Cell Culture Protocol
All experiments were carried out using aseptic techniques. All culturing of the human podocytes was performed as per guidelines from Celprogen Inc., CA. Media was acquired from Celprogen and contains all growth factors and nutrients necessary for culturing and sub-culturing human podocyte primary cells. Experiments were carried out with cells from passage 7 to 12. Expression of WT-1 was measured by western blot before initiation of the experiments to determine whether cells preserve podocyte characteristics.
Cell Culture
Human podocytes were obtained from Celprogen Inc., Torrance, Ca (cat # E36036-08). Cells were thawed in 37°C water bath and 500 μL of warmed human podocyte complete growth media was added. A complete growth media made specifically for the proliferation and culturing of human podocytes (Celprogen Inc., Torrance, CA, cat# M36036-08S) that contains 10% fetal bovine serum (FBS) as well as a standard mixture of Penicillin and Streptomycin (10,000U/ mL) was used. The cells were gently re-suspended and transferred to a 15 mL conical tube, a process that was repeated two more times to ensure full transfer. Cells were then centrifuged for five minutes at 100 G, supernatant was discarded, and cells re-suspended in 500 μL of media and transferred to a T75 flasks coated with human podocyte cell culture extracellular matrix (ECM) (Celprogen Inc., Torrance, CA, cat# E36036-08-T25 or T75). Cells were then allowed to grow to >80% confluency in 5% CO2 and 19% O2 water-jacketed incubator at 37°C before splitting in a 1:3 ratio to ensure enough viable cells for the experiments, as per company guidelines.
We expanded in equal concentrations into 110 flasks. Briefly, cells were then divided for proliferative purposes and at the time of experimentation the cells were at passage 9–10 and morphology of cells was monitored throughout (sub-culturing). Cells were washed twice with 1X PBS for 3 minutes each and incubated in 5 mL of 0.05% trypsin for 5 minutes or until they were fully detached. 500 μL of fresh media were added and supernatant was transferred to a 10 mL conical tube and centrifuged at 100 G for 5 minutes. Supernatant was removed and cells then re-suspended in 3 mL of media. 1 mL was transferred into ECM coated T75 flask containing 9 mL of warmed Human Podocyte Complete Growth Media with Serum and Antibiotics.
Cells were grown to >80% confluency in ECM coated T75 flasks. Cells were washed twice with 1X PBS for 3 minutes and then 5 mL of 0.05% trypsin added to release the cells from the flask. The flasks were incubated at 37°C for 5 minutes and trypsinization was stopped by addition of 5 mL of serum-enriched growth media and cell suspension transferred into a 15 mL conical tube and centrifuged at 100 G for 5 minutes. Supernatant was removed from the conical tube and cells were re-suspended in 5 mL of growth media for distribution into new ECM coated flasks for experimentation. Then, 500 μL of cell suspension was added to an ECM coated T25 flask along with 7.5 mL of media.
Cell counting
Cells were washed twice with 1X PBS for 3 minutes each and incubated in 5 mL of 0.05% trypsin for 5 minutes or until they were fully detached. 500 μL of fresh media were added and supernatant was transferred to a 10 mL conical tube and centrifuged at 100 G for 5 minutes. The cell supernatant was discarded and cells were suspended in 100 uL of media. To this 10 uL of Trpyan Blue 0.04% (Fisher Scientific) was added. 10 uL of the cell suspension was then added on to a hemocytometer and four areas were counted and averaged in each sample, and multiplied by 10/11 dilution factor to correct for the cell to Trypan ratio and then by 104 to find cells per mL, as described56. This was done to the 0-hour control and in all 96-hour cell groups. This process was carried out within 5 minutes of detaching the cells from growing flasks to reduce the reduction of cell viability. The cells were then centrifuge for 5 minutes and supernatant was removed. Cells were then lysed in the same way as the other groups. All cells counts for each square were above 100. Very few dead or dying cells were seen possibly be due to the dead cells releasing from the flask or being removed during the washing process. The average of four fields per sample was used for cell counting and expressed as mean+/−SD (please see results). All cell groups were counted in this manner.
Experimental protocol
General characteristics
A total of 110 flasks of cells were created, 108 flasks were used for the experiments and 2 used for cell count at 0 hour. Cells were incubated overnight, allowing cells to attach to the flasks before the media was changed. Flasks were then randomly designated as hypoxic injury or normoxic cells after overnight incubation.
Hypoxic injury
Normoxic conditions were generated by incubation of the cells at 5% C02 with 19% O2, whereas severe hypoxic conditions were generated by incubation of the cells at 5% CO2 and 1% O2. Oxygen levels were controlled using nitrogen gas injection in accordance with previous protocols57.
Treatment with endothelin-receptor antagonists
We first performed a pilot study to determine the dose of ET antagonist to be used in our experiments. Dosage was determined after performing a cell survival experiment using concentrations of 1 mg/mL, 0.1 mg/mL, and 0.01 mg/mL of either ET-A or ET-B antagonist. This study allowed us to determine that concentration of ET-A or ET-B antagonists of 1 mg/mL and 0.1 mg/mL were unsuitable for the experimentation due to significant decreased cell-count and low protein content at time points of interest. Cells were observed under normoxic and hypoxic conditions for 1, 24, and 96 hours to determine the point when the cells became nonviable. These experiments required 48 flasks of cells. The cells were collected and lysed using 0.0125% Triton X. The protein content of each was analyzed using a standard Bicinchromatic Acid (BCA) Protein Assay Kit (Fisher Thermoscientific Product# 23227).
After identification of the appropriate dose, hypoxic and normoxic groups were subsequently split into control, treated with endothelin receptor (ET)-A antagonist, or treated with ET-B antagonist. ET-A (ABT-627, atrasentan) and ET-B (A-192621) antagonists (Abbvie, North Chicago, IL) were suspended in 10% DMSO and 90% DI water to a final concentration of 10 mg/mL, DMSO was added to the solutions in order to make the antagonists soluble. 7.5 μL of drug suspension was added to each flask to bring the final drug concentration per flask to 0.01 mg/mL. These experiments required 21 flasks of cells (20 for the experiments and 1 for the cell count at baseline)
Addition of vascular endothelial growth factor neutralizing antibody
In a separate set of experiments following the above protocols, additional groups were incubated with a VEGF neutralizing antibody (VEGF-NA, R&D Systems, AF-293-NA). The purpose of these studies was to determine whether ET-A blockade protects podocytes by restoring VEGF bioavailability. Anti-VEGF antibody was reconstituted according to instructions included with antibody. Anti-VEGF antibody was added to a final concentration of 0.1 ug/mL of cell culture media, as previously shown58. Groups were as follows: Normoxic, Normoxic+VEGF-NA treated, Hypoxic, Hypoxic+VEGF-NA treated, Hypoxic ET-A treated, Hypoxic ET-A+VEGF-NA treated, Hypoxic ET-B treated, and Hypoxic ET-B+VEGF NA treated. These experiments required 41 flasks (40 for the experiments and 1 for the cell count at baseline). Cell homogenates and supernatant were collected as before at 1, 24, and 96 hours and stored at −80 °C.
The treatments with ET receptor antagonists and the addition of VEGF-NA were performed using a one-time dose. This strategy was decided to reduce chances of variability, keep total DMSO concentration low, to have the ability to quantify total soluble factors produced during the study, and to account effects of soluble markers produced during drug induction and hypoxic injury.
Podocyte counts
To determine the impact of hypoxia and treatments, cell counting was performed in all groups at 0 (average original number of plated podocytes for all the groups: 2.6 × 107) and 96 hours for comparisons.
Images of cells were taken at the 96-hour time point (x100) before being counted and lysed. Only one time point was chosen to image in order to reduce the amount of time the groups spent outside of their hypoxic or normoxic environments and reduce the chance of variability due to temperature change or infection. The 0 hour and 96 hour groups were re-suspended in 1 mL of media and 100 μL of Trypan Blue 0.04% (Fisher Scientific). Cells were counted using a hemocytometer following an established protocol56 and as we previously described33. Following counting, cells were centrifuged and lysed by adding 1 mL of 0.0125% TritonX to each cell pellet.
Sample Collection
Baseline or 0 hour for the experiment for all groups was considered the time at which the ET-A or ET-B antagonists were introduced. Time points of 1, 24 and 96 hours after drug introduction were collected in order to determine the time-dependent changes in podocyte number and subsequent expression or concentration of apoptotic and survival factors, as described below. Cell supernatant and homogenates were collected from each group at 1, 24, and 96 hours after drug introduction. In general, 1 and 24 were chosen to show how quickly the cells began to decline after hypoxic insult. The 96 hours time point was chosen to show an exacerbation of hypoxic injury over an extended period. Samples were then transferred to microcentrifuge tubes and stored at −80°C for subsequent measure of protein expression via western blot or concentration of growth factors via ELISA.
Proteome Profiler Arrays
In order to determine whether and how the apoptotic cascade plays a role in hypoxia-induced podocyte damage and how ET antagonism may protect podocytes, a Proteome Profiler Human Apoptosis Array Kit (R&D Systems, Minneapolis, MN, cat#ARY009) was obtained to identify apoptotic factors. Two control and 2 treated groups were chosen: Normoxic 72 hour control, hypoxic 72 hour control, hypoxic ET-A treated 72 hour group, and hypoxic ET-B 72 hour treated group. The 72-hour time point was chosen in order to comply with each kit’s recommended minimum protein loading. All reagents and membranes were provided with each kit and experiment performed following vendors’ instructions.
Briefly, membranes were blocked using blocking buffer provided with the kit for one hour on the bench top at room temperature, then washed three times (using kit’s buffer), and 250 μg of protein was loaded into each well and incubated overnight at 4C on a shaker plate. After overnight incubation, a secondary antibody cocktail was reconstituted and membranes incubated for one hour at room temperature, then washed three times and then incubated in streptavidin-HRP for 30 minutes at room temperature. After a single wash, membranes were incubated for 1 minute in a chemiluminescence reagent mix and then developed. Biomarkers that expressed the highest were then quantified by western blot as described below.
Westen Blotting
Total protein concentration of each sample was determined by BCA, as previously described7. A Bio-Rad Criterion TGX Precast Gel Any Kd or 18% agarose (if the target molecular weight was low) were then loaded with samples of 8 ug of total protein with 4x reducing Laemmli SDS-Sample Buffer (Boston Bio products, Ashland, MA, cat #BP-110R). Gels were run using Bio-Rad Criterion Cell (Bio-Rad, Hercules, CA, cat# 165-6001) at 100 volts for 1 hour or until adequate separation was achieved. Gels were removed and transferred to nitrocellulose using Criterion Blotter (Bio-Rad, Hercules, CA, cat# 170-407) at 100 volts at 4C for 45 minutes. Nitrocellulose membranes were incubated in Ponceau S Solution (Sigma Aldrich, St Louis, MO, cat # P7170) to visual total protein and to assess transfer.
Membranes were washed using tris buffered saline with 0.1% volume/volume tween (TBST) added and blocked using 5% weight/volume nonfat dry milk reconstituted in TBST. All primary and secondary antibodies were diluted using 5% nonfat dry milk. Primary antibodies for VEGF, podocin, nephrin, pro-apoptotic apoptosis induced factor (AIF), BAX, p53, SMAC, Cytochrome C, and HSP60, and anti-apoptotic p-AKT and BCL-2 (Santa Cruz Biotechnologies, Santa Cruz, CA) were used. All primary antibodies were diluted to 1:200 in 5% milk and incubated overnight in 4 °C. β-Actins was used as a loading control and diluted 1:500 in 5% milk and incubated at 1 hour at room temperature or overnight at 4°C. The following day membranes were washed three times in TBST followed by incubation with goat anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (HRP) for 1 hour diluted to 1:5000 (Santa Cruz Biotechnologies, CA). After one hour incubation gels were washed 3 times in TBST and SuperSignal West Pico Chemiluminescent Substrate (Fisher Thermoscientific Products, cat# 34078) was added according to manufacturer’s instructions for 3–5 minutes. Membranes were developed using Carestream Kodak BioMax light film (Sigma-Aldrich, St. Louis, MO, cat. # 8194540) with exposure lengths varying from 1–20 minutes. Protein expression of each factor (relative to β-actins, 2 representative bands per group are shown in each figure) was quantified and the average expression (experiments were performed in triplicates) was used for comparisons.
Enzyme Linked Immunosorbent Assay (ELISA)
ET-1 in media from normoxic and hypoxic cells was quantified by ELISA (R&D Systems, Minneapolis, MN). Furthermore, human VEGF and s-Flt1 ELISAs (R&D Systems, Minneapolis, MN) were quantified in media from normoxic, hypoxic, and ET antagonist-treated groups. Samples were collected and stored following manufacturer’s guidelines. 96-well tray provided with the ELISA was loaded and analyzed following vendors’ instructions. Results for VEGF and s-Flt1 were expressed in pg of VEGF or s-Flt1/μg of total protein after 1, 24 and 96 hours of hypoxia, whereas for ET-1 were expressed in pg of ET-1/μg of total protein after 96 hours of hypoxia.
Statistical Analysis
Results are expressed as mean ± SD. Comparisons within groups were performed using paired student’s t-test, and among groups using one-way ANOVA. Statistical significance was accepted for p<0.05.
Acknowledgments
Acknowledgments-Sources of support
This work was supported by grant HL095638, HL51971, and GM104357 (ARC); and by grant 18490005 from the American Heart Association (ARC). ET-A and ET-B antagonists were a generous gift by Abbvie Laboratories, IL.
Footnotes
Disclosures: ARC serves as a consultant for Actelion Pharmaceuticals US, Inc.
References
- 1.US Renal Data System U and Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States NIoH. National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2012. [Google Scholar]
- 2.Vupputuri S, Kimes TM, Calloway MO, Christian JB, Bruhn D, Martin AA, Nichols GA. The economic burden of progressive chronic kidney disease among patients with type 2 diabetes. J Diabetes Complications. 2014;28:10–6. doi: 10.1016/j.jdiacomp.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 3.Ritchie J, Green D, Chrysochou C, Chalmers N, Foley RN, Kalra PA. High-risk clinical presentations in atherosclerotic renovascular disease: prognosis and response to renal artery revascularization. Am J Kidney Dis. 2014;63:186–97. doi: 10.1053/j.ajkd.2013.07.020. [DOI] [PubMed] [Google Scholar]
- 4.Textor SC, Lerman LO. Reality and renovascular disease: when does renal artery stenosis warrant revascularization? Am J Kidney Dis. 2014;63:175–7. doi: 10.1053/j.ajkd.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Textor SC, Misra S, Oderich GS. Percutaneous revascularization for ischemic nephropathy: the past, present, and future. Kidney international. 2013;83:28–40. doi: 10.1038/ki.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chade AR, Rodriguez-Porcel M, Grande JP, Krier JD, Lerman A, Romero JC, Napoli C, Lerman LO. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation. 2002;106:1165–71. doi: 10.1161/01.cir.0000027105.02327.48. [DOI] [PubMed] [Google Scholar]
- 7.Chade AR, Rodriguez-Porcel M, Grande JP, Zhu X, Sica V, Napoli C, Sawamura T, Textor SC, Lerman A, Lerman LO. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler Thromb Vasc Biol. 2003;23:1295–301. doi: 10.1161/01.ATV.0000077477.40824.52. [DOI] [PubMed] [Google Scholar]
- 8.Chade AR, Stewart NJ, Peavy PR. Disparate effects of single endothelin-A and -B receptor blocker therapy on the progression of renal injury in advanced renovascular disease. Kidney international. 2014;85:833–44. doi: 10.1038/ki.2013.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chade AR, Tullos N, Stewart NJ, Surles B. Endothelin-a receptor antagonism after renal angioplasty enhances renal recovery in renovascular disease. Journal of the American Society of Nephrology : JASN. 2015;26:1071–80. doi: 10.1681/ASN.2014040323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chade AR, Best PJ, Rodriguez-Porcel M, Herrmann J, Zhu X, Sawamura T, Napoli C, Lerman A, Lerman LO. Endothelin-1 receptor blockade prevents renal injury in experimental hypercholesterolemia. Kidney international. 2003;64:962–9. doi: 10.1046/j.1523-1755.2003.00170.x. [DOI] [PubMed] [Google Scholar]
- 11.Chade AR, Krier JD, Textor SC, Lerman A, Lerman LO. Endothelin-a receptor blockade improves renal microvascular architecture and function in experimental hypercholesterolemia. Journal of the American Society of Nephrology : JASN. 2006;17:3394–403. doi: 10.1681/ASN.2006060635. [DOI] [PubMed] [Google Scholar]
- 12.Hargrove GM, Dufresne J, Whiteside C, Muruve DA, Wong NC. Diabetes mellitus increases endothelin-1 gene transcription in rat kidney. Kidney international. 2000;58:1534–45. doi: 10.1046/j.1523-1755.2000.00315.x. [DOI] [PubMed] [Google Scholar]
- 13.Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM. Endothelin receptor A-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia. 2011;54:979–88. doi: 10.1007/s00125-010-2021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelsen S, Hall JE, Chade AR. Endothelin-A receptor blockade slows the progression of renal injury in experimental renovascular disease. American journal of physiology Renal physiology. 2011;301:F218–25. doi: 10.1152/ajprenal.00089.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartner A, Klanke B, Cordasic N, Amann K, Schmieder RE, Veelken R, Hilgers KF. Statin treatment reduces glomerular inflammation and podocyte damage in rat deoxycorticosterone-acetate-salt hypertension. J Hypertens. 2009;27:376–85. doi: 10.1097/hjh.0b013e32831997d6. [DOI] [PubMed] [Google Scholar]
- 16.Opocensky M, Kramer HJ, Backer A, Vernerova Z, Eis V, Cervenka L, Certikova Chabova V, Tesar V, Vaneckova I. Late-onset endothelin-A receptor blockade reduces podocyte injury in homozygous Ren-2 rats despite severe hypertension. Hypertension. 2006;48:965–71. doi: 10.1161/01.HYP.0000245117.57524.d6. [DOI] [PubMed] [Google Scholar]
- 17.Wagner SJ, Craici IM, Grande JP, Garovic VD. From placenta to podocyte: vascular and podocyte pathophysiology in preeclampsia. Clinical nephrology. 2012;78:241–9. doi: 10.5414/cn107321. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Shon E, Kim CS, Kim JS. Renal podocyte injury in a rat model of type 2 diabetes is prevented by metformin. Exp Diabetes Res. 2012;2012:210821. doi: 10.1155/2012/210821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tufro A, Veron D. VEGF and podocytes in diabetic nephropathy. Semin Nephrol. 2012;32:385–93. doi: 10.1016/j.semnephrol.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barton M, Tharaux PL. Endothelin and the podocyte. Clin Kidney J. 2012;5:17–27. doi: 10.1093/ckj/sfs001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB J. 2006;20:1706–8. doi: 10.1096/fj.05-5680fje. [DOI] [PubMed] [Google Scholar]
- 22.Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, Lerman LO. Cortical microvascular remodeling in the stenotic kidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1854–9. doi: 10.1161/01.ATV.0000142443.52606.81. [DOI] [PubMed] [Google Scholar]
- 23.Gomez SI, Warner L, Haas JA, Bolterman RJ, Textor SC, Lerman LO, Romero JC. Increased hypoxia and reduced renal tubular response to furosemide detected by BOLD magnetic resonance imaging in swine renovascular hypertension. American journal of physiology Renal physiology. 2009;297:F981–6. doi: 10.1152/ajprenal.90757.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Funk J, Ott V, Herrmann A, Rapp W, Raab S, Riboulet W, Vandjour A, Hainaut E, Benardeau A, Singer T, Jacobsen B. Semiautomated quantitative image analysis of glomerular immunohistochemistry markers desmin, vimentin, podocin, synaptopodin and WT-1 in acute and chronic rat kidney disease models. Histochem Cell Biol. 2015 doi: 10.1007/s00418-015-1391-6. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, Dong J, Yuan L, Liang C, Ren K, Zhang W, Fang F, Shen J. Nephrin and podocin loss is prevented by mycophenolate mofetil in early experimental diabetic nephropathy. Cytokine. 2008;44:85–91. doi: 10.1016/j.cyto.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Dhaun N, Melville V, Blackwell S, Talwar DK, Johnston NR, Goddard J, Webb DJ. Endothelin-A receptor antagonism modifies cardiovascular risk factors in CKD. Journal of the American Society of Nephrology : JASN. 2013;24:31–6. doi: 10.1681/ASN.2012040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White WM, Garrett AT, Craici IM, Wagner SJ, Fitz-Gibbon PD, Butters KA, Brost BC, Rose CH, Grande JP, Garovic VD. Persistent urinary podocyte loss following preeclampsia may reflect subclinical renal injury. PLoS One. 2014;9:e92693. doi: 10.1371/journal.pone.0092693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhaun N, Webb DJ, Kluth DC. Endothelin-1 and the kidney--beyond BP. Br J Pharmacol. 2012;167:720–31. doi: 10.1111/j.1476-5381.2012.02070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rebibou JM, He CJ, Delarue F, Peraldi MN, Adida C, Rondeau E, Sraer JD. Functional endothelin 1 receptors on human glomerular podocytes and mesangial cells. Nephrol Dial Transplant. 1992;7:288–92. doi: 10.1093/oxfordjournals.ndt.a092130. [DOI] [PubMed] [Google Scholar]
- 30.Morigi M, Buelli S, Angioletti S, Zanchi C, Longaretti L, Zoja C, Galbusera M, Gastoldi S, Mundel P, Remuzzi G, Benigni A. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: implication for permselective dysfunction of chronic nephropathies. Am J Pathol. 2005;166:1309–20. doi: 10.1016/S0002-9440(10)62350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morigi M, Buelli S, Zanchi C, Longaretti L, Macconi D, Benigni A, Moioli D, Remuzzi G, Zoja C. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol. 2006;169:1965–75. doi: 10.2353/ajpath.2006.051331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barton M. Therapeutic potential of endothelin receptor antagonists for chronic proteinuric renal disease in humans. Biochim Biophys Acta. 2010;1802:1203–13. doi: 10.1016/j.bbadis.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Chade AR, Kelsen S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: a novel potential therapeutic approach. American journal of physiology Renal physiology. 2012;302:F1342–50. doi: 10.1152/ajprenal.00674.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudnicki M, Perco P, Enrich J, Eder S, Heininger D, Bernthaler A, Wiesinger M, Sarkozi R, Noppert SJ, Schramek H, Mayer B, Oberbauer R, Mayer G. Hypoxia response and VEGF-A expression in human proximal tubular epithelial cells in stable and progressive renal disease. Lab Invest. 2009;89:337–46. doi: 10.1038/labinvest.2008.158. [DOI] [PubMed] [Google Scholar]
- 35.Iliescu R, Fernandez SR, Kelsen S, Maric C, Chade AR. Role of renal microcirculation in experimental renovascular disease. Nephrol Dial Transplant. 2010;25:1079–87. doi: 10.1093/ndt/gfp605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–45. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 37.Bertuccio C, Veron D, Aggarwal PK, Holzman L, Tufro A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. J Biol Chem. 2011;286:39933–44. doi: 10.1074/jbc.M111.241620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foster RR, Satchell SC, Seckley J, Emmett MS, Joory K, Xing CY, Saleem MA, Mathieson PW, Bates DO, Harper SJ. VEGF-C promotes survival in podocytes. American journal of physiology Renal physiology. 2006;291:F196–207. doi: 10.1152/ajprenal.00431.2005. [DOI] [PubMed] [Google Scholar]
- 39.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T Animal Models of Diabetic Complications C. Mouse models of diabetic nephropathy. Journal of the American Society of Nephrology : JASN. 2009;20:2503–12. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brosius FC, Khoury CC, Buller CL, Chen S. Abnormalities in signaling pathways in diabetic nephropathy. Expert Rev Endocrinol Metab. 2010;5:51–64. doi: 10.1586/eem.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.George EM, Cockrell K, Adair TH, Granger JP. Regulation of sFlt-1 and VEGF secretion by adenosine under hypoxic conditions in rat placental villous explants. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1629–33. doi: 10.1152/ajpregu.00330.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chappell JC, Taylor SM, Ferrara N, Bautch VL. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell. 2009;17:377–86. doi: 10.1016/j.devcel.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaisbuch E, Whitty JE, Hassan SS, Romero R, Kusanovic JP, Cotton DB, Sorokin Y, Karumanchi SA. Circulating angiogenic and antiangiogenic factors in women with eclampsia. Am J Obstet Gynecol. 2011;204:152, e1–9. doi: 10.1016/j.ajog.2010.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsui M, Takeda Y, Uemura S, Matsumoto T, Seno A, Onoue K, Tsushima H, Morimoto K, Soeda T, Okayama S, Somekawa S, Samejima K, Kawata H, Kawakami R, Nakatani K, Iwano M, Saito Y. Suppressed soluble Fms-like tyrosine kinase-1 production aggravates atherosclerosis in chronic kidney disease. Kidney international. 2014;85:393–403. doi: 10.1038/ki.2013.339. [DOI] [PubMed] [Google Scholar]
- 45.Khan S, Cleveland RP, Koch CJ, Schelling JR. Hypoxia induces renal tubular epithelial cell apoptosis in chronic renal disease. Lab Invest. 1999;79:1089–99. [PubMed] [Google Scholar]
- 46.Tanaka T, Miyata T, Inagi R, Kurokawa K, Adler S, Fujita T, Nangaku M. Hypoxia-induced apoptosis in cultured glomerular endothelial cells: involvement of mitochondrial pathways. Kidney international. 2003;64:2020–32. doi: 10.1046/j.1523-1755.2003.00301.x. [DOI] [PubMed] [Google Scholar]
- 47.Artus C, Boujrad H, Bouharrour A, Brunelle MN, Hoos S, Yuste VJ, Lenormand P, Rousselle JC, Namane A, England P, Lorenzo HK, Susin SA. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29:1585–99. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luu Y, Bush J, Cheung KJ, Jr, Li G. The p53 stabilizing compound CP-31398 induces apoptosis by activating the intrinsic Bax/mitochondrial/caspase-9 pathway. Experimental cell research. 2002;276:214–22. doi: 10.1006/excr.2002.5526. [DOI] [PubMed] [Google Scholar]
- 49.Hsiao WT, Tsai MD, Jow GM, Tien LT, Lee YJ. Involvement of Smac, p53, and caspase pathways in induction of apoptosis by gossypol in human retinoblastoma cells. Mol Vis. 2012;18:2033–42. [PMC free article] [PubMed] [Google Scholar]
- 50.Eskes R, Antonsson B, Osen-Sand A, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou J-C. Bax-induced Cytochrome C Release from Mitochondria Is Independent of the Permeability Transition Pore but Highly Dependent on Mg2+ Ions. J Cell Biol. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding G, Reddy K, Kapasi AA, Franki N, Gibbons N, Kasinath BS, Singhal PC. Angiotensin II induces apoptosis in rat glomerular epithelial cells. American journal of physiology Renal physiology. 2002;283:F173–80. doi: 10.1152/ajprenal.00240.2001. [DOI] [PubMed] [Google Scholar]
- 52.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney international. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 53.Hara A, Wada T, Furuichi K, Sakai N, Kawachi H, Shimizu F, Shibuya M, Matsushima K, Yokoyama H, Egashira K, Kaneko S. Blockade of VEGF accelerates proteinuria, via decrease in nephrin expression in rat crescentic glomerulonephritis. Kidney international. 2006;69:1986–95. doi: 10.1038/sj.ki.5000439. [DOI] [PubMed] [Google Scholar]
- 54.Ziyadeh FN. Different roles for TGF-beta and VEGF in the pathogenesis of the cardinal features of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82(Suppl 1):S38–41. doi: 10.1016/j.diabres.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 55.Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, Chander PN, Goligorsky MS. Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. Journal of the American Society of Nephrology : JASN. 2015;26:817–29. doi: 10.1681/ASN.2013101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stephenson E, Jacquet L, Miere C, Wood V, Kadeva N, Cornwell G, Codognotto S, Dajani Y, Braude P, Ilic D. Derivation and propagation of human embryonic stem cell lines from frozen embryos in an animal product-free environment. Nat Protoc. 2012;7:1366–81. doi: 10.1038/nprot.2012.080. [DOI] [PubMed] [Google Scholar]
- 57.Lu H, Kapur G, Mattoo TK, Lyman WD. Hypoxia decreases podocyte expression of slit diaphragm proteins. Int J Nephrol Renovasc Dis. 2012;5:101–7. doi: 10.2147/IJNRD.S27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eirin A, Zhu XY, Li Z, Ebrahimi B, Zhang X, Tang H, Korsmo MJ, Chade AR, Grande JP, Ward CJ, Simari RD, Lerman A, Textor SC, Lerman LO. Endothelial outgrowth cells shift macrophage phenotype and improve kidney viability in swine renal artery stenosis. Arterioscler Thromb Vasc Biol. 2013;33:1006–13. doi: 10.1161/ATVBAHA.113.301164. [DOI] [PMC free article] [PubMed] [Google Scholar]