

Abstract
Traumatic brain injury (TBI) elicits immediate neuroinflammatory events that contribute to acute cognitive, motor, and affective disturbance. Despite resolution of these acute complications, significant neuropsychiatric and cognitive issues can develop and progress after TBI. We and others have provided novel evidence that these complications are potentiated by repeated injuries, immune challenges and stressors. A key component to this may be increased sensitization or priming of glia after TBI. Therefore, our objectives were to determine the degree to which cognitive deterioration occurred after diffuse TBI (moderate midline fluid percussion injury) and ascertain if glial reactivity induced by an acute immune challenge potentiated cognitive decline 30 days post injury (dpi). In post-recovery assessments, hippocampal-dependent learning and memory recall were normal 7 dpi, but anterograde learning was impaired by 30 dpi. Examination of mRNA and morphological profiles of glia 30 dpi indicated a low but persistent level of inflammation with elevated expression of GFAP and IL-1β in astrocytes and MHCII and IL-1β in microglia. Moreover, an acute immune challenge 30 dpi robustly interrupted memory consolidation specifically in TBI mice. These deficits were associated with exaggerated microglia-mediated inflammation with amplified (IL-1β, CCL2, TNFα) and prolonged (TNFα) cytokine/chemokine expression, and a marked reactive morphological profile of microglia in the CA3 of the hippocampus. Collectively, these data indicate that microglia remain sensitized 30 dpi after moderate TBI and a secondary inflammatory challenge elicits robust microglial reactivity that augments cognitive decline.
Keywords: Traumatic Brain Injury, Fluid percussion injury, Microglia, Astrocytes, Cognitive decline, Lipopolysaccharide, Neuroinflammation
1. Introduction
TBI is the leading cause of neurological disability in the United States and there is significant risk of neuropsychiatric illness after injury (Centers for Disease Control and Prevention, 2015). For instance, depressive complications and cognitive decline often occur in humans after TBI (Rosenthal et al., 1998, Hart et al., 2012). Indeed, 15–30% of TBI patients experience cognitive decline over time (Ruff et al., 1991, Himanen et al., 2006, Salmond et al., 2006, Till et al., 2008, Almeida et al., 2015) that may continue to progress after injury (Millis et al., 2001, Wang et al., 2012). For example, in a 5 year study, 27% of the TBI patients presented with reduced verbal fluency and verbal list learning (Till et al., 2008). The underlying cause of these neuropsychiatric complications after TBI is unclear but may be related to ongoing neuroinflammatory processes (Norden et al., 2014b).
Mounting evidence indicates that microglia-mediated inflammation persists long after TBI. For instance, clinical studies report elevated metabolic activity, white matter abnormalities and microglial activation persisting long after injury (Brooks et al., 2000, Ramlackhansingh et al., 2011). These findings are paralleled in rodent models of both penetrating and diffuse brain injury. For example, CD68+ microglia were detected in the lesion site 1 year after controlled cortical impact injury (CCI) (Loane et al., 2014) and increased major histocompatibility complex (MHC)II and CD68 were also detected 14–30 days after diffuse brain injury in rats (Ziebell et al., 2012) and mice (Fenn et al., 2014). These studies provide evidence that microglia maintain a primed profile after TBI (Witcher et al., 2015). In the context of cognition, enhanced glial inflammation and concomitant cognitive decline are evident after TBI. For example, repeated closed head injury (CHI) in mice resulted in increased GFAP (astrocytes) and Iba-1 (microglia) labeling, and evidence of white matter damage that corresponded with deficits in hippocampal-dependent learning 12–18 months after injury (Mouzon et al., 2014). Clinical studies also link glial reactivity with cognitive impairment after injury. For example, PET scan shows amplified microglial activation in the thalamus that was associated with lower performance on tests of processing speed (Ramlackhansingh et al., 2011). Thus, inflammation is likely a contributing factor of cognitive decline after TBI.
Microglia and astrocytes have dynamic roles in coordinating responses between the immune system and the brain (Norden et al., 2014a, Norden et al., 2015). Therefore, persistent glial reactivity after TBI (Fenn et al., 2014, Hazra et al., 2014) may impair immune surveillance and cause maladaptive behavioral responses (Norden et al., 2014b). We and others have identified a population of “primed” microglia in the aged brain that become hyper-reactive following acute immune challenge (lipopolysaccharide (LPS) or E.coli) (Chen et al., 2008, Barrientos et al., 2009b, Henry et al., 2009). These primed microglia have elevated MHCII expression and produce exaggerated levels of pro-inflammatory cytokines (Henry et al., 2009) that correspond with impaired cognitive performance (Chen et al., 2008, Barrientos et al., 2009b, Barrientos et al., 2010) and prolonged depressive-like behavior (Godbout et al., 2008). Similarly, 30 days after a diffuse brain injury in mice, an acute immune challenge (LPS) caused a hyper-reactive microglial inflammatory response that triggered the development of depressive-like behavior (Fenn et al., 2014). Thus, a primed profile of microglia and astrocytes after TBI may set the stage for exaggerated responses to acute challenges precipitating the development of neuropsychiatric complications.
The goals of this study were to determine the extent of cognitive decline after diffuse TBI and ascertain if immune challenge potentiates cognitive decline by augmenting glia-mediated inflammation. The midline fluid percussion injury (FPI) model was used because it causes mild neuronal pathology including diffuse axonal injury and transient neurological deficits that recapitulate complications after mild to moderate concussive head trauma (Morales et al., 2005, Lifshitz et al., 2007, Lifshitz, 2009). Here we extend our previous findings to show that mice recover from TBI and have normal learning and memory 7 dpi, but begin to develop cognitive deficits by 30 dpi. Moreover, microglia and astrocytes have heightened inflammatory gene expression 30 dpi and induction of an immune response 30 dpi further amplifies microglial activation and exacerbates deficits in memory recall.
2. Materials and Methods
2.1 Mice and LPS Injections
Adult (3 mo) male BALB/c mice were bred at The Ohio State University (OSU). Mice were individually housed and maintained at 25° C under a 12 h light/12 h dark cycle with ad libitum access to food and water. For injections, mice were intraperitoneally (i.p.) injected 30 dpi with saline or LPS (0.33mg/kg; serotype 0127:B8, Sigma) 1–2 h before the start of the dark phase (Godbout et al., 2005, Fenn et al., 2012). The LPS dosage was selected because it elicits a pro-inflammatory cytokine response in the brain resulting in a transient sickness response (24 h) in uninjured adult mice (Berg et al., 2004, Godbout et al., 2005). To control for sickness induction and subsequent recovery, food intake and body weight were determined over a 72 hour time course (data not shown). All procedures were in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and were approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee.
2.2 Midline Fluid Percussion Injury
Mice received a midline diffuse TBI using a fluid percussion injury (FPI) apparatus (Custom Design & Fabrication, Richmond, VA) as previously described (Fenn et al., 2014, Fenn et al., 2015). This diffuse injury occurs in the absence of a contusion, does not induce tissue cavitation or gross neuronal loss, and causes diffuse axonal injury in the neocortex, hippocampus, and dorsolateral thalamus (Kelley et al., 2006, Kelley et al., 2007, Bachstetter et al., 2013, Fenn et al., 2014). To prepare for FPI, mice were anesthetized with 5% isoflurane and were stabilized using head ear bars in a stereotaxic frame. Anesthesia was maintained with continuous inhalation of isoflurane (1.5–3%) through a nose-cone. Next, a midline craniectomy between bregma and lambda was performed with a 3 mm outer diameter trephine. A rigid Luer-loc needle hub was secured over the craniectomy and capped. After 4–6 h, injury was induced by filling the injury hub with saline and imposing a 10 ms pulse of saline (1.2 atmospheres; 670–720 mV) onto the dura through the hub (Witgen et al., 2006, Lifshitz et al., 2007, Fenn et al., 2014). All Sham controls received the same procedure without the fluid pulse. Immediately after injury, the injury hub was removed, dural integrity was confirmed, and mice were evaluated for injury severity using the self-righting test (Lifshitz et al., 2007). Mice with a confirmed dura breach were euthanized immediately after injury and excluded from the study. Self-righting inclusion criteria were based on our previous work with BALB/c mice (Fenn et al., 2014). Only mice with a moderate TBI were used: Sham ≤ 60 s; 60 s < mild ≤ 200 s; 200 s < moderate ≤ 540 s; severe > 540 s.
2.3 Recovery and Nesting Behavior
For all Sham and TBI mice, recovery after injury was monitored post-operatively (e.g. grooming, body weight, nesting behavior) for 7 consecutive days and then weekly until the 30 day end point. After Sham or TBI, mice were singly housed. Body weight was determined daily and expressed as percentage of weight change from baseline (body weight prior to injury). In addition, mice were provided a compressed cotton square bedding (2 inches × 2 inches) after Sham or TBI. The percentage of the bedding torn was determined on each day (24 hour intervals) for 7 days post TBI. This tearing was assessed in 25% increments and nesting behavior was expressed as percentage of bedding torn.
2.4 Barnes Maze
Hippocampal-dependent learning and memory recall were determined using the Barnes maze paradigm as previously described (Bach et al., 1995) with a few modifications. These modifications include using dim light and white noise to serve as an aversive stimulus, to facilitate movement of BALB/c in the maze. The acclimation phase consisted of 2 trials: 1) mice were guided to the escape hole and allowed 2 minutes in the escape hole, and 2) with the addition of white noise, mice were guided to an incorrect or “dummy” hole and then guided to the escape hole and allowed 2 min in the escape hole without the aversive stimulus. Trials were initiated 30 min apart for each mouse. Following the acclimation day, the position of the escape box was shifted 180° to its permanent location for the remainder of the paradigm. The acquisition phase consisted of four trials per day for four consecutive days. Mice were placed in the center of the maze under a semi-opaque container for 10 seconds (s). Recording began when the container was lifted and white noise was simultaneously added. The white noise ceased once the mouse entered the escape hole. Each trial lasted 180 s or until the mouse entered the escape hole. If the mouse did not reach the escape hole in the allotted time then it was guided to the hole. Speed, distance traveled, time to find the escape hole, and the number of primary errors (errors made before encountering escape hole) were determined. To evaluate memory recall, mice were subjected to a probe trial. The probe trial consisted of one 60 s trial, 1, 3 or 7 days after the completion of acquisition phase. In the probe trial, the escape hole was replaced with a dummy hole and mice were allowed to explore the maze for 60 s. Speed, distance, time to find the dummy hole in the previous location of the escape hole, time spent in the escape quadrant, and the number of primary errors were determined. Mice in the acquisition phase and probe trial were tracked, recorded, and analyzed using the EthoVision XT 8.5 tracking software (Noldus Information Technology).
2.5 Percoll-Enrichment of Microglia and Astrocytes
Microglia and astrocytes were isolated from whole brain homogenates using a Percoll density gradient as previously described (Norden et al., 2014a). In brief, tissues were homogenized and cell pellets were re-suspended in 70% isotonic Percoll. A discontinuous Percoll density gradient (70%, 50%, 35%, 0%) was layered and centrifuged for 20 min at 2000 × g. Enriched microglia were collected from the 70% and 50% interphase, where approximately 90% of the cells are CD11b+ microglia (Henry et al., 2009). Enriched astrocytes were collected from the 50% and 35% interphase, where approximately 75% of the cells are GLAST-1+ astrocytes (Norden et al., 2014a).
2.6 RNA Isolation and RT-PCR
For Percoll enriched microglia and astrocytes, mRNA was isolated using the PrepEase kit (USB, CA). RNA was reverse transcribed to cDNA and real-time (RT)-PCR was performed using the Applied Biosystems Taqman® Gene Expression Assay-on-Demand Gene Expression protocol. In brief, experimental cDNA was amplified by real-time PCR. Target cDNA (e.g., IL-1β, TNFα, CCL2) and reference cDNA (glyceraldehyde-3-phosphate dehydrogenase; GAPDH) were amplified simultaneously using an oligonucleotide probe with a 5′ fluorescent reporter dye (6-FAM). Fluorescence was determined on an ABI PRISM 7300-sequence detection system (Applied Biosystems). Data were analyzed using the comparative threshold cycle (Ct) method and results are expressed as fold difference from Sham or Sham-Saline.
2.7 Flow Cytometric Assessment of Brain Macrophages
Cells were percoll-enriched from the brain and were assayed for surface protein expression of CD11b and CD45 as previously described (Henry et al., 2009, Fenn et al., 2014). In brief, Fc receptors were blocked with anti-CD16/CD32 antibody (eBioscience, San Diego, CA). Cells were labelled with rat anti-mouse CD11b-APC and CD45-PerCP antibodies (eBioscience, San Diego, CA). Surface protein expression was determined using a Becton-Dickinson FACSCaliber four color cytometer (BD, Franklin Lakes, NJ). Ten thousand events characterized as microglia/macrophages were recorded. For each antibody, gating was determined based on appropriate negative isotype stained controls from the representative cell population. Macrophages were differentiated from microglia based on CD45High labeling (Wohleb et al., 2012, Wohleb et al., 2014). Flow data were analyzed using FlowJo software (Tree Star, San Carlos, CA).
2.8 Doublecortin and 5′Bromodeoxyuridine Labeling
The presence of proliferating cells was determined using 5′bromodeoxyuridine (BrdU) labeling and immature neurons were identified by doublecortin (DCX) labeling. In brief, BrdU (Roche, 10mg/ml) was dissolved in warm PBS and then filtered. Mice were injected with 50mg/kg BrdU (Sahinkaya et al., 2014) 20 and 24 hours after LPS injection and perfused 48 hours after the last injection. For quantification of BrdU+ and DCX+ cells, every 6th section (30 μm) throughout the hippocampus was collected. For BrdU labeling, sections were washed, denatured in 2N HCL at 37° C for 30 min, and blocked (1% BSA/PBS, 5% NGS, 0.1% TX). Sections were then incubated in primary antibody (rat anti-BrdU, AbD Serotec, mAb 1:1000) at 4° C for 24 hours and then secondary (Alexa Fluor 488) at room temperature for 1 hour. For DCX labeling, sections were washed, blocked (1% BSA/PBS, 5% donkey serum, 0.5% TX) for 1 hour, then incubated in primary antibody (goat anti-DCX, Santa Cruz Biotechnology, mAb 1:200) at 4° C for 48 hours and then secondary (Alexa Fluor 488) at 4°C overnight. Sections were mounted on slides, then cover-slipped with Fluoromount G (Beckman Coulter, Inc.), and stored at −20°C.
Fluorescent sections were visualized using an epi-fluorescent Leica DM5000B microscope. Images were captured using a Leica DFC300 FX camera and imaging software. Total number of BrdU+ or DCX+ cells in the granule cell layer of the dentate gyrus (DG) in the hippocampus was estimated by multiplying the number of cells counted in each section by six.
2.9 Amyloid Beta Precursor Protein (APP) Labeling
To identify neuronal injury or degradation, tissue sections were labeled with APP. Free-floating sections were washed in PBS, then blocked (5% NGS, 1% BSA, in PBS) and incubated with rabbit anti-mouse APP (1:125; Life Technologies) overnight at 4°C. Next, sections were washed in PBS and incubated with a fluorochrome-conjugated secondary antibody (Alexa Fluor 488). Sections were mounted on slides, then cover-slipped with Fluoromount G (Beckman Coulter, Inc.), and stored at −20°C.
2.10 Immunohistochemistry and Digital Image Analysis for Iba1 and GFAP
Iba-1 and GFAP labeling was performed as previously described (Fenn et al., 2014). In brief, the brain was collected after transcardial perfusion with sterile PBS (PBS, pH 7.4) and 4% formaldehyde. Brains were post-fixed in 4% formaldehyde for 24 hours and incubated in 30% sucrose for 48 hours. Fixed brains were frozen with isopentane (−78° C) and sectioned (30 μm) using a Microm HM550 cryostat. Brain regions within the hippocampus (DG, CA1, CA3) were identified by reference markers in accordance with the stereotaxic mouse brain atlas (Paxinos and Franklin, 2004). To label for Iba-1 or GFAP, sections were placed free-floating in cryoprotectant (30% polyethylene glycol, 30% ethylene glycol, 40% 0.2M phosphate buffer) until staining. Next, sections were washed in PBS, then blocked (5% NGS, 1% BSA, 0.5% TritonX in PBS) and incubated with rabbit anti-mouse Iba-1 (1:1000; Wako Chemicals) or rabbit anti-mouse GFAP antibody (1:500; Dako) overnight at 4°C. Next, sections were washed in PBS and incubated with a fluorochrome-conjugated secondary antibody (Alexa Fluor 594 or Alexa Fluor 588). Sections were mounted on slides, then cover-slipped with Fluoromount G (Beckman Coulter, Inc.), and stored at −20°C.
Fluorescent images were visualized using an epifluorescent Leica DM5000B microscope and captured using a Leica DFC300 FX camera and imaging software. To quantify the phenotypic changes of microglia, digital image analysis of labeling was performed (Donnelly et al., 2009). For each mouse 10–12 representative images were taken at 20× magnification in the dentate gyrus, CA1, and CA3 regions of the hippocampus. Adobe Photoshop was used for image conversion and Metamorph for thresholding functions and analysis of proportional area. Proportional area was reported as the average percent area in the positive threshold for all representative pictures.
2.11 Microglia and Astrocyte Analysis and Reconstruction
Glia Reconstruct was used to analyze GFAP and Iba-1 labeled images as previously described (Kongsui et al., 2014a, Kongsui et al., 2014b, Walker et al., 2014) but with modifications. In brief, images were captured using a Leica DFC300 FX (20X) as described above. 3–5 representative hippocampal sections per experimental mouse were used. Prior to analysis, the signal range was defined as occurring between X–Y that included all cellular material but no background, a cumulative spectra (CTS) analysis (Kongsui et al., 2014b) was undertaken (Johnson and Walker, 2015). CTS analysis involves identifying the number of pixels within an image that occur at each of the 256 possible pixel intensities and then, expressing the number of pixels occurring at each pixel intensity as a percentage of the total number of pixels within the image. This method provides a significantly more transparent method for quantifying immuno-labelled material (Johnson and Walker, 2015). Following CTS analysis, which provides information only on shifts in the density of immune-labelled material, both astrocyte and microglial morphologies were digitally reconstructed using Matlab (The MathWorks, Inc.) executed algorithms that use variance minimization strategy to extract the signal from background. Size filtering was additionally used to eliminate non-cellular material (Radler et al., 2015). Using this approach the key morphological features of glia can be examined. For example, maximum cell perimeter, total cell length, cell body perimeter, number of primary processes, number of nodes (branch points), total length of all processes, and total volume of all processes can be measured. In addition, the area encompassed by the entire cell was measured as the convex hull area, determined from the polygon created from straight lines connecting the most distal points of the microglial processes. Pixel information was converted into μm values to provide information on relative size, area or volume of microglia and astrocytes.
2.12 Statistical Analysis
To ensure a normal distribution, data were subjected to the Shapiro-Wilk test using Statistical Analysis Systems (SAS) statistical software (Cary, NC). Observations greater than 3 interquartile ranges from the first and third quartile were considered outliers and were excluded in the subsequent analysis. To determine significant main effects and interactions between main factors, data were analyzed using one-way (i.e., TBI and LPS) or two-way (i.e., TBI × LPS) ANOVA using the General Linear Model procedures of SAS. When appropriate, differences between treatment means were evaluated by an F-protected t-test using the Least-Significant Difference procedure of SAS. All data are expressed as treatment means + standard error of the mean (SEM). Values were considered to be significantly different at p-values < 0.05.
3. Results
3.1 Mice Recover to Baseline Nesting Behavior by 7 Days after Diffuse TBI
We have reported that exposure to midline fluid percussion injury induces a moderate and diffuse brain injury in which mice return to baseline locomotor activity and motor coordination by 7 dpi (Fenn et al., 2014). To confirm this, the average time to self-right (i.e., righting reflex), body weight and nesting behavior were determined (Fig.1). Fig.1A shows that mice subjected to TBI had an increased time to regain consciousness and self-right after injury (314.5±15.6 s) compared to Sham controls (18.0±2.3 s) (main effect of TBI, F(1,85) = 403.6, p < 0.0001). In addition, nesting behavior and body mass were monitored each day after injury until day 7. Fig.1B shows that nesting behavior was reduced after TBI (F(5,50) = 86.6, p < 0.0001) and this was dependent on time (F(5,50) = 5.9, p < 0.0001). For instance, post-hoc analysis indicates that nesting behavior was reduced on days 1–4 (p < 0.01, for each day) after TBI, but returned to baseline levels by 6 dpi (Fig.1B). Body mass was also reduced following TBI in a time dependent manner (Fig.1C, F(6,52) = 217.0, p < 0.0001). Body mass approached baseline by 7 dpi (Fig.1C), and returned to baseline by 14 dpi (not shown). Overall, these data along with our previous findings (Fenn et al., 2014, Fenn et al., 2015) indicate that the mice received a moderate injury and that the injured mice return to baseline activity, motor coordination, and nesting behavior by 7 dpi.
Figure 1. Mice recover to baseline nesting behavior by 7 days after diffuse TBI.
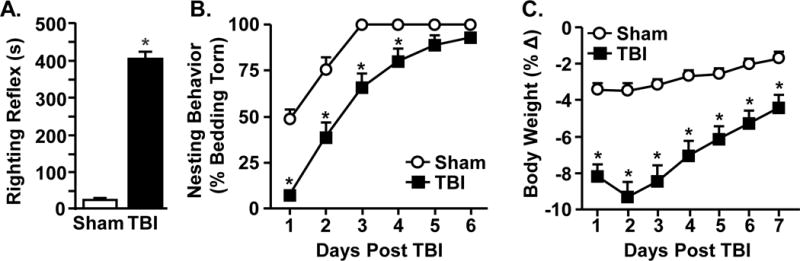
Adult mice were subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). B) The average time to self-right after injury was determined immediately after injury (n = 40–46). After injury, mice were monitored daily for 6 days and C) nesting behavior and D) percent change in body weight were determined. These data include all mice used in this study. Graphs represent the mean ± SEM (n = 25–31). Means with (*) are different from Sham controls (p<0.05)
3.2 TBI Induces Acute Deficits in Retrograde Memory Recall
In the first assessment of cognition after TBI, retrograde memory recall was determined in adult mice. In this experiment, mice were trained for 4 days to find the escape (acquisition), received a sham or TBI, and memory recall was probed 7 dpi (Fig.2A). Fig.2B–D shows that mice acquired learning with time-dependent increases in velocity (Fig.2B, main effect of time, F(3,9) = 12.2, p < 0.0001), decreases in the time to find the escape hole (Fig.2C, main effect of time, F(3,9) = 38.2, p < 0.0001), and decreases in the number of errors (Fig.2D, main effect of time, F(3,9) = 7.3, p < 0.001).
Figure 2. TBI induces acute deficits in retrograde memory recall.
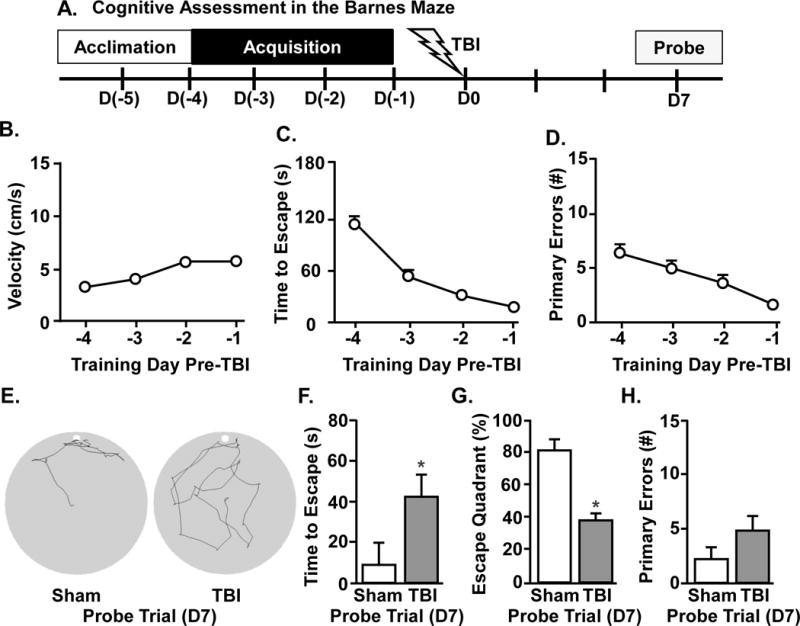
A) Representative time line of cognitive assessment in the Barnes maze. Naïve mice were acclimated to the Barnes maze, then trained for 4d to find the escape hole and then subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). Prior to TBI, B) velocity, C) time to find escape hole, and D) number of primary errors were determined during the acquisition phase. E) Representative search paths for Sham and TBI mice during the 60 s probe trial at 7 days following injury. Memory recall was determined during the probe trial by F) time to find escape, G) percent of time spent in escape quadrant, and H) number of primary errors. Graphs represent the mean ± SEM (n = 6). Means with (*) are different from Sham controls (p<0.05)
Next, mice received Sham or TBI and memory recall was determined in the probe trial at 7 dpi. As highlighted above, locomotor function (Fenn et al., 2014) and nesting behavior (Fig.1B) of TBI mice returns to baseline by 7 dpi. Fig.2E shows representative search patterns for Sham and TBI mice 7 dpi in the probe trial. The time to find the escape hole was increased during the probe trial in TBI mice compared to Sham controls (Fig.2F, main effect of TBI, F(1,9) = 9.4, p < 0.02). Moreover, TBI mice spent less time in the escape quadrant than shams (Fig.2G, main effect of TBI, F(1,10) = 24.2, p < 0.001). There was no difference in the number of errors made during the probe trial (Fig.2H), but TBI mice had a reduced percentage of correct head pokes into the escape hole compared to Sham mice (data not shown, F(1,9) = 12.6, p < 0.008). Taken together, these data indicate that TBI negatively affects retrograde memory recall.
3.3 TBI Does Not Interfere Acutely with Anterograde Learning and Recall
Our findings indicate that TBI disrupts retrograde memory recall. Therefore, anterograde learning and memory after TBI was assessed. Mice were subjected to TBI and acquisition in the Barnes maze started 7 dpi (day 7–10). Memory recall was probed 11 dpi (Fig.3A). These time points after injury were selected because injured mice return to baseline locomotor activity, motor coordination (Fenn et al., 2014) and nesting behavior by 7 dpi (Fig.1B). The ability to locate the escape was not impaired 7 dpi (Fig.3A–C) and both groups had similar escape times (Fig.3C) and made the similar number of errors (Fig.3D). Representative search patterns during the probe trial are shown (Fig.3E). Latency to escape, time spent in the escape quadrant, and errors during the probe trail were similar for both Sham and TBI mice (Fig.3E–H). These data indicate that anterograde learning and recall were unaffected 7 dpi.
Figure 3. TBI does not interfere acutely with anterograde learning and recall.
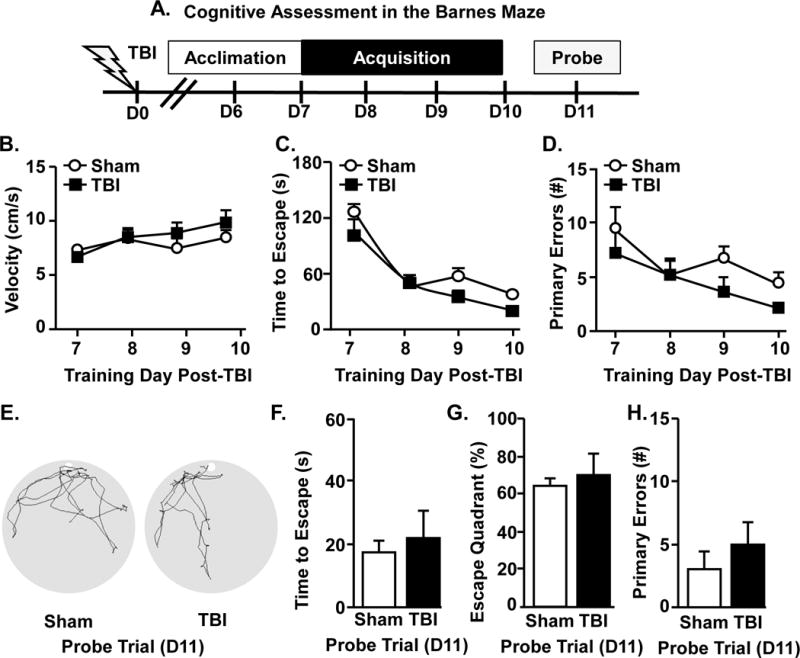
A) Representative timeline of cognitive assessment in the Barnes maze 7 days after injury. In this experiment, Sham and TBI mice were acclimated to the Barnes maze (6 dpi), then trained for 4 days to find the escape hole. 7 days after memory acquisition was determined over 4 training days and B) velocity, C) time to find escape hole, and D) number of primary errors were determined during the acquisition phase. E) Representative search paths of Sham and TBI mice during the 60 s probe trial at 11d following TBI. Memory recall was determined during the probe trial by F) time to find escape hole, G) percent of time spent in escape quadrant, and H) number of primary errors. Graphs represent the mean ± SEM (n = 6). Means with (*) are different from Sham controls (p<0.05).
3.4 Immune Challenge Exacerbates Deficits in Anterograde Learning that Develop 30 Days after TBI
We reported that microglial priming persisted 30 days post-TBI and that immune challenge exaggerated microglial activation and depressive-like behavior (Fenn et al., 2014). Here, diffuse TBI disrupted retrograde (Fig.2) but not anterograde learning and recall 7 dpi (Fig.3). Therefore, we assessed if cognitive decline was evident 30 dpi and if acute immune challenge 30 dpi impaired cognition. The acquisition in the Barnes maze started 27 dpi (day 27–30) for sham and TBI mice. After the completion of training, mice were challenged with LPS and memory recall was probed 33 dpi (Fig.4A).
Figure 4. Immune challenge exacerbates deficits in anterograde learning that develop 30 days after TBI.
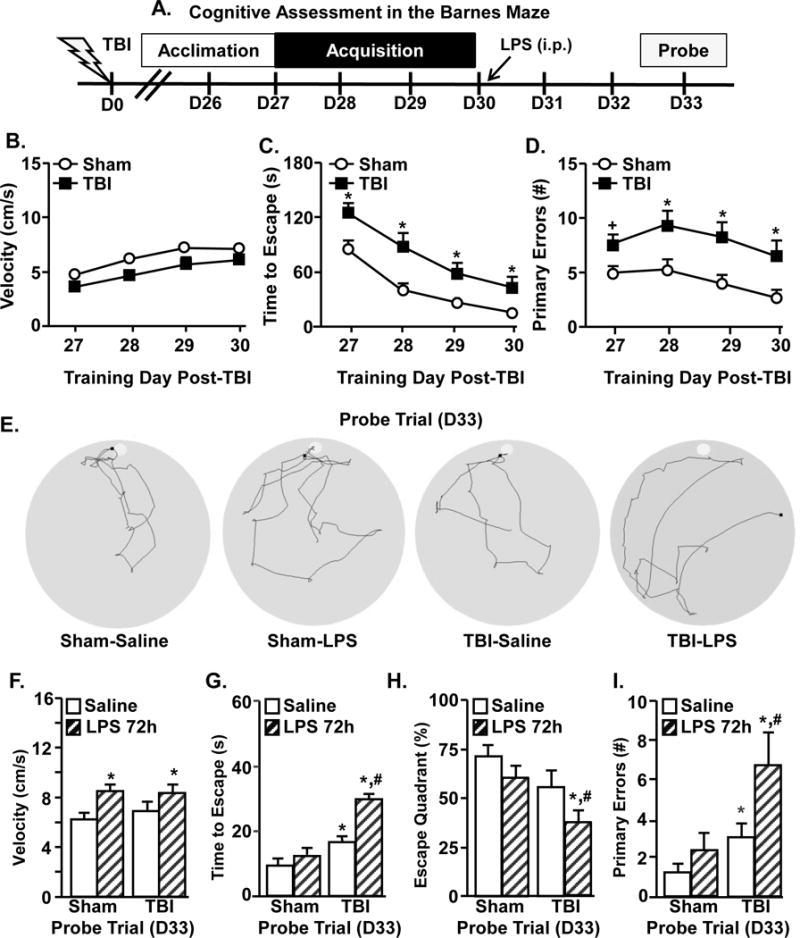
A) Representative time line of cognitive assessment in the Barnes maze starting 27 dpi. Mice were subjected to Sham or TBI and memory acquisition was determined over 4 training days (27–30 dpi) by B) velocity, C) time to find escape hole, and D) number of primary errors. Graphs represent the mean ±SEM (n = 19–25). Means with (*) are different from Sham controls (p<0.05) and means with (+) tend to be different from saline controls (p=0.06–0.10). Next, Sham and TBI mice received LPS or saline (0.33mg/kg) i.p. at the end of the 4th day of training (30 dpi). Memory recall was determined 72 hours after injection in the probe trial. E) Representative search paths for the probe trial. Recall was assessed by F) velocity, G) time to find escape hole, H) percent of time spent in escape quadrant, and I) number of primary errors. Bars represent the mean ±SEM (n = 8–12). Means with (*) are different from Sham-Saline controls (p<0.05) and means with (#) are different from all other treatment groups (p<0.05).
First, hippocampal learning was assessed in Sham and TBI mice 30 days after injury. During the acquisition phase of testing, the relative velocity (cm/s) was similar between the Sham and TBI mice (Fig.4B). While both Sham and TBI mice had daily improvement in finding the escape holes during the acquisition phase, there were significant differences between these groups. For example, the time to find the escape during the acquisition phase was increased by TBI (Fig.4C, main effect of TBI, F(3,41) = 38.2, p < 0.0001). Moreover, the number of errors made during the acquisition phase was increased by TBI (Fig.4D, main effect of TBI, F(3,41) = 28.56, p < 0.0001). Overall, these data indicate that there are deficits in anterograde learning by 30 days after TBI that were not present 7 days after injury.
Next, memory recall was determined in the same sham and TBI mice 72 hours after induction of an immune response (D33). Representative search patterns from the probe trial are shown (Fig.4E). This time point (72 h after LPS) was selected because, independent of age or injury, mice are neither sick nor lethargic (Godbout et al., 2008, Fenn et al., 2014). Fig.4F shows that walking velocity during the probe trial was increased by LPS (main effect of LPS, F(1,39)= 10.3, p < 0.005) but this increase was independent of TBI. These velocity data and our previous data (Godbout et al., 2008) indicate that LPS-injected adult mice return to baseline locomotion and activity by 72 hours. Thus, the probe trial data are not confounded by either lethargy or malaise.
The data from the probe trial indicates that LPS injection further augmented cognitive impairment in TBI mice. For instance, time to find the escape hole was increased by TBI (Fig.4G, main effect of TBI, F(1,39) = 9.14, p < 0.005) and this increase was exacerbated with LPS challenge (TBI × LPS interaction, F(1,39) = 4.6, p < 0.04). These effects were paralleled in the percent time spent in the escape quadrant and the number of primary errors. Post-hoc analysis revealed that the TBI-LPS group had the longest escape latency compared to all groups (Fig.4G, p < 0.05). In addition, TBI-LPS mice spent the least amount of time in the escape quadrant (Fig.4H, p < 0.05) and made the most errors (Fig.4I, p < 0.05) during the probe trial. Therefore, induction of an immune response 30 dpi further impairs cognition beyond TBI alone.
3.5 Neurogenesis in the Hippocampus is Not Altered by Immune Challenge 30 Days after TBI
We show that immune challenge further impairs hippocampal memory 30 dpi. Hippocampal neurogenesis is associated with enhanced neuronal plasticity and cognitive performance (Kheirbek and Hen, 2011). Moreover, neuroinflammatory processes can reduce neurogenesis (Valero et al., 2014) and damage neurons (Mouzon et al., 2014). Thus, neurogenesis and neuronal damage were examined 30 dpi in mice challenged with LPS. Representative images of BrdU+ cells in the dentate gyrus (DG) of the hippocampus 72 h after immune challenge are shown (Fig.5A). The number of BrdU+ cells in the DG was the same for TBI and LPS groups (Fig.5B). In addition, immature neurons in the hippocampus were labeled with DCX 72 h after LPS (Fig.5C). There were no significant differences in the number of DCX+ cells with TBI or LPS (Fig.5D). Furthermore, the positive control (ischemic brain injury, left panel) shows APP+ cells adjacent to the lesion (Fig.5E). In contrast, TBI mice showed limited APP expression after LPS challenge (Fig.5E). Last, hippocampal neuron loss 30 dpi was not detected by Fluorojade C staining in any of the treatment groups (data not shown). Collectively, induction of an immune response 30 dpi neither influenced neurogenesis nor caused significant neuronal damage in the HPC.
Figure 5. Neurogenesis in the hippocampus is not altered by immune challenge 30 days after TBI.
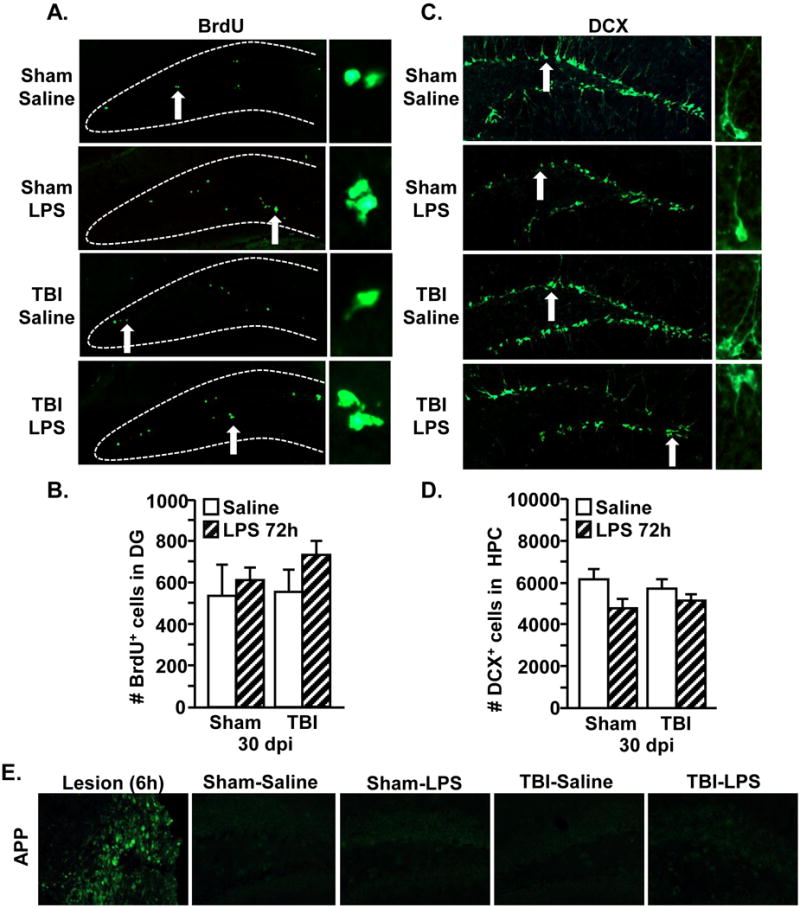
Adult mice were subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). At 30 days following TBI, mice were injected i.p. with saline or LPS (0.33mg/kg). Next, mice were injected i.p. twice with BrdU (50mg/kg), at 20 and 24 hours after LPS. Mice were sacrificed 72 hours after LPS and brains were collected, fixed and the hippocampus was sectioned. A) Representative images of BrdU labeling in the DG of the four treatment groups. Arrow depicts BrdU+ cell shown in the enlarged image (right). B) The number of BrdU+ cells was determined in the DG (n = 3–5). C) Representative images of DCX labeling in the hippocampus of the four treatment groups. Arrow depicts DCX+ cell shown in the enlarged image. D) The number of DCX+ cells was determined in the hippocampus. Bars represent the mean + SEM (n = 3–5). E) Representative images of APP labeling of ischemic injury positive control (Lesion) and APP labeling in the DG of the four treatment groups.
3.6 Amplified Microglia Activation in TBI (30 dpi) Mice after Immune Challenge
Neuroinflammation mediated by resident glia may underlie cognitive deficits 30 dpi. In support of this, a heightened inflammatory or primed profile of microglia and astrocytes persists months after TBI (Loane et al., 2014, Mouzon et al., 2014, Weil et al., 2014). In addition, primed (MHCII+) microglia that persist in the brain after TBI are hyper-reactive after peripheral immune challenge (Fenn et al., 2014). Therefore, the degree to which immune challenge elicited microglial reactivity 30 dpi and 24 hours after LPS was examined. As expected, MHCII mRNA expression was increased in TBI mice compared to sham controls (Fig.6C, main effect of TBI, F(1,36) = 7.1, p < 0.04), but was not influenced by LPS (not shown). Fig.6D–E show that mRNA expression of IL-1β, CCL2, and TNFα in microglia were increased 24 hours after LPS (main effect of LPS, F(1,36) = 7.8, p < 0.01, for each). Moreover, post-hoc analysis indicated that microglial expression of these cytokines was highest in the TBI-LPS mice compared to all other groups (Fig.6B–E, p < 0.05, for each). Notably, while there was a sham-saline control group represented in Figs.6D–E, there was not a TBI-saline group included. Nonetheless, these data highlight the LPS effect on microglial mRNA expression of IL-1β, CCL2, and TNFα between Sham and TBI (30 dpi) mice 24 h after immune challenge. Taken together, the neuroinflammatory profile of microglia was enhanced in mice that received a TBI 30 days prior to the LPS challenge.
Figure 6. Amplified Microglia Activation in TBI (30 dpi) mice after Immune Challenge.
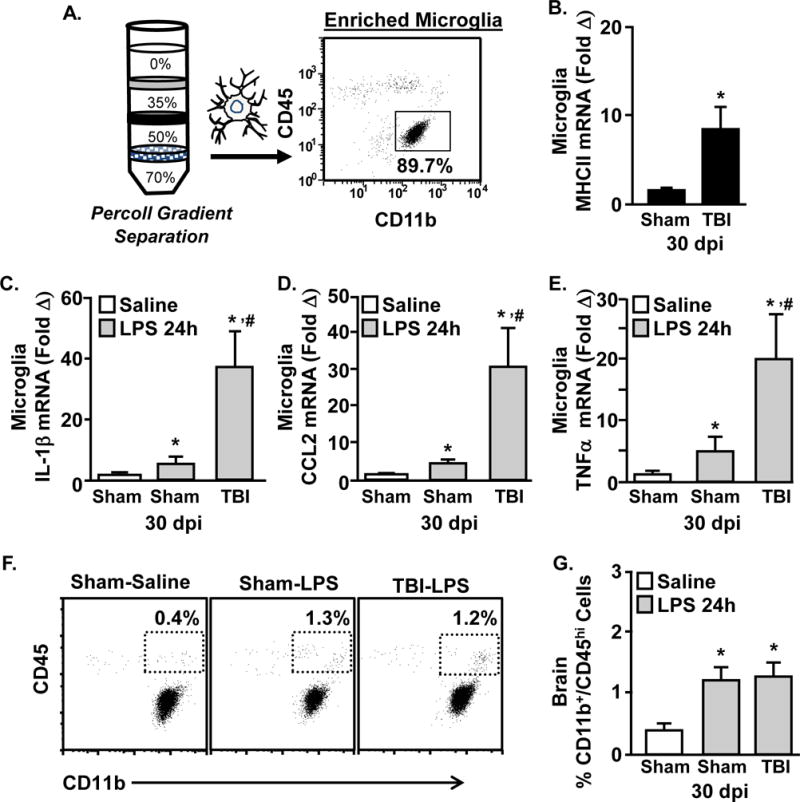
Adult mice were subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). 30 days following TBI, mice were injected i.p. with saline or LPS (0.33 mg/kg) and percoll-enriched cells were collected 24 hours later. A) Representative analysis of CD11b and CD45 labeling shows that 85% of the cells recovered from the 50–70% phase of Percoll are microglia (CD11b+/CD45low). The mRNA levels of B) MHCII, C) IL-1β, D) CCL2, and E) TNFα were determined in enriched microglia. Bars represent the mean + SEM (n = 9–12). Means with (*) are different from Sham-Saline and means with (#) are different from Sham-LPS mice (p<0.05). In a related study, mice were subjected to TBI and then a LPS injection 30 days later. Brain microglia and macrophages were percoll-enriched 24 h after LPS injection. F) Representative bivariate dot plots of CD11b and CD45 labeling are shown. G) The percentage of percoll-enriched cells from the brain that were CD11b+/CD45high. Bars represent the mean + SEM (n = 9–12). Means with (*) are different from Sham-Saline.
Because increased monocyte/macrophage (CD11b+/CD45high) recruitment to the brain with neuroinflammation can influence behavior (Wohleb et al., 2012), we also determined the percentage of CD45high monocytes/macrophages in the brain 24 h after LPS. The representative bivariate dot plots show a modest increase (0.5% to 1.25%) in brain macrophages (CD11b+/CD45high) 24 h after LPS challenge (Fig.6G, main effect of LPS, F(1,27) = 14.8, p < 0.001). Nonetheless, the increased presence of monocytes in the brain after LPS was not further enhanced in mice that received a TBI 30 days prior to immune challenge. Taken together, microglia in the brain of mice 30 days after TBI have an amplified neuroinflammatory mRNA profile after LPS challenge that was not associated with enhanced monocyte trafficking to the brain.
3.7 Prolonged Microglia Activation in TBI (30 dpi) Mice after Immune Challenge
Next, glial activation profiles were assessed 72 hours after LPS. Microglia (CD11b+/CD45low) and astrocytes (Glast-1+/CD11b−) were percoll-enriched 72 hours after LPS (Henry et al., 2009, Norden et al., 2014a) (Fig.7A) and inflammatory gene expression was determined. In astrocytes, GFAP mRNA expression was increased 30 dpi (Fig.7B, main effect of TBI, F(1,28) = 17.7, p < 0.0005), but were not further influenced by LPS (not shown). Vimentin mRNA was also increased 30 days after TBI (main effect of TBI, F(1,27) = 5.8, p < 0.05). Post-hoc analysis revealed that vimentin mRNA tended to be higher in TBI-LPS mice compared to all other groups (Fig.7C, p = 0.1). Fig.7D shows IL-1β mRNA in astrocytes was increased by TBI (main effect of TBI, F(1,28) = 6.1, p < 0.05) and tended to be higher with LPS (tendency of LPS, F(1,28) = 3.6, p =0.08). There was not an interaction between TBI and LPS for IL-1β expression in astrocytes. CCL2 mRNA in astrocytes was unaffected by either TBI or LPS (Fig.7E). Overall, there was increased mRNA expression of GFAP and IL-1β in enriched astrocytes 30 days after TBI, but these levels were not further enhanced 72 hours after LPS challenge.
Figure 7. Prolonged Microglia Activation in TBI (30 dpi) mice after Immune Challenge.
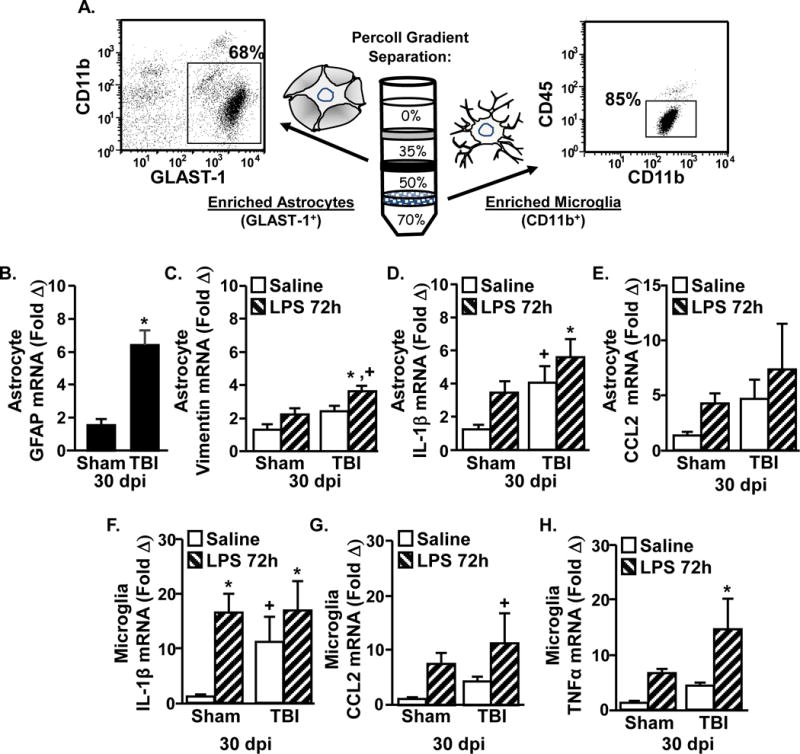
Adult mice were subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). 30 days following TBI, mice were injected i.p. with saline or LPS (0.33 mg/kg) and enriched astrocytes and microglia were collected 72 hours later. A) Representative analysis of Glast-1, CD11b, and CD45 labeling shows that 68% of the cells recovered from the 35–50% phase of Percoll are astrocytes (Glast-1+/CD11b−) and 85% of the cells recovered from the 50–70% phase of Percoll are microglia (CD11b+/CD45low). The mRNA levels of B) GFAP, C) vimentin, D) IL-1β, and E) CCL2 were determined in enriched astrocytes. The mRNA levels of F) IL-1β, G) CCL2, and H) TNFα were determined in enriched microglia. Bars represent the mean + SEM (n = 9–12). Means with (*) are different from Sham-Saline controls (p<0.05), and (+) tend to be different from Sham controls (p<0.1).
Microglial IL-1β mRNA was increased by LPS (main effect of LPS, F(1,33) = 7.2, p < 0.01). Post hoc analysis revealed that there was no difference in TBI-LPS mice compared with Sham-LPS mice (Fig.7F). In addition, microglia from TBI-Saline mice tended to have higher IL-1β mRNA expression than Sham-Saline controls (p = 0.06). Microglial CCL2 mRNA was increased by LPS (Fig.7G, main effect of LPS, F(1,36) = 7.8, p < 0.01). CCL2 mRNA tended to be highest in microglia from LPS-TBI mice (p = 0.08), but there was not a significant interaction between LPS and TBI at this time point. Microglial TNFα mRNA expression was also increased by LPS (Fig.7H, main effect of LPS, F(1,33) = 8.1, p < 0.01) and this increase tended to be influenced by TBI (LPS × TBI interaction, F(1,33) = 3.9, p = 0.06). For instance, post-hoc analysis confirmed that TNFα mRNA was highest in microglia from TBI-LPS mice compared to all other groups (p < 0.05). Collectively, there was prolonged microglial expression of TNFα in TBI mice after immune challenge.
3.8 Increased GFAP Immunoreactivity of Astrocytes 30 Days after TBI
Astrocytes maintain a more inflammatory mRNA profile 30 dpi with increased GFAP and IL-1β expression. To examine astrocytes specifically in the hippocampus (DG, CA1 and CA3, Fig.8A&B), GFAP immunoreactivity was determined 30 dpi. Consistent with the GFAP mRNA data, GFAP immunoreactivity was increased by TBI in the DG (Fig.8C, main effect of TBI, F(1,30) = 6.3, p < 0.05) and the CA1 (Fig.8D, main effect of TBI, F(1,30) = 19.1, p = 0.0001). GFAP labeling was not increased in the CA3 of the hippocampus 30 dpi (Fig.8E). Overall there was increased GFAP immunoreactivity in the hippocampus 30 dpi, but it was not further enhanced by immune challenge.
Figure 8. Increased GFAP immunoreactivity of astrocytes 30 days after TBI.
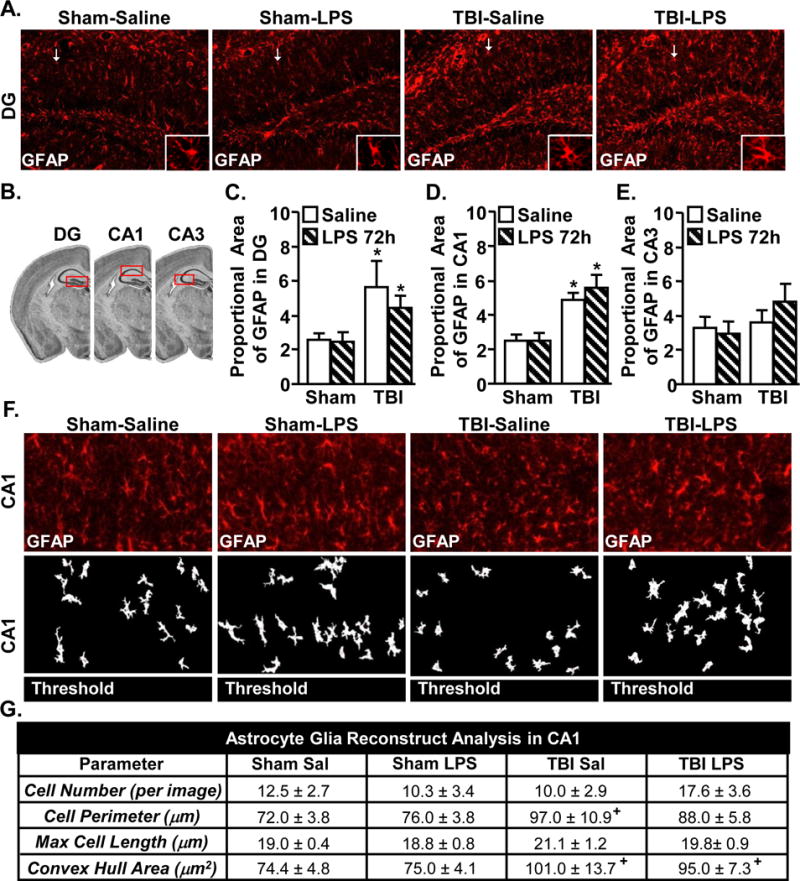
Adult mice were subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). 30 days following TBI, mice were injected i.p. with saline or LPS (0.33mg/kg) and mice were perfused and PFA fixed 72 hours later. Brains were post-fixed, frozen, and sectioned and GFAP expression was determined. A) Representative images of labeling for GFAP in the DG of the hippocampus. White arrow depicts the GFAP+ cell shown in the enlarged image. B) Illustration of DG, CA1, and CA3 regions within the hippocampus that were analyzed. Proportional area of GFAP labeling in the C) DG, D) CA1, and E) CA3. Bars represent mean ± SEM (n = 3–5). Means with (*) are different from Sham controls (p<0.05). F) Glia reconstruct representative CA1 images of GFAP labeling (top) and subsequent thresholded image (bottom). G) Glia Reconstruct images analyses of astrocytes in CA1 (n = 9–13). Table represents the mean + SEM (n = 3–5). Means with (+) are different from Sham controls (p=0.07).
To better understand cellular changes in astrocytes in the CA1, Glia Reconstruct was used to determine cell perimeter, length, and convex hull area. First, cumulative spectra analysis was used to find the appropriate pixel intensity for analysis. Images were then thresholded and analyzed. Representative GFAP labeling (top) and the corresponding thresholded images (bottom) from the CA1 are shown (Fig.8F). Analysis of the thresholded images in the CA1 (Fig.8G) indicated that TBI increased astrocyte perimeter (main effect of TBI, F(1,32) = 4.7, p < 0.05) and astrocyte area (main effect of TBI, F(1,32) = 6.1, p < 0.05). Cell number and max length were not significantly affected by TBI (30 dpi). These alterations in astrocyte morphology, however, were not further enhanced by LPS challenge. Similar Glia Reconstruct analysis revealed no significant morphological changes in the DG (data not shown). Overall, astrocytes retained a more reactive morphology in the hippocampus 30 dpi, but these alterations were not further enhanced by immune challenge.
3.9 Reactive Microglia in CA3 of TBI Mice after Immune Challenge
Consistent with microglial priming after TBI, Fig.6–7 indicate that microglia exhibited an amplified and prolonged inflammatory gene expression profile 30 dpi with amplified expression of IL-1β, CCL2 and TNFα. Iba-1 labeling was used to examine microglia after TBI and immune challenge in the hippocampus (DG, CA1 and CA3, Fig.9A). Notably, Iba-1 proportional area was not increased 30 dpi in the DG, CA1, or CA3 as a function of TBI alone (Fig.9C–E). Iba-1 immunoreactivity was increased by LPS in the CA3 (Fig.9E, main effect of LPS, F(1,28) = 5.6, p < 0.05). Moreover, post-hoc analysis revealed that Iba-1 immunoreactivity was highest in the TBI-LPS group compared to all groups (p < 0.05).
Figure 9. Reactive microglia in CA3 of TBI mice after immune challenge.
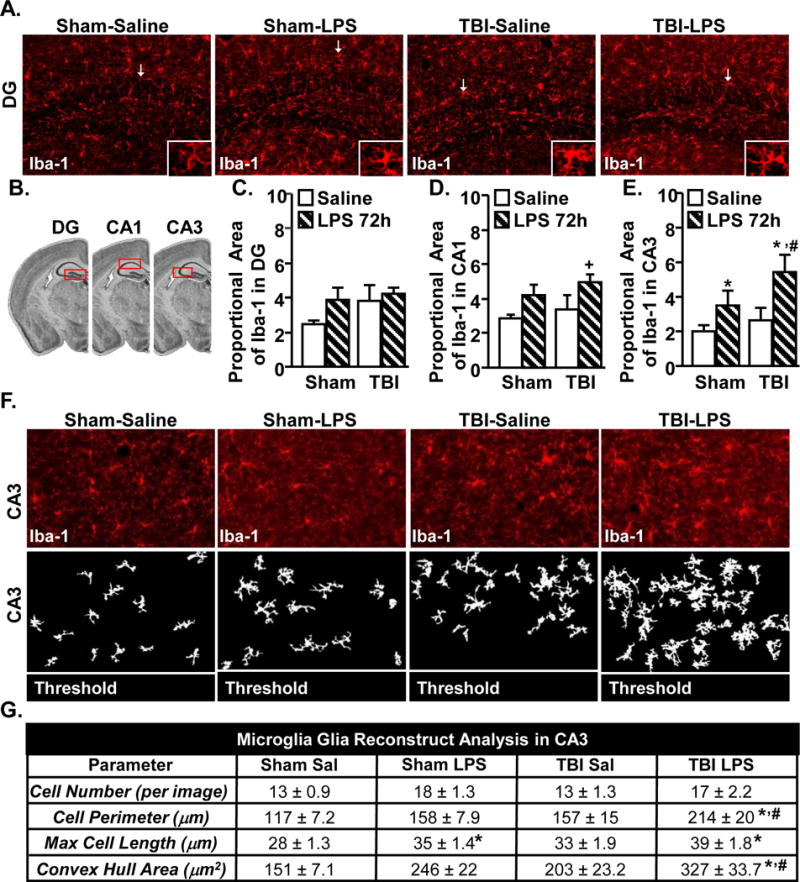
Adult mice were subjected to a sham injury (sham) or a moderate midline fluid percussion injury (TBI). 30 days following TBI, mice were injected i.p. with saline or LPS (0.33mg/kg) and mice were perfused and PFA fixed 24 hours later. Brains were post-fixed, frozen, and sectioned and 1ba-1 expression was determined. A) Representative images of labeling for Iba-1 in the DG of the hippocampus. B) Illustration of DG, CA1, and CA3 regions within the hippocampus that were analyzed. C–E) Proportional area of Iba-1 labeling in the DG, CA1, CA3 was determined. F) “Glia reconstruct” representative CA3 images of Iba-1 labeling (top) and subsequent thresholded image (bottom). G) “Glia Reconstruct” analyses of microglia in the CA3. Table represents the mean + SEM (n = 3–5). Means with (*) are different from Sham-Saline controls (p<0.05) and means with (#) are different from all other groups (p<0.05).
Next, several key morphological parameters of microglia were determined in the CA3 using Glia reconstruct. Representative Iba-1 labeling of microglia (top) and the corresponding thresholded images (bottom) from the CA3 region are shown (Fig.9F). The analysis of the Glia reconstruction is shown in Fig.9G. There was no significant effect of either LPS or TBI on the number of microglia 72 h after LPS. Microglia cell perimeter in the CA3 was increased by LPS (main effect of LPS, F(1,31) = 8.1, p < 0.01). In addition, post-hoc analysis revealed that microglial perimeter was highest in the TBI-LPS mice compared to all other groups (p < 0.05, Fig.9G). The max cell was increased 72 hours after LPS (main effect of LPS, F(1,31) = 5.1, p < 0.05) but was not further enhanced in TBI mice. Convex hull area of microglia was increased by LPS (main effect of LPS, F(1,31) =13.4, p < 0.005). Post hoc analysis revealed that microglia from TBI-LPS mice had the highest convex hull area compared to all other groups (p < 0.05). Overall, immune challenge elicited hyper-reactivity of microglia specifically in the CA3 of the hippocampus of TBI mice.
4. Discussion
We have previously reported that primed and immune reactive microglia persists in the brain 30 days after diffuse TBI (Fenn et al., 2014). We extend these findings to show that while mice recovered to baseline behavior by 7 dpi, astrocytes and microglia with elevated inflammatory profile persisted in the hippocampus 30 dpi coinciding with deterioration of hippocampal-dependent learning. Moreover, induction of an immune response 30 dpi augmented deficits in memory recall in TBI mice alone. These deficits were associated with exaggerated microglia-mediated inflammation shown by amplified cytokine expression and hyper-reactive morphological profiles. These novel data indicate that diffuse TBI primes microglia and that immune challenge exacerbates inflammation and cognitive impairment.
One important finding was that TBI disrupts retrograde memory acutely (7 dpi), while anterograde memory was unaffected. For example, mice had deficits in recalling a task learned prior to the injury. When mice were tested 7 dpi, however, there were no deficits in hippocampal-dependent learning of new tasks or recall. This is consistent with other studies showing acute effects of brain injury on anterograde memory (O’Connor et al., 2003, Whiting and Hamm, 2008, Hylin et al., 2013). In addition, 7 dpi reflects the return to baseline nesting behavior, motor coordination, and home cage activity after TBI. Overall, all mice showed significant functional recovery by 7 days after diffuse brain injury.
A novel aspect of the cognitive assessments was that mice had intact anterograde learning ability, but hippocampal-dependent learning deficits were evident by 30 dpi. For example, all mice showed improved learning of the location of the escape hole 30 dpi, but TBI mice took more time to acquire the memory and made more errors compared to Sham. The retention of anterograde learning 7 dpi followed by reduced cognitive ability 30 dpi indicates that cognitive decline occurs over time. The comparison of 7 dpi to 30 dpi memory recall represents a cross sectional analysis. Nonetheless, at 7 dpi there was no difference between Sham and TBI in memory recall. At 30 dpi, however, there was a difference between Sham and TBI in memory recall with reduced time spent in the target quadrant and increased errors. These data are relevant because there is evidence of cognitive decline in longitudinal studies of human patients after TBI (Ruff et al., 1991, Till et al., 2008, Ramlackhansingh et al., 2011). Moreover, several studies report that focal brain injury in rodents was associated with cognitive decline. For instance, mice had impaired spatial learning in the Morris water maze 2 months after CCI (Hsieh et al., 2014). Furthermore, mice tested in the Barnes maze at 6, 12, and 18 months after single mild CHI had cognitive deterioration at 18 months which was augmented with repeated injury (Mouzon et al., 2014). These cognitive differences were associated with white matter damage and increased GFAP and Iba-1 labeling but were not associated with neurodegenerative pathology 12 and 18 months after injury. Here we show that cognitive decline occurred within 30 days after a single and diffuse TBI. Thus, there is a critical injury by time interaction on memory impairment.
An important finding of this study is that deficits in hippocampal-dependent learning that develop 30 dpi coincided with inflammation mediated by glia. For instance, astrocytes isolated from whole brain 30 days after TBI had increased mRNA levels of GFAP, vimentin and IL-1β compared to shams. There was also TBI associated increases in GFAP immunoreactivity in the DG and CA1 of the hippocampus. Furthermore, analysis on the morphology of astrocytes in the in the CA1 revealed larger astrocytes with increased cell area and cell perimeter. This is consistent with a “reactive” astrocyte profile detected after injury (Hazra et al., 2014, Kabadi et al., 2014, Mouzon et al., 2014). Moreover, it is possible the increased GFAP immunoreactive profile of astrocytes represents a reduction in the support of neuronal synapse activity and contributes to the development of longer term cognitive deficits. For instance, a recent paper showed that reactive astrocytes in the brain of Alzheimer’s disease mice aberrantly produced high levels of the inhibitory gliotransmitter GABA, which directly impaired hippocampal-dependent memory (Jo et al., 2014). It is unknown if this GABA gliotransmitter increase is the mechanism of impairment here, but there is mounting support for the idea that reactive astrocytes can interfere with normal CNS homeostasis (Molofsky et al., 2012). Thus, it is important to delineate the consequence of a persistent reactive astrocyte population after TBI and their role in precipitating cognitive decline.
Along with the enhanced reactive profile of astrocytes 30 dpi microglia also had a more inflammatory profile. In microglia, there was increased microglial expression of MHCII and IL-1β mRNA in TBI mice 30 dpi compared to sham controls. This is consistent with our previous work (Fenn et al., 2014) showing persistent inflammatory profiles of microglia after a single and diffuse TBI. The differences in microglial Iba-1 labeling 30 dpi, however, were not as evident with TBI. For example, Iba-1 was higher in the DG after TBI but it was not significantly different from shams. Microglial immunoreactivity in the CA1 and CA3 were also not different than shams. Nonetheless, we previously detected increased Iba-1 immunoreactivity in the DG in TBI mice compared to shams (Fenn et al., 2014). Thus, glial mRNA and morphological data in the hippocampus indicate a pro-inflammatory astrocytic and microglial profile that persists 30 dpi. These data are consistent with other studies and provide a link between persistent glial changes and cognitive impairment (Bao et al., 2012, Briones et al., 2013, Mouzon et al., 2014).
Neurogenesis in the hippocampus is associated with enhanced neuronal plasticity and cognitive performance (Sahay et al., 2011) and is negatively influenced by inflammation (Valero et al., 2014). Furthermore, microglia and astrocytes play an integral role in neural and synaptic functioning (Battista et al., 2006, Ben Menachem-Zidon et al., 2011). Therefore, we expected that acute immune challenge would exaggerate glial responses and reduce the number of newly formed cells in the hippocampus of TBI mice. Surprisingly, there was not a reduction in the number of newly generated cells (BrdU+) or immature neurons (DCX+) in the dentate gyrus of TBI mice 72 hours after LPS. The fate of the BrdU+ cells was not determined, so it is possible that the higher inflammation would influence the development of neuronal precursors into mature neuron (Wang et al., 2011). Nonetheless, memory recall was impaired by induction of immune response in TBI mice. Notably, this cognitive impairment was evident without significant neuronal damage (APP) or death (Fluorojade labelling, not shown). Thus, inflammation can negatively influence cognitive processes acutely and independent of reduced neurogenesis or increased cell death.
A novel component of this study was the immune challenge provided 30 dpi and the determination of glial profiles and cognition. We have previously shown that microglia from TBI mice (30 dpi) have amplified expression of IL-1β and TNFα after LPS (24 h) challenge. This amplified microglial response corresponded with prolonged social withdrawal, resignation, and anhedonia (Fenn et al., 2014). Immune challenge is relevant because individuals are likely to experience infection or illness over a lifespan at a higher frequency than repeated brain injuries (Norden et al., 2014b). Here we show novel data that increased immune reactivity of microglia to acute immune challenge exacerbates deficits in memory retention and recall in TBI mice compared to Sham. The robust deficits in memory in TBI mice after LPS were paralleled by an increased pro-inflammatory mRNA profile of microglia. Immune challenge amplified microglial mRNA expression of IL-1β, TNFα, CCL2 (24 h after LPS) in TBI mice and higher TNFα mRNA levels were maintained by microglia even 72 h after LPS. Notably, while amplified CCL2 and IL-1β mRNA expression by microglia has been linked to increased monocyte recruitment to the brain in models of repeated social defeat (Wohleb et al., 2013) and traumatic CNS injury (Semple et al., 2010), the presence of Cd11b+/CD45high macrophages in the brain after LPS was not enhanced in the TBI mice (30 dpi). Thus, an enhanced recruitment of monocytes to the brain in the TBI mice is not a major determinant in the LPS-associated augmentation in cognitive impairment at 30 dpi. In addition, a higher inflammatory profile of astrocytes persisted 30 days after TBI but this was not augmented by LPS challenge. We interpret these data to indicate that resident microglia are the primary drivers of the amplified and prolonged cytokine response after immune challenge in mice 30 days after TBI. Moreover a myriad of studies show the impact of either amplified of prolonged exposure to IL-1β, TNFα and CCL2 on behavior, cognition, neurochemistry and plasticity (Cunningham et al., 1996, Chen et al., 2008, Barrientos et al., 2009a, Barrientos et al., 2010, Semple et al., 2010, Tobinick et al., 2012). Collectively, we interpret these data to indicate that the further disruption in hippocampal dependent memory of TBI mice after LPS challenge is caused by the heightened inflammatory response of microglia.
Related to the points above, our data show increased microglial reactivity specifically in the CA3 of the hippocampus. Microglia in the CA3 of TBI mice had the highest Iba-1 proportional area, perimeter and convex cell area after immune challenge compared to controls. It is unclear why hyper-reactivity of microglia was detected selectively in the CA3. This may be related to the higher capillary density of the CA3 compared to CA1 (Cavaglia et al., 2001). Here we provided a systemic immune challenge and these signals are communicated to the brain, in part, by humoral pathways. Thus, the CA3 may intrinsically have more immune sensing by microglia based on increased access to the blood. Overall, immune challenge caused amplified microglia activation within the hippocampus of TBI mice that was associated with cognitive impairment.
In summary, we extend our previous findings to show that while mice recover from diffuse TBI, cognitive issues develop 30 days later. Because these deficits were not evident 7 dpi, these data suggest that after a single diffuse injury, inflammatory processes in glia are either maintained or continue to progress 30 days after injury. We show evidence that microglia and astrocytes maintain a pro-inflammatory profile in the hippocampus 30 days after TBI. Moreover an acute innate immune challenge augmented microglial reactivity within the hippocampus and further exacerbated the deficits in hippocampal-dependent memory.
Significance Statement.
Traumatic brain injury (TBI) is a major risk factor in development of neuropsychiatric problems long after injury, negatively affecting quality of life. Mounting evidence indicates that inflammatory processes worsen with time after a brain injury and are likely mediated by glia. Here, we show that primed microglia and astrocytes developed in mice 1 month following moderate diffuse TBI, coinciding with cognitive deficits that were not initially evident after injury. Additionally, TBI-induced glial priming may adversely affect the ability of glia to appropriately respond to immune challenges, which occur regularly across the lifespan. Indeed, we show that an acute immune challenge augmented microglial reactivity and cognitive deficits. This idea may provide new avenues of clinical assessments and treatments following TBI.
Mounting evidence indicates that inflammatory processes worsen with time after a brain injury and are likely mediated by glia.
Primed microglia and astrocytes developed in mice 1 month following diffuse TBI, coinciding with cognitive deficits that were not initially evident after injury.
An acute immune challenge augmented microglial reactivity and cognitive deficits. This idea may provide new avenues of clinical assessments and treatments following TBI.
Acknowledgments
This research was supported by an NIA grant (R01-AG-033028 to J.P.G.), College of Medicine Dean’s Discovery Grant (to J.P.G.), Funding from the Center for Brain and Spinal Cord Repair (to J.P.G), an American Surgical Society Fellowship (to D.S.E). In addition, this work was supported by a P30 Core grant NINDS P30-NS045758. M.M.M. was supported by a National Institute of Dental and Craniofacial Research Training Grant (T32-DE-01-4320), A.M.F. was supported by the OSU Presidential Fellowship, and P.G.P was supported by a Ray W. Poppleton Endowment
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no competing financial interests.
References
- Almeida OP, Hankey GJ, Yeap BB, Golledge J, Flicker L. Prevalence, associated factors, mood and cognitive outcomes of traumatic brain injury in later life: the health in men study (HIMS) International journal of geriatric psychiatry. 2015 doi: 10.1002/gps.4276. [DOI] [PubMed] [Google Scholar]
- Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–915. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:6143–6153. doi: 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Shultz SR, Hepburn JD, Omana V, Weaver LC, Cain DP, Brown A. A CD11d monoclonal antibody treatment reduces tissue injury and improves neurological outcome after fluid percussion brain injury in rats. Journal of neurotrauma. 2012;29:2375–2392. doi: 10.1089/neu.2012.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain, behavior, and immunity. 2009a;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Watkins LR, Maier SF. Memory impairments in healthy aging: Role of aging-induced microglial sensitization. Aging and disease. 2010;1:212–231. [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Watkins LR, Rudy JW, Maier SF. Characterization of the sickness response in young and aging rats following E. coli infection. Brain, behavior, and immunity. 2009b;23:450–454. doi: 10.1016/j.bbi.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. The European journal of neuroscience. 2006;23:83–93. doi: 10.1111/j.1460-9568.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- Ben Menachem-Zidon O, Avital A, Ben-Menahem Y, Goshen I, Kreisel T, Shmueli EM, Segal M, Ben Hur T, Yirmiya R. Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain, behavior, and immunity. 2011;25:1008–1016. doi: 10.1016/j.bbi.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Berg BM, Godbout JP, Kelley KW, Johnson RW. Alpha-tocopherol attenuates lipopolysaccharide-induced sickness behavior in mice. Brain, behavior, and immunity. 2004;18:149–157. doi: 10.1016/S0889-1591(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Briones TL, Woods J, Rogozinska M. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta neuropathologica communications. 2013;1:57. doi: 10.1186/2051-5960-1-57. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brooks WM, Stidley CA, Petropoulos H, Jung RE, Weers DC, Friedman SD, Barlow MA, Sibbitt WL, Jr, Yeo RA. Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. Journal of neurotrauma. 2000;17:629–640. doi: 10.1089/089771500415382. [DOI] [PubMed] [Google Scholar]
- Cavaglia M, Dombrowski SM, Drazba J, Vasanji A, Bokesch PM, Janigro D. Regional variation in brain capillary density and vascular response to ischemia. Brain research. 2001;910:81–93. doi: 10.1016/s0006-8993(01)02637-3. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention C, National Center for Injury Prevention and Control, and National Institute of Health, (NIH) Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation 2015 [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain, behavior, and immunity. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neuroscience letters. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry. 2014;76:575–584. doi: 10.1016/j.biopsych.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Henry CJ, Huang Y, Dugan A, Godbout JP. Lipopolysaccharide-induced interleukin (IL)-4 receptor-alpha expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain, behavior, and immunity. 2012;26:766–777. doi: 10.1016/j.bbi.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP. Methylene Blue Attenuates Traumatic Brain Injury-associated Neuroinflammation and Acute Depressive-like Behavior in Mice. Journal of neurotrauma. 2015;32:127–138. doi: 10.1089/neu.2014.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, J OC, Castanon N, Kelley KW, Dantzer R, Johnson RW. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Hoffman JM, Pretz C, Kennedy R, Clark AN, Brenner LA. A longitudinal study of major and minor depression following traumatic brain injury. Archives of physical medicine and rehabilitation. 2012;93:1343–1349. doi: 10.1016/j.apmr.2012.03.036. [DOI] [PubMed] [Google Scholar]
- Hazra A, Macolino C, Elliott MB, Chin J. Delayed thalamic astrocytosis and disrupted sleep-wake patterns in a preclinical model of traumatic brain injury. Journal of neuroscience research. 2014;92:1434–1445. doi: 10.1002/jnr.23430. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain, behavior, and immunity. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen L, Portin R, Isoniemi H, Helenius H, Kurki T, Tenovuo O. Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology. 2006;66:187–192. doi: 10.1212/01.wnl.0000194264.60150.d3. [DOI] [PubMed] [Google Scholar]
- Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J, Nakamura MC. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. Journal of neurotrauma. 2014;31:1677–1688. doi: 10.1089/neu.2013.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylin MJ, Orsi SA, Zhao J, Bockhorst K, Perez A, Moore AN, Dash PK. Behavioral and histopathological alterations resulting from mild fluid percussion injury. Journal of neurotrauma. 2013;30:702–715. doi: 10.1089/neu.2012.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee da Y, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nature medicine. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SJ, Walker FR. Strategies to improve quantitative assessment of immunohistochemical and immunofluorescence labelling. Scientific Reports. 2015 doi: 10.1038/srep10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Loane DJ, Luo T, Faden AI. CR8, a novel inhibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014;34:502–513. doi: 10.1038/jcbfm.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BJ, Farkas O, Lifshitz J, Povlishock JT. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Experimental neurology. 2006;198:350–360. doi: 10.1016/j.expneurol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. Journal of neuropathology and experimental neurology. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- Kheirbek MA, Hen R. Dorsal vs ventral hippocampal neurogenesis: implications for cognition and mood. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2011;36:373–374. doi: 10.1038/npp.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongsui R, Beynon SB, Johnson SJ, Mayhew J, Kuter P, Nilsson M, Walker FR. Chronic stress induces prolonged suppression of the P2X7 receptor within multiple regions of the hippocampus: a cumulative threshold spectra analysis. Brain, behavior, and immunity. 2014a;42:69–80. doi: 10.1016/j.bbi.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Kongsui R, Beynon SB, Johnson SJ, Walker FR. Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. Journal of neuroinflammation. 2014b;11:182. doi: 10.1186/s12974-014-0182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz J. Fluid percussion injury model. In: Chen J, Xu ZC, Xu X-M, Zhang JH, editors. Animal Models of Acute Neurological Injuries. 2009. p. 369. [Google Scholar]
- Lifshitz J, Witgen BM, Grady MS. Acute cognitive impairment after lateral fluid percussion brain injury recovers by 1 month: evaluation by conditioned fear response. Behavioural brain research. 2007;177:347–357. doi: 10.1016/j.bbr.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. Journal of neuropathology and experimental neurology. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millis SR, Rosenthal M, Novack TA, Sherer M, Nick TG, Kreutzer JS, High WM, Jr, Ricker JH. Long-term neuropsychological outcome after traumatic brain injury. The Journal of head trauma rehabilitation. 2001;16:343–355. doi: 10.1097/00001199-200108000-00005. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. Astrocytes and disease: a neurodevelopmental perspective. Genes & development. 2012;26:891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, Longhi L, Laurer H, Maegele M, Neugebauer E, Graham DI, Stocchetti N, McIntosh TK. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Annals of neurology. 2014;75:241–254. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- Norden DM, Fenn AM, Dugan A, Godbout JP. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia. 2014a;62:881–895. doi: 10.1002/glia.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, Godbout JP. Microglial Priming and Enhanced Reactivity to Secondary Insult in Aging, and Traumatic CNS injury, and Neurodegenerative Disease. Neuropharmacology. 2014b;96:29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2015 doi: 10.1002/glia.22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor C, Heath DL, Cernak I, Nimmo AJ, Vink R. Effects of daily versus weekly testing and pre-training on the assessment of neurologic impairment following diffuse traumatic brain injury in rats. Journal of neurotrauma. 2003;20:985–993. doi: 10.1089/089771503770195830. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. 2nd 2004. [Google Scholar]
- Radler ME, Wright BJ, Walker FR, Hale MW, Kent S. Calorie restriction increases lipopolysaccharide-induced neuropeptide Y immunolabeling and reduces microglial cell area in the arcuate hypothalamic nucleus. Neuroscience. 2015;285:236–247. doi: 10.1016/j.neuroscience.2014.11.014. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Annals of neurology. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Rosenthal M, Christensen BK, Ross TP. Depression following traumatic brain injury. Archives of physical medicine and rehabilitation. 1998;79:90–103. doi: 10.1016/s0003-9993(98)90215-5. [DOI] [PubMed] [Google Scholar]
- Ruff RM, Young D, Gautille T, Marshall LF, Barth J, Jane JA, Kreutzer J, Marmarou A, Levin HS, Eisenberg HM, Foulkes MA. Verbal-Learning Deficits Following Severe Head-Injury - Heterogeneity in Recovery over 1 Year. Journal of neurosurgery. 1991;75:S50–S58. [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahinkaya FR, Milich LM, McTigue DM. Changes in NG2 cells and oligodendrocytes in a new model of intraspinal hemorrhage. Experimental neurology. 2014;255:113–126. doi: 10.1016/j.expneurol.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond CH, Menon DK, Chatfield DA, Pickard JD, Sahakian BJ. Changes over time in cognitive and structural profiles of head injury survivors. Neuropsychologia. 2006;44:1995–1998. doi: 10.1016/j.neuropsychologia.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till C, Colella B, Verwegen J, Green RE. Postrecovery cognitive decline in adults with traumatic brain injury. Archives of physical medicine and rehabilitation. 2008;89:S25–34. doi: 10.1016/j.apmr.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS drugs. 2012;26:1051–1070. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- Valero J, Mastrella G, Neiva I, Sanchez S, Malva JO. Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Frontiers in neuroscience. 2014;8:83. doi: 10.3389/fnins.2014.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FR, Beynon SB, Jones KA, Zhao Z, Kongsui R, Cairns M, Nilsson M. Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain, behavior, and immunity. 2014;37:1–14. doi: 10.1016/j.bbi.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Wang HK, Lin SH, Sung PS, Wu MH, Hung KW, Wang LC, Huang CY, Lu K, Chen HJ, Tsai KJ. Population based study on patients with traumatic brain injury suggests increased risk of dementia. Journal of neurology, neurosurgery, and psychiatry. 2012;83:1080–1085. doi: 10.1136/jnnp-2012-302633. [DOI] [PubMed] [Google Scholar]
- Wang Y, Neumann M, Hansen K, Hong SM, Kim S, Noble-Haeusslein LJ, Liu J. Fluoxetine increases hippocampal neurogenesis and induces epigenetic factors but does not improve functional recovery after traumatic brain injury. Journal of neurotrauma. 2011;28:259–268. doi: 10.1089/neu.2010.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil ZM, Gaier KR, Karelina K. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiology of disease. 2014;70:108–116. doi: 10.1016/j.nbd.2014.06.016. [DOI] [PubMed] [Google Scholar]
- Whiting MD, Hamm RJ. Mechanisms of anterograde and retrograde memory impairment following experimental traumatic brain injury. Brain research. 2008;1213:69–77. doi: 10.1016/j.brainres.2008.01.107. [DOI] [PubMed] [Google Scholar]
- Witcher KG, Eiferman DS, Godbout JP. Priming the Inflammatory Pump of the CNS after Traumatic Brain Injury. Trends in neurosciences. 2015;38:609–620. doi: 10.1016/j.tins.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witgen BM, Lifshitz J, Grady MS. Inbred mouse strains as a tool to analyze hippocampal neuronal loss after brain injury: a stereological study. Journal of neurotrauma. 2006;23:1320–1329. doi: 10.1089/neu.2006.23.1320. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. 2012;37:1491–1505. doi: 10.1016/j.psyneuen.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, Godbout JP. Reestablishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 2014;75:970–981. doi: 10.1016/j.biopsych.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J. Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. Journal of neuroinflammation. 2012;9:247. doi: 10.1186/1742-2094-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]