

Abstract
The discovery of 3-methoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-ol (a benzosuberene-based analogue referred to as KGP18) was originally inspired by the natural products colchicine and combretastatin A-4 (CA4). The relative structural simplicity and ease of synthesis of KGP18, coupled with its potent biological activity as an inhibitor of tubulin polymerization and its cytotoxicity (in vitro) against human cancer cell lines, has resulted in studies focused on new analogue design and synthesis. Our goal was to probe the relationship of structure to function in this class of anticancer agents. A series of twenty-two new benzosuberene-based analogues of KGP18 was designed and synthesized. These compounds vary in their methoxylation pattern and separately incorporate trifluoromethyl groups around the pendant aryl ring for the evaluation of the effect of functional group modifications on the fused six-membered aromatic ring. In addition, the 8,9-saturated congener of KGP18 has been synthesized to assess the necessity of unsaturation at the carbon atom bearing the pendant aryl ring. Six of the molecules from this benzosuberene-series of compounds were active (IC50 < 5 μM) as inhibitors of tubulin polymerization while four analogues were comparable (IC50 approximately 1 μM) in their tubulin inhibitory activity to CA4 and KGP18. The potency of a bis-trifluoromethyl analogue 74 and the unsaturated KGP18 derivative 73 as inhibitors of tubulin assembly along with their moderate cytotoxicity suggested the potential utility of these compounds as vascular disrupting agents (VDAs) to selectively target microvessels feeding tumors. Accordingly, water-soluble and DMSO-soluble phosphate prodrug salts of each were synthesized for preliminary in vivo studies to assess their potential efficacy as VDAs.
Keywords: inhibitors of tubulin polymerization, benzosuberene analogues, vascular disrupting agents, small-molecule synthesis
Graphical Abstract
1. Introduction
As solid tumors grow beyond approximately 1 mm3 in size, they require an ever larger vascular network to supply oxygen and nutrients to the cells and remove cellular waste products.1 Since the vasculature feeding tumors tends to grow rapidly to keep up with tumor expansion, it has a tendency to vary in diameter and incorporate bulges and blind ends, rendering it somewhat fragile and chaotic in character.2–4 The primitive nature of the vascular network in tumors makes it a promising target for cancer therapy.
There are two types of antivascular therapies: angiogenesis inhibiting agents (AIAs) and vascular disrupting agents (VDAs).5–7 AIAs inhibit the formation of new vasculature in developing tumors, while VDAs damage the already existing tumor vasculature.6,8–9 VDAs are further subdivided into biologics and small-molecule anticancer agents. Inhibitors of tubulin polymerization represent one class of small-molecule VDAs. These compounds disrupt the tubulin-microtubule protein system and cause structural changes to the endothelial cells lining the vasculature, in response to cell signaling events. These morphological changes eventually lead to irreversible damage to the tumor vasculature, thus starving the tumor of nutrients and oxygen, ultimately leading to tumor necrosis.10–19
One class of VDAs interact with the colchicine site on β-tubulin near the α,β-tubulin heterodimer interface. Two of the most potent colchicine site binding VDAs are the natural products combretastatin A-4 (CA4)20 and combretastatin A-1 (CA1)21 originally isolated from Combretum caffrum, the South African bushwillow tree (Fig. 1). The corresponding phosphate prodrug salts combretastatin A-4P (CA4P)22–23 and combretastatin A-1P (CA1P)15 have improved water solubility and have been extensively evaluated in both pre-clinical experiments and clinical studies in humans.24–29
Figure 1.

Representative small-molecule inhibitors of tubulin polymerization: colchicine, combretastatins (CA4, CA1),20–21 dihydronaphthalene analogue (OXi6196),30–32 benzosuberene analogues (KGP18 and KGP156),30,33,34 indole analogue (OXi8006)35 and benzo[b]furan analogue (BNC105).36
Significant structural similarities exist among the natural products colchicine, CA4, and CA1 (Fig. 1), including a trimethoxy phenyl ring, a separate p-methoxy phenyl moiety, and bridging functionality connecting the two rings at a comparable centroid to centroid distance. The relative structural simplicity of CA4 has inspired the synthesis of a vast array of synthetic analogues and derivatives in which both aryl rings and the ethylene bridge have been structurally modified. An early initial molecular design paradigm led us to utilize clinically relevant non-steroidal, selective estrogen receptor modulators (SERMs) and related compounds as molecular templates modified to mimic colchicine and CA4.37–39 This led us to the discovery and establishment of benzo[b]thiophene,40–42 benzofuran,42–43 dihydronaphthalene, 30–32 benzosuberene,30–34 and indole-based35 analogues as potent inhibitors of tubulin polymerization (Fig. 1). Two benzosuberene analogues (referred to as KGP1830,33 and its amino congener KGP15631,34) are especially promising anticancer agents based, in part, on their pronounced cytotoxicity against human cancer cell lines and their efficacy as inhibitors of tubulin polymerization. Our previous studies in this area30,33,34 resulted in two separate synthetic strategies towards the pendant 9-aryl, fused six-seven ring system present in the benzosuberene analogues KGP18 and KGP156. These studies included a variety of functional group modifications designed to probe structural diversity as it relates to biological function. Inspired by our original work with these and related benzosuberene analogues, Maderna and colleagues at Pfizer developed a separate synthetic approach utilizing a ring-closing metathesis (RCM) reaction to form the benzosuberene molecular core and a Suzuki coupling to install the pendant aryl ring.44 They prepared and evaluated a series of structurally diverse benzosuberene analogues.45 Using KGP18 and KGP156 as models, we developed a series of analogues to analyze further functional group modifications for their effects on cytotoxicity and inhibition of tubulin polymerization.
2. Results and Discussion
2.1. Synthesis
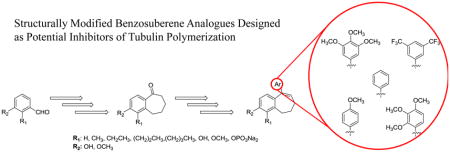
Twenty-two benzosuberene analogues (Fig. 2) were synthesized and evaluated for both their ability to inhibit tubulin polymerization and for their in vitro cytotoxicity against selected human cancer cell lines. Structural modifications to the R1 and R2 positions of the fused aryl ring as well as the pendant aryl ring were explored in order to evaluate their impact on tubulin dynamics and cytotoxicity. The synthesis of each benzosuberene analogue involved a Wittig olefination reaction followed by hydrogenation to afford carboxylic acid derivatives 7-12. An intramolecular Friedel-Crafts annulation facilitated by Eaton’s reagent (7.7 weight percent P2O5 in CH3SO3H)46–47 yielded benzosuberone analogues 13-18, which were subsequently treated with requisite aryl-lithium reagents to generate tertiary alcohols 19-27, which upon dehydration afforded the final benzosuberene analogues 28-36 (Scheme 1).
Figure 2.

Compilation of synthesized benzosuberene analogues.
Scheme 1.

Synthesis of benzosuberene analogues 28-36.
Concomitant elimination accompanied the addition of 4-methoxyphenyl lithium, which resulted in benzosuberene analogue 37 (Scheme 2).
Scheme 2.
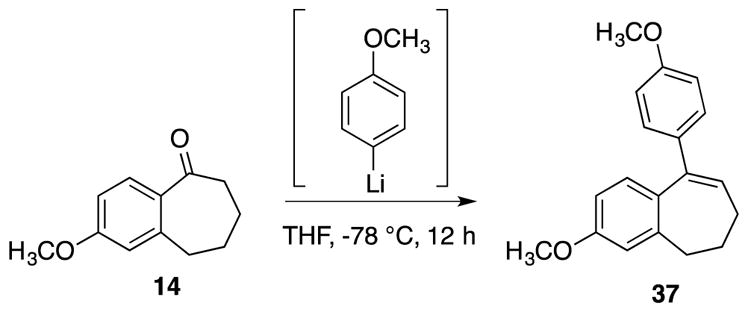
Synthesis of benzosuberene analogue 37.
Benzosuberone analogues 13 and 14 were also subjected to a demethylation reaction using the ionic liquid [TMAH][Al2Cl7]48 to afford phenolic derivatives 38-40. We previously demonstrated the regioselective demethylation of compound 13.33,48 Protection of the phenolic moieties as their corresponding tert-butyldimethylsilyl (TBS) ethers followed by nucleophilic addition with an appropriately substituted lithiated aryl ring produced tertiary alcohols 44-50, which were subsequently dehydrated to yield TBS protected benzosuberene analogues 51-57 (Scheme 3). Removal of the TBS protecting groups upon treatment with TBAF resulted in benzosuberene analogues 64-70 (Scheme 4).
Scheme 3.

Synthesis of TBS protected analogues 51-57
Scheme 4.

Synthesis of benzosuberene analogues 61, 62, and 64-71.
In our hands, desired benzosuberene analogues 61, 62, and 71 were not accessible by the methodology involving aryl lithium addition, instead yielding recovered starting material. It is possible that competing enolate formation was faster than 1,2-carbonyl addition in these cases. Alternatively, the synthesis of analogues 61, 62, and 71 was accomplished using a Suzuki coupling to attach the pendant aryl ring, similar to the methodology of Maderna et. al.44 Vinyl triflates 58, 59, and 60 were reacted with the corresponding boronic acids to generate target benzosuberene analogues 61 and 62 and TBS protected compound 63, which, after deprotection with TBAF, afforded benzosuberene analogue 71 (Scheme 4).
The double bond of benzosuberene 69 (KGP18) was reduced to afford benzosuberane analogue 72, which was subsequently converted to its corresponding phosphate salt 73. Benzosuberene analogue 65 was also converted to its phosphate prodrug salt 74 under analogous reaction conditions (Scheme 5).
Scheme 5.

Synthesis of reduced benzosuberane analogue 72 and phosphate salts 73 and 74.
2.2. Biological Evaluation
Each of the twenty-two analogues was evaluated biologically for its ability to inhibit tubulin polymerization (cell free assay), as well as its cytotoxicity against human cancer cell lines [SK-OV-3 (ovarian), NCI-H460 (lung), and DU-145 (prostate)]. It is important to note that these two assays, while providing complementary structure activity relationship (SAR) information, represent very different approaches to analyzing the biological activity of the target benzosuberene analogues. It is common for biologically active, small-molecule inhibitors of tubulin polymerization to demonstrate IC50 values in the low micromolar range (in this type of cell free assay) while demonstrating sub-micromolar to nanomolar GI50 values in terms of cytotoxicity against human cancer cell lines. This activity differential may be due, in part, to requisite stoichiometry differences in regard to the number of molecules of inhibitor bound to tubulin in a cell-based assay versus a pure protein assay, as well as practical assay limits (in the low micromolar range) in the pure protein assay (no cells and no additional microtubule-associated proteins). Inhibitor generated interference within the dynamic that is inherent to the tubulin-microtubule protein system in cells may influence signal transduction leading to an amplification of activity in cell-based assays.35,49 An initial structure-activity analysis for the 22 compounds for which we obtained biological data (Table 1), is perhaps best accomplished by focusing on the studies on the presumptive intracellular target, tubulin. The most extensive data were obtained for effects on tubulin assembly, in part because we have rarely observed substantial inhibition of colchicine binding (> 50%, with 5 μM inhibitor) if the assembly IC50 is > 3 μM.
Table 1.
Inhibition of tubulin polymerization, percent inhibition of colchicine binding, and cytotoxicity of the target benzosuberene analogues
| Compound | Inhibition of tubulin polymerization IC50 (μM) ± SD | % Inhibition of colchicine binding ± SD | GI50 (μM) SRB assaya | ||
|---|---|---|---|---|---|
|
| |||||
| SK-OV-3 | NCI-H460 | DU-145 | |||
| CA4 | 1.0b | 84 ± 3 (1 μM), 98 ± 0.007 (5 μM) | 0.00455 | 0.00223c | 0.00327c |
| CA4P | >40b | nr | 0.00119 | 0.00194c | 0.00323c |
| KGP18 | 1.4d | nr | 0.0000543e | 0.0000418e | 0.0000249e |
| 28 | >20 | nr | 32.7 | 37.5 | 89.3 |
| 29 | 1.0 ± 0.02 | 37 ± 5 (1 μM), 72 ± 0.8 (5 μM) | 0.0516 | 0.0527 | 0.0619 |
| 30 | 1.6 ± 0.2 | 65 ± 0.6 (5 μM) | 0.330 | 0.422 | 0.644 |
| 31 | >20 | nr | 0.568 | 0.763 | 1.51 |
| 32 | >20 | nr | 2.96 | 3.32 | 6.03 |
| 33 | >20 | nr | 11.5 | 16.1 | 12.2 |
| 34 | >20 | nr | 31.1 | 25.5 | 52.1 |
| 35 | 3.1 ± 0.03 | 30 ± 4 (5 μM), 56 ± 4 (50 μM) | 0.277 | 0.593 | 0.708 |
| 36 | >20 | nr | 20.5 | 33.4 | 48.3 |
| 37 | >20 | nr | 40.7 | 57.7 | 68.7 |
| 61 | >20 | nr | 6.96 | 10.5 | 26.2 |
| 62 | 1.2 ± 0.007 | 36 ± 5 (1 μM), 69 ± 3 (5 μM) | 0.0432 | 0.120 | 0.0562 |
| 64 | >20 | nr | 0.557 | 0.652 | 4.40 |
| 65 | 3.8 ± 0.3 | 8.5 ± 4 (5 μM), 37 ± 5 (50 μM) | 4.81 | 4.39 | 4.92 |
| 66 | >20 | nr | 16.8 | 25.0 | 21.8 |
| 67 | 7.4 ± 0.06 | nr | 18.4 | 10.6 | 8.59 |
| 68 | 2.7 ± 0.1 | 27 ± 5 (5 μM) | 0.527 | 0.647 | 1.02 |
| 70 | 7.7 ± 0.2 | nr | 0.346 | 0.691 | 1.53 |
| 71 | 11 ± 0.4 | nr | 3.53 | 4.24 | 7.54 |
| 72 | 0.70 ± 0.1 | 21 ± 0.9 (1 μM), 67 ± 0.6 (5 μM) | 0.408 | 0.141 | 0.570 |
| 73 | >20 | nr | 0.357 | 0.145 | 0.753 |
| 74 | >20 | nr | 17.2 | 16.3 | 17.5 |
Ten of the evaluated compounds were active inhibitors of tubulin polymerization (IC50 values < 20 μM). Structural variability was tolerated (in terms of retained tubulin inhibitory activity) by the incorporation of several groups (H, OCH3, CH3) at R1 while the pendant 3,4,5-trimethoxyaryl motif reminiscent of CA4 and colchicine was maintained. This mirrored our previous observations with other functional groups [OH (parent KGP18), NH2 (parent KGP156), Br, Cl) situated at R1 in this same molecular template. Replacement of the R2 methoxy group with a hydroxyl group, while either maintaining R1 = OH or modifying R1 to be a hydrogen atom, led to analogues (70 and 68, respectively) that were also active inhibitors of tubulin polymerization. Variation of the methoxylation pattern (2,3,4-trimethoxy) within the pendant aryl ring also led to active inhibitors of tubulin polymerization when R1 was H or OH (62 and 71, respectively). Replacement of the trimethoxyaryl ring with either a 3,5-bis-trifluoromethyl aryl ring or a 4-methoxyaryl ring with maintenance of R1 = OH (compounds 65 and 67, respectively) resulted in benzosuberene analogues that were still inhibitory against tubulin polymerization. Intriguingly, the double-bond reduced analogue 72 was the most active inhibitor of tubulin assembly within the entire series of benzosuberene analogues analyzed. Loss of inhibition of tubulin polymerization was observed when the trimethoxy aryl substituent was replaced with an unsubstituted phenyl ring (28 and 64). While the R1 methyl analogue 30 functioned as an inhibitor of tubulin polymerization, extension of the alkyl chain to ethyl, propyl, and butyl resulted in the loss of inhibitory activity (as observed with compounds 31, 32, and 33). While ten of the analogues inhibited tubulin assembly, only 29, 30, 62, and 72 demonstrated inhibition values (1.0 μM, 1.6 μM, 1.2 μM, and 0.70 μM, respectively) comparable to those observed with lead compounds KGP18 and CA4.
Molecular docking studies were carried out on several analogues that were active inhibitors of tubulin polymerization (compounds 29, 62 and 72), and compared to compound 33 with an IC50 > 20 μM (inactive) in this assay. Docking placed the trimethoxyphenyl ring of all three active analogues in a similar position to that of the trimethoxyphenyl moiety of N-deacetyl-N-(2-mercaptoacetyl)-colchicine in the structure co-crystallized with tubulin. In contrast, modeling placed multiple top conformations of compound 33 with its trimethoxyphenyl ring outside of this pocket (see Supplementary Data).
Among the twenty-two benzosuberene analogues investigated in this study, the most cytotoxic agents were compounds 29 and 62 (for example, GI50 = 0.0516 μM and 0.0432 μM, respectively, against the SK-OV-3 cell line). Both of these compounds bear trimethoxy aryl groups, but, unlike KGP18, they each contain a hydrogen atom at the R1 position, rather than a hydroxyl group. The cytotoxicity, inhibition of colchicine binding, and the inhibition of tubulin polymerization correlate well for these compounds. Compounds 31 and 64 were not inhibitors of tubulin polymerization (IC50 > 20 μM), but they were found to be cytotoxic (GI50 < 1 μM) against two of the three cell lines utilized in this study. Although these compounds are structurally similar to others in this library, they may have an alternate mechanism of inhibiting cell growth. We note the strong antitubulin activity of compound 72, in which the double bond in the seven-membered fused ring was reduced, although this modification appears to be associated with reduced cytotoxicity. This reduction in cytotoxicity (for compound 72) correlates with a decrease in the percent inhibition of colchicine binding (at 1 μM) relative to compounds 29 and 62, although all three compounds (29, 62, and 72) are comparable in the colchicine binding assay at 5 μM..
Two compounds, 65 and the strong tubulin inhibitor 72, were selected for conversion to prodrugs (74 and 73, respectively) by phosphorylation, in order to improve water-solubility and potentially bioavailability. As with CA4 (phosphorylated to CA4P), this synthetic transformation eliminated (IC50 > 20 μM) the ability to inhibit tubulin polymerization (cell-free assay), while the cytotoxicity was maintained presumably due to phosphatase activity present in the cell-based assay.
We previously demonstrated that dynamic bioluminescence imaging (BLI) provides a facile indication of vascular disruption in luciferase transfected tumors.7,51–52 Analysis is noninvasive, and each tumor acts as its own control. Specifically, the substrate luciferin normally diffuses into the blood stream following subcutaneous injection, and, when it reaches a tumor, light emission occurs. Vascular disruption impairs delivery, and reduced light emission is observed. Analogue 73 showed no obvious acute toxicity over 24 h to breast tumor bearing SCID mice following IP administration of saline solutions delivering doses up to 40 mg/kg. Doses of 20 or 30 mg/kg showed a similar modest reduction of BLI signal 4 h after administration. At 40 mg/kg there was approximately a 50% reduction of the emitted signal. In each case, the signal generally returned to its original level within 24 h. By comparison, saline controls showed a highly reproducible signal, and CA4P (a well-established VDA in clinical development) showed greater than 90% reduction at 4 h, which remained depressed (>75%, after 24 h). These data are presented in Figures 3 and 4. Patterns of light emission are presented in Figure 5. The signal intensity for saline emphasizes reproducibility of signal (hence assessment of vasculature) for repeat measurements. As reported previously, CA4P7,51–52 caused significant vascular impairment at 4 h. Compound 73 had little effect at 20 or 30 mg/kg, but caused about 50% reduction in light emission at 40 mg/kg. While these initial, preliminary studies at 20, 30, and 40 mg/kg suggested a potential VDA mechanism for analogue 73, future dose escalation studies will be necessary to establish a maximum tolerated dose (MTD) in this mouse model and to confirm the extent to which analogue 73 is capable of disrupting tumor-associated vasculature.
Figure 3.

BLI assessment of vascular disruption caused in MDA-MB-231-luc orthotopic human tumor xenografts by analogue 73. Dynamic BLI was performed at baseline (bottom row), 4 h after VDA administration (middle), and after 24 h (top), and images are shown for representative mice 17 min after administering fresh luciferin substrate on each occasion to each animal. Images show bioluminescent signal intensity overlaid on photographs of the mice. Analogue 73 was administered at 20, 30 or 40 mg/kg IP in saline, and additional mice received saline control or CA4P (120 mg/kg) for comparison. Analogue 73 caused a reduced signal at all doses at 4 h with substantial recovery by 24 h.
Figure 4.

Vascular disruption caused by analogue 73
Relative signal intensity is plotted for the mice shown in Fig. 3.
Figure 5.

Dynamic bioluminescence with respect to vascular disruption.
Graphs show evolution of light emission from individual MDA-MB-231-luc tumors following administration of luciferin substrate subcutaneously in the fore-back region of each mouse at baseline (blue) and 4 h after administration of agent (red). Analogue 73 had a modest effect at 30 mg/kg and a greater effect at 40 mg/kg. By comparison control saline showed a high degree of reproducibility and CA4P showed >90% reduction in light emission.
3. Conclusion
In summary, the results of these experiments have expanded our SAR knowledge regarding effects that modifications of the benzosuberene skeleton play in relationship to cytotoxicity and antitubulin activity. The most promising analogues evaluated in this study demonstrated inhibition of tubulin assembly comparable to CA4 and KGP18, but these compounds had reduced inhibitory effects on colchicine binding and on cell growth. Preliminary in vivo BLI evaluation of the VDA capability of benzosuberene analogue 73 against an MDA-MB-231-luc xenograft (in a SCID mouse model) showed efficacy, but, at the doses examined, the effect of 73 was less pronounced than that obtained with an established VDA (in this case, CA4P) currently in clinical development. Future studies with compound 73 and related benzosuberene analogues involving dose escalation and other tumor models to assess vascular damage appear warranted.
4. Experimental Section
4.1. Chemistry
General Materials and Methods
Tetrahydrofuran (THF), dichloromethane, ethanol, methanol, dimethylformamide (DMF), and acetonitrile were used in their anhydrous forms. Reactions were performed under nitrogen gas, unless otherwise specified. Thin-layer chromatography (TLC) plates (precoated glass plates with silica gel 60 F254, 0.25 mm thickness) were used to monitor reactions. Purification of intermediates and products was carried out with a Biotage Isolera or Teledyne Combiflash flash purification system using silica gel (200–400 mesh, 60 Å) or RP-18 pre-packed columns or manually in glass columns. Intermediates and products synthesized were characterized on the basis of their 1H NMR (500 or 300 MHz), 13C NMR (125 or 75 MHz), 19F (470 MHz) and 31P NMR (200 or 120 MHz) spectroscopic data using a Varian VNMRS 500 MHz or a Bruker DPX 300 MHz instrument. Spectra were recorded in CDCl3, D2O, (CD3)2CO, or CD3OD. All chemical shifts are expressed in ppm (δ), and peak patterns are reported as broad (br), singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), sextet (sext), septet (sept), double doublet (dd), double double doublet (ddd), and multiplet (m).
Purity of the final compounds was further analyzed at 25 °C using an Agilent 1200 HPLC system with a diode-array detector (λ = 190–400 nm), a Zorbax XDB-C18 HPLC column (4.6 mm Å~ 150 mm, 5 μm), and a Zorbax reliance cartridge guard-column; Method A: solvent A, acetonitrile, solvent B, 0.1% TFA in H2O; or Method B: solvent A, acetonitrile, solvent B, H2O; gradient, 10% A/90% B to 100% A/0% B over 0 to 40 min; post-time 10 min; flow rate 1.0 mL/min; injection volume 20 μL; monitored at wavelengths of 210, 230, 254, 280, and 320 nm. Mass spectrometry was carried out under positive or negative ESI (electrospray ionization) using a Thermo Scientific LTQ OrbitrapDiscovery instrument.
4.1.1. 5-(2′,3′-Dimethoxyphenyl)pent-4-enoic acid (1).31,33
To dissolved 3-(carboxypropyl)triphenyl phosphonium bromide (13.04 g, 30.39 mmol) in THF (500 mL) was added potassium tert-butoxide (7.43 g, 66.2 mmol), and the reaction mixture was stirred at room temperature for 1 h. 2,3-Dimethoxybenzaldehyde (5.02 g, 30.1 mmol) dissolved in THF (100 mL) was added, and the mixture was stirred at room temperature for 12 h. The THF was removed under reduced pressure, and the resulting material was quenched with 2 M HCl (75 mL) and extracted with EtOAc (3 × 100 mL). The combined organic layers were evaporated under reduced pressure, and the crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford compound 1 (5.39 g, 21.5 mmol, 72%) as a yellow oil. NMR characterization was conducted after the next step.
4.1.2. 5-(2′,3′-Dimethoxyphenyl)pentanoic acid (7).31,33,53
To dissolved carboxylic acid 1 (5.39 g, 21.5 mmol) in methanol (100 mL) was added 10% palladium on carbon (0.43 g) and hydrogen gas. The reaction mixture was stirred at room temperature for 12 h and filtered through Celite®, and the Celite® was washed with EtOAc (3 × 50 mL). The combined organic phase (MeOH and EtOAc) was evaporated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 7 (4.45 g, 18.7 mmol, 82%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 11.67 (1H, s), 6.99 (1H, t, J = 8 Hz), 6.78 (2H, m), 3.86 (3H, s), 3.84 (3H, s), 2.68 (2H, t, J = 8 Hz), 2.41 (2H, t, J = 7.5 Hz), 1.70 (4H, m). 13C NMR (125 MHz, CDCl3) δ 180.2, 152.7, 147.1, 135.8, 123.8, 121.9, 110.2, 60.6, 55.6, 34.0, 30.8, 29.4, 24.5.
4.1.3. 1,2-Dimethoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (13).31,33,54
To carboxylic acid 7 (3.55 g, 14.9 mmol) was added Eaton’s reagent (29 mL, 3 g per mmol of compound 7), and the reaction mixture was stirred at room temperature for 12 h. It was then poured over ice and neutralized with sodium bicarbonate. The reaction mixture was extracted with EtOAc (3 × 50 mL), and the combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 13 (2.43 g, 11.0 mmol, 74%) as a light yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.33 (1H, d, J = 8.5 Hz), 6.67 (1H, d, J = 9 Hz), 3.72 (3H, s), 3.62 (3H, s), 2.83 (2H, t, J = 6 Hz), 2.50 (2H, t, J = 6 Hz), 1.66 (2H, p, J = 6.5 Hz), 1.59 (2H, p, J = 6.5 Hz). 13C NMR (125 MHz, CDCl3) δ 204.2, 155.9, 145.8, 135.5, 132.6, 125.2, 109.6, 60.8, 55.6, 40.4, 24.7, 23.0, 20.7.
4.1.4. [TMAH][Al2Cl7]48
To dry dichloromethane (150 mL) was added AlCl3 (19.84 g, 149.1 mmol), and the mixture was stirred and cooled to 0 °C. Trimethylamine hydrochloride (7.11 g, 74.5 mmol) was added, and the reaction mixture was allowed to stir for 2 h at room temperature. The resulting liquid was stored at room temperature under nitrogen.
4.1.5. 1-Hydroxy-2-dimethoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (38).31,33,55
To benzosuberone 13 (1.01 g, 4.54 mmol) in a 20 mL microwave vial was added [TMAH][Al2Cl7] (18.3 mL, 9.08 mmol), and the mixture was subjected to microwave irradiation for 1 h at 80 °C on high absorbance. The solution was then poured into water (50 mL) and extracted with EtOAc (3 x 25 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 38 (0.61 g, 3.0 mmol, 65%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.24 (1H, d, J = 8.5 Hz), 6.67 (1H, d, J = 9 Hz), 6.26 (1H, s), 3.78 (3H, s), 2.93 (2H, t, J = 5.5 Hz), 2.61 (2H, t, J = 6.5 Hz), 1.71 (4H, m). 13C NMR (125 MHz, CDCl3) δ 205.0, 149.5, 142.5, 133.0, 127.8, 120.7, 107.9, 55.9, 40.6, 24.4, 23.0, 21.2.
4.1.6. 1-((tert-Butyldimethylsilyl)oxy)-2-dimethoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (41).31,33
Benzosuberone 38 (2.16 g, 10.5 mmol) was dissolved in dimethylformamide (50 mL). TBSCl (3.16 g, 21.0 mmol) and DIPEA (5.50 mL, 31.6 mmol) were added, and the solution was stirred for 12 h at room temperature. The reaction mixture was washed with water and extracted with EtOAc (5 x 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford TBS protected analogue 41 (2.37 g, 7.38 mmol, 71%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.23 (1H, d, J = 8.5 Hz), 6.62 (1H, d, J = 9 Hz), 3.67 (3H, s), 2.88 (2H, t, J = 5.5 Hz), 2.53 (2H, t, J = 5.5 Hz), 1.64 (4H, m), 0.89 (9H, s), 0.06 (6H, s). 13C NMR (125 MHz, CDCl3) δ 204.3, 152.9, 141.6, 132.9, 132.7, 122.2, 108.6, 54.6, 40.5, 26.0, 24.6, 23.8, 21.1, 18.8, −4.0.
4.1.7. 4-((tert-Butyldimethylsilyl)oxy)-3-methoxy-6,7-dihydro-5H-benzo[7]annulen-9-yl trifluoromethanesulfonate (60)
To an oven-dried flask, diisopropylamine (0.18 mL, 1.3 mmol) dissolved in THF (50 mL) was added and cooled to −78 °C, and n-BuLi (0.51 mL, 1.3 mmol) was added. The reaction mixture was stirred for 15 min. TBS protected 41 (0.37 g, 1.2 mmol) dissolved in THF (10 mL) was added dropwise, and the reaction mixture was stirred for 2 h at −78 °C. N-Phenyl-bis(trifluoromethanesulfonimide) (0.45 g, 1.3 mmol) dissolved in THF (10 mL) was then added dropwise, and the reaction mixture was stirred for 12 h while warming from −78 °C to room temperature. After 12 h, the THF was evaporated under reduced pressure, and the resulting solid was washed with water (50 mL) and extracted with EtOAc (3 x 50 mL). The organic layer was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford triflate 60 (0.17 g, 0.38 mmol, 33%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.10 (1H, d, J = 8.5 Hz), 6.79 (1H, d, J = 8.5 Hz), 6.09 (1H, t, J = 6.5 Hz), 3.82 (3H, s), 2.88 (2H, t, J = 6 Hz), 2.15 (2H, q, J = 6.5 Hz), 2.03 (2H, p, J = 6.5 Hz), 1.03 (9H, s), 0.20 (6H, s). 13C NMR (125 MHz, CDCl3) δ 150.8. 146.5, 132.9, 130.9, 129.9, 126.0, 120.9, 119.7, 108.8, 54.7. 30.5, 26.0, 25.0, 24.5, 18.9, 4.0.
4.1.8. tert-Butyl((3-methoxy-9-(2′,3′,4′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)dimethylsilane (63)
Triflate 60 (0.17 g, 0.38 mmol) was dissolved in THF (25 mL) and 2,3,4-trimethoxyphenyl boronic acid (0.09 g, 0.41 mmol), barium hydroxide octahydrate (0.18 g, 0.57 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.013 g, 0.011 mmol) were added to the solution and refluxed at 80 °C for 2 h. The solution was then filtered through Celite®, and the Celite® was washed with dichloromethane. The organic solution (dichloromethane and THF) was evaporated under reduced pressure. The crude reaction product was purified using flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 63 (0.07 g, 0.15 mmol, 41%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.91 (1H, d, J = 8.5 Hz), 6.64 (1H, d, J = 8.5 Hz), 6.60 (1H, d, J = 8 Hz), 6.44 (1H, d, J = 8 Hz), 6.10 (1H, t, J = 7 Hz), 3.87 (3H, s), 3.83 (3H, s), 3.76 (3H, s), 3.38 (3H, s), 2.89 (2H, t, J = 6.5 Hz), 2.12 (2H, p, J = 7 Hz), 1.95 (2H, q, J = 7 Hz), 1.05 (9H, s), 0.22 (6H, s). 13C NMR (125 MHz, CDCl3) δ 153.0, 151.7, 148.5, 142.4, 140.3, 135.8, 132.5, 131.1, 128.2, 124.9, 120.7, 108.0, 106.6, 105.2, 60.6, 60.4, 55.9, 54.6, 33.8, 26.2, 25.5, 24.2, 19.0, −3.9.
4.1.9. 3-Methoxy-9-(2′,3, ′4′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-ol (71)
TBS-protected benzosuberene 63 (0.068 g, 0.146 mmol) was dissolved in THF (5 mL), and tetrabutylammonium fluoride (0.18 mL, 0.18 mmol) was added. The reaction was stirred at room temperature for 12 h, washed with water, extracted with EtOAc (3 x 25 mL), dried over sodium sulfate, and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene analogue 71 (0.053 g, 0.15 mmol, 96%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 6.91 (1H, d, J = 8.5 Hz), 6.63 (1H, d, J = 8.5 Hz), 6.61 (1H, d, J = 8.5 Hz), 6.37 (1H, d, J = 8.5 Hz), 6.10 (1H, t, J = 7 Hz), 5.71 (1H, s) 3.87 (3H, s), 3.85 (3H, s), 3.82 (3H, s), 3.42 (3H, s), 2.87 (2H, t, J = 7 Hz), 2.15 (2H, p, J = 7 Hz), 1.96 (2H, q, J = 7 Hz). 13C NMR (125 MHz, CDCl3) δ 153.0, 151.8, 144.7, 142.39, 142.36, 140.0, 136.2, 131.0, 128.6, 126.8, 125.0, 119.0, 107.3, 106.6, 60.7, 60.5, 55.94, 55.90, 33.6, 25.6, 23.4. HRMS: Obsvd 379.1516 [M + Na+], Calcd for C21H24O5Na: 379.1516. HPLC (Method B): 16.18 min.
4.1.10. 3,4-Dimethoxy-6,7-dihydro-5H-benzo[7]annulen-9-yl trifluoromethanesulfonate (58)
Diisopropylamine (0.84 mL, 6.0 mmol) was dissolved in THF (20 mL) and cooled to −78 °C. n-BuLi (2.4 mL, 6.0 mmol) was added dropwise, and the solution was stirred for 15 min. Benzosuberone 13 (1.2 g, 5.4 mmol) dissolved in THF (10 mL) was added dropwise, and the reaction was stirred for 2 h at −78 °C. N-Phenyl-bis(trifluoromethanesulfonimide) (2.14 g, 5.99 mmol) dissolved in THF (10 mL) was then added dropwise, and the reaction mixture was stirred for 12 h while warming from −78 °C to room temperature. After 12 h, the THF was evaporated under reduced pressure, and the resulting solid was washed with water (50 mL) and extracted with EtOAc (3 x 50 mL). The organic layer was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford triflate 58 (1.01 g, 2.87 mmol, 50%) as a yellow oil. NMR characterization was performed after the next step.
4.1.11. 3,4-Dimethoxy-9-(2′,3′,4′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (61)
Triflate 58 (0.46 g, 1.3 mmol) was dissolved in THF (25 mL) and 2,3,4-trimethoxyphenyl boronic acid (0.31 g, 1.4 mmol), barium hydroxide octahydrate (0.62 g, 2.0 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.05 g, 0.04 mmol) were added to the solution, which was refluxed at 80 °C for 2 h. The solution was then filtered through Celite®, and the Celite® was washed with dichloromethane. The organic solution (dichloromethane and THF) was evaporated under reduced pressure. The crude reaction product was purified using flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 45%A / 55%B (10 CV), 45%A / 55%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 61 (0.07 g, 0.19 mmol, 15%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 6.91 (1H, d, J = 8.5 Hz), 6.64 (2H, overlapping d, J = 8.5 Hz), 6.58 (1H, d, J = 8.5 Hz), 6.11 (1H, t, J = 7 Hz), 3.86 (3H, s), 3.84 (3H, s), 3.82 (3H, s). 3.81 (3H, s), 3.38 (3H, s), 2.86 (2H, t, J = 7 Hz), 2.15 (2H, p, J = 6 Hz), 1.95 (2H, q, J = 7 Hz). 13C NMR (125 MHz, CDCl3) δ 153.1, 151.7, 151.0, 146.2, 142.4, 140.1, 135.9, 134.9, 130.8, 128.4, 124.9, 123.5, 108.9, 106.7, 61.2, 60.6, 60.3, 55.9, 55.6, 34.4, 25.4, 23.9. HRMS: Obsvd 393.1740 [M + Na+], Calcd for C22H26O5Na: 393.1672. HPLC (Method B): 18.25 min.
4.1.12. 1-((tert-Butyldimethylsilyl)oxy)-2-methoxy-5-phenyl-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (44)
To an oven dried flask, THF (50 mL) and phenyl bromide (0.69 mL, 6.5 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (2.74 mL, 6.86 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. TBS-protected 41 (1.55 g, 4.83 mmol) in THF (25 mL) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 44 (0.80 g, 2.01 mmol, 42%) as a clear oil. NMR characterization was performed after the next step.
4.1.13 tert-Butyl((3-methoxy-9-phenyl-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)dimethylsilane (51)
Acetic acid (10 mL) was added to alcohol 44 (0.80 g, 2.0 mmol), and the reaction mixture was stirred for 12 h at room temperature. The mixture was washed with water and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over sodium sulfate. The organic phase was evaporated under reduced pressure, and the crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 51 (0.38 g, 1.0 mmol, 49%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.28 (5H, m), 6.70 (1H, d, J = 8 Hz), 6.60 (1H, d, J = 8.5 Hz), 6.37 (1H, t, J = 7.5 Hz), 3.81 (3H, s), 2.79 (2H, t, J = 7 Hz), 2.13 (2H, p, J = 7 Hz). 1.98 (2H, q, J = 7.5 Hz), 1.07 (9H, s), 0.26 (6H, s). 13C NMR (125 MHz, CDCl3) δ 148.6, 143.0, 142.8, 141.6, 134.1, 133.3, 128.0, 127.3, 126.8, 122.1, 108.8, 108.4, 54.7, 33.8, 26.2, 25.6, 24.2, 19.0, −3.80.
4.1.14. 3-Methoxy-9-phenyl-6,7-dihydro-5H-benzo[7]annulen-4-ol (64)
TBS-protected benzosuberene 51 (0.38 g, 1.0 mmol) was dissolved in THF (25 mL), TBAF (1.20 mL, 1.20 mmol) was added, and the reaction mixture was stirred at room temperature for 12 h. The solution was washed with water and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 3%A / 97%B (1 CV), 3%A / 97%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene analogue 64 (0.12 g, 0.45 mmol, 44%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.28 (5H, m), 6.71 (1H, d, J = 8.5 Hz), 6.54 (1H, d, J = 8.5 Hz), 6.38 (1H, t, J = 7.5 Hz), 5.77 (1H, s), 3.91 (3H, s), 2.79 (2H, t, J = 7 Hz), 2.16 (2H, p, J = 7 Hz), 1.99 (2H, q, J = 7Hz). 13C NMR (125 MHz, CDCl3) δ 145.0, 142.8, 142.7, 142.4, 134.6, 128.02, 128.00, 127.8, 127.6, 126.9, 120.6, 107.7, 55.9, 33.5, 25.7, 23.5. HRMS: Obsvd 267.1385 [M + H+], Calcd for C18H19O2: 267.1380. HPLC (Method B): 17.89 min.
4.1.15. ((5-(3′, 5′-Bis(trifluoromethyl)phenyl)-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-1-yl)oxy)(tert-butyl)dimethylsilane (45)
1-Bromo-3,5-bis(trifluoromethyl)benzene (0.36 g, 1.2 mmol) was dissolved in THF (13 mL), and the reaction flask was cooled to −78 °C. n-BuLi (0.55 mL, 2.5 M) was added to the reaction mixture, which was stirred for 1 h. Ketone 41 (0.29 g, 0.91 mmol) was dissolved in THF (5 mL) and slowly added to the reaction mixture over a period of 15 min. The reaction mixture was stirred for 20 h while warming from −78 °C to room temperature. The reaction mixture was diluted with H2O (25 mL) and extracted with EtOAc (2 × 25 mL), and the organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford alcohol 45 (0.40 g, 0.75 mmol, 82%) as a yellow solid. 1H NMR (CDCl3, 500 MHz) δ 7.77 (1H, s), 7.74 (2H, s), 6.95 (1H, d, J = 8.7 Hz), 6.70 (1H, d, J = 8.7 Hz), 3.80 (3H, s), 3.32 (1H, ddd, J = 14.8, 7.6, 2.0 Hz), 2.57 (1H, ddd, J = 14.2, 7.6, 3.0 Hz), 2.41 (1H, s), 2.34 – 2.21 (1H, m), 2.16 (1H, ddd, J = 13.8, 10.3, 3.1 Hz), 1.97 – 1.90 (1H, m), 1.75 – 1.68 (1H, m), 1.67 – 1.57 (1H, m), 1.56 – 1.48 (1H, m), 0.99 (9H, s), 0.18 (3H, s), 0.17 (3H, s). 13C NMR (CDCl3, 125 MHz) δ 149.9, 149.7, 142.5, 137.1, 132.7, 131.6 (q, J = 33.2 Hz), 127.3 (q, J = 3 Hz), 123.5 (q, J = 272.8 Hz), 121.2 (hept, J = 3.8 Hz), 120.3, 108.6, 79.8, 54.8, 41.6, 26.7, 26.2, 25.3, 25.3, 19.1, −3.94, −3.95.
4.1.16. ((9-(3′,5′-Bis(trifluoromethyl)phenyl)-3-methoxy-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)(tert-butyl)dimethylsilane (52)
2 M HCl (8 mL, 16 mmol) was added to a well-stirred solution of alcohol 45 (0.68 g, 1.3 mmol) in EtOH (5 mL), and the reaction mixture was stirred for 12 h at ambient temperature. The reaction mixture was then extracted with EtOAc (4 × 15 mL), and the organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 15%A / 85%B (10 CV), 15%A / 85%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 52 (0.43 g, 0.84 mmol, 66%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.77 (1H, s), 7.74 (2H, s), 6.73 (1H, d, J = 8.5 Hz), 6.50 (1H, t, J = 7.1 Hz), 6.49 (1H, d, J = 8.5 Hz), 3.84 (3H, s), 2.80 (2H, t, J = 6.9 Hz), 2.16 (2H, p, J = 7.0 Hz), 2.04 (2H, q, J = 7.1 Hz), 1.07 (9H, s), 0.27 (6H, s). 13C NMR (CDCl3, 125 MHz) δ 149.3, 145.0, 142.1, 141.1, 133.5, 132.5, 131.5 (q, J = 33.3 Hz), 130.8, 128.0 (q, J = 3.4 Hz), 123.7 (q, J = 272.7 Hz), 121.9, 120.6 (hept, J = 3.7 Hz), 109.0, 54.8, 33.8, 26.3, 26.0, 24.4, 19.2, −3.6.
4.1.17. 9-(3′,5′-Bis(trifluoromethyl)phenyl)-3-methoxy-6,7-dihydro-5H-benzo[7]annulen-4-ol (65)
To a solution of TBS-protected analogue 52 (0.43 g, 0.84 mmol) in THF (5 mL) was added TBAF (1.0 mL, 1 M in THF), and the reaction mixture was stirred for 18 h at ambient temperature and then concentrated under reduced pressure. The reaction mixture was washed with water (5 mL) and extracted with EtOAc (3 x 10 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 6%A / 94%B (1 CV), 6%A / 94%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford phenolic benzosuberene analogue 65 (0.26 g, 0.66 mmol, 78%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.77 (1H, s), 7.74 (2H, s), 6.74 (1H, d, J = 8.3 Hz), 6.51 (1H, t, J = 7.3 Hz), 6.45 (1H, d, J = 8.4 Hz), 5.83 (1H, s), 3.93 (3H, s), 2.79 (2H, t, J = 6.9 Hz), 2.19 (2H, p, J = 7.1 Hz), 2.05 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz) δ 145.8, 144.9, 142.9, 140.8, 133.0, 131.5 (q, J = 33.4 Hz), 131.2, 128.03, 127.99, 123.6 (q, J = 272.8 Hz), 120.6 (hept, J = 3.9 Hz), 120.4, 108.2, 56.1, 33.4, 26.1, 23.7. 19F NMR (CDCl3, 470 MHz) δ −62.80. HRMS: Obsvd 401.0963 [M − H]−, Calcd for C20H15F6O2: 401.0976. HPLC (Method B): 20.39 min.
4.1.18. 9-(3′,5′-Bis(trifluoromethyl)phenyl)-3-methoxy-6,7-dihydro-5H-benzo[7]annulen-4-yl disodium phosphate (74)
To a well-stirred solution of phenol 65 (0.10 g, 0.25 mmol) in CH2Cl2 (10 mL), POCl3 (153.3 mg, 1.00 mmol) and pyridine (70.8 mg, 0.9 mmol) were added to the reaction flask. After the reaction mixture was stirred for 15 h at ambient temperature, the solvent was evaporated under reduced pressure. Saturated aqueous Na2CO3 (20 mL) was added to the flask, and the reaction mixture was stirred for another 2 h. The reaction mixture was concentrated to dryness with a stream of N2 gas and purified by flash chromatography using a prepacked C-18 30 g reversed phase column [solvent A: acetonitrile; solvent B: water; gradient: 30%A / 70%B (1 CV), 30%A / 70%B → 100%A / 0%B (10 CV), 100%A / 0%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford phosphate salt 74 (0.043 g, 0.082 mmol, 33 %) as a white solid. 1H NMR (D2O, 500 MHz) δ 7.96 (1H, s), 7.87 (2H, s), 6.91 (1H, d, J = 8.6 Hz), 6.72 (1H, d, J = 8.4 Hz), 6.62 (1H, t, J = 7.4 Hz), 3.84 (3H, s), 2.76 (2H, t, J = 7.0 Hz), 2.17 (2H, p, J = 7.1 Hz), 1.97 (2H, q, J = 7.2 Hz). 13C NMR (CD3OD, 125 MHz) δ 153.1, 146.6, 142.0, 141.9, 137.2, 133.3, 132.6 (q, J = 33.0 Hz), 132.4, 129.0 (q, J = 2.5 Hz), 124.9 (q, J = 271.3 Hz), 125.2 (hept, J = 3.8 Hz), 121.2, 111.1, 56.4, 34.6, 26.9, 25.9. 19F NMR (D2O, 470 MHz) δ −62.8. 31P NMR (D2O, 200 MHz) δ −3.5. HRMS: Obsvd 527.0429 [M + H]+, Calcd for C19H16F6O2Na2O5P: 527.0429. HPLC (Method A): 13.79 min.
4.1.19. 1-((tert-Butyldimethylsilyl)oxy)-5-(4-hydroxyphenyl)-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (47)
To a solution of 4-methoxyphenyl bromide (0.52 g, 2.8 mmol) in THF (40 mL) at −78 °C was added n-BuLi (0.34 mL, 2.5 M in hexanes), and the reaction mixture was stirred for 30 min. Benzosuberone 41 (0.61 g, 1.9 mmol) in THF (20 mL) was added dropwise over a period of 15 min. The reaction mixture was allowed to reach room temperature over 12 h. Upon completion, water was added, and the mixture was extracted with EtOAc (3 x 20 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford alcohol 47 (0.32 g, 0.75 mmol, 39%) as a clear oil. 1H NMR (CDCl3, 500 MHz) δ 7.26 (1H, d, J = 8.6 Hz), 7.15 (2H, d, J = 8.8 Hz), 6.81 (2H, d, J = 8.8 Hz), 6.73 (1H, d, J = 8.7 Hz), 3.81 (3H, s), 3.79 (3H, s), 3.30 (1H, dd, J = 14.6, 7.5 Hz), 2.67 – 2.59 (1H, m), 2.14 – 2.04 (2H, m), 1.77 – 1.65 (2H, m), 1.35 – 1.25 (2H, m), 0.99 (9H, s), 0.20 (3H, s), 0.14 (3H, s). 13C NMR (CDCl3, 125 MHz) δ 158.7, 149.2, 141.8, 139.0, 137.6, 132.7, 128.3, 119.1, 113.6, 107.8, 79.4, 55.2, 54.6, 41.2, 27.1, 26.7, 26.1, 25.5, −3.86, −4.22.
4.1.20. tert-Butyl((3-methoxy-9-(4′-methoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)dimethylsilane (54)
Tertiary alcohol 47 (0.53 g, 1.2 mmol) was dissolved in acetic acid (5 mL) and refluxed for 5 h. No apparent change in TLC was observed, so water (15 mL) was added to the reaction mixture, which was refluxed for 2 h. The solvents were evaporated, and the resulting product was extracted with EtOAc (3 x 15 mL). The combined organic extracts were washed with sat. NaHCO3, brine, dried over Na2SO4, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford a clear oil that solidified as a colorless solid of TBS-protected benzosuberene analogue 54 (0.43 g, 1.1 mmol, 85%). 1H NMR (CDCl3, 500 MHz) δ 7.20 (2H, d, J = 8.7 Hz), 6.83 (2H, d, J = 8.8 Hz), 6.68 (1H, d, J = 8.4 Hz), 6.58 (1H, d, J = 8.4 Hz), 6.26 (1H, t, J = 7.3 Hz), 3.81 (3H, s), 3.79 (3H, s), 2.75 (2H, t, J = 6.9 Hz), 2.09 (2H, p, J = 7.1 Hz), 1.93 (2H, q, J = 7.2 Hz), 1.04 (9H, s), 0.23 (6H, s). 13C NMR (CDCl3, 126 MHz) δ 158.7, 148.5, 142.4, 141.5, 135.5, 134.3, 133.3, 129.0, 125.7, 122.0, 113.4, 108.3, 55.3, 54.7, 33.94, 26.2, 25.5, 24.2, 19.0, −3.9.
4.1.21. 3-Methoxy-9-(4′-methoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-ol (67)
The TBS-protected analogue 54 (0.33 g, 0.8 mmol) was dissolved in THF (5 mL). To the solution, TBAF-3 H2O (0.96 mmol) was added, and the mixture was stirred for 3 h at room temperature. The reaction was quenched with water (15 mL), followed by the evaporation of organic solvent under reduced pressure. The resultant aqueous phase was then extracted with EtOAc (3 x 20 mL). The combined organic extracts were washed with brine solution, dried over Na2SO4, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 40 g silica column [solvent A, EtOAc, solvent B, hexanes; gradient 0%A/ 100%B →100%A/ 0%B over 9.0 min; flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford the benzosuberene analogue 67 (0.21 g, 0.71 mmol, 89%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.21 (2H, d, J = 8.7 Hz), 6.83 (2H, d, J = 8.7 Hz), 6.70 (1H, d, J = 8.4 Hz), 6.54 (1H, d, J = 8.4 Hz), 6.28 (1H, t, J = 7.4 Hz), 5.72 (1H, s), 3.90 (3H, s), 3.81 (3H, s), 2.75 (2H, t, J = 7.0 Hz), 2.13 (2H, p, J = 7.0 Hz), 1.95 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 126 MHz) δ 158.8, 144.9, 142.4, 142.2, 135.3, 134.8, 129.0, 127.8, 126.1, 120.6, 113.4, 107.6, 55.9, 55.3, 33.7, 25.6, 23.5. HRMS: Obsvd 297.1492 [M + H+], Calcd for C19H21O3: 297.1485. HPLC (Method B): 17.42 min.
4.1.22. 1-((tert-Butyldimethylsilyl)oxy)-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (49).31,33
To an oven dried flask, THF (50 mL) and 3,4,5-trimethoxyphenyl bromide (2.46 g, 9.97 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (4.2 mL, 10 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 41 (2.37 g, 7.38 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78° C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 80%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford compound 49 (1.80 g, 3.70 mmol, 50%) as a clear oil. NMR characterization was performed after the next step.
4.1.23. tert-Butyl((3-methoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)dimethylsilane (56)
Acetic acid (20 mL) was added to tertiary alcohol 49 (1.80 g, 3.70 mmol), and the reaction mixture was stirred for 12 h at room temperature. The mixture was washed with water and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford TBS-protected benzosuberene 56 (1.43 g, 8.38 mmol, 83%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 6.67 (1H, d, J = 8.5 Hz), 6.61 (1H, d, J = 8.5 Hz), 6.50 (2H, s), 6.32 (1H, t, J = 7.5 Hz), 3.85 (3H, s), 3.78 (3H, s), 3.77 (6H, s), 2.78 (2H, t, J = 7 Hz), 2.11 (2H, p, J = 7 Hz), 1.95 (2H, q, J = 7.5 Hz), 1.06 (9H s), 0.25 (6H, s). 13C NMR (125 MHz, CDCl3) δ 152.8, 148.6, 143.1, 141.5, 138.6, 137.3, 133.8, 133.2, 126.7, 122.4, 108.4, 105.3, 60.7, 56.0, 54.5, 34.0, 26.2, 25.6, 24.2, 19.0, −3.8.
4.1.24. 3-Methoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-ol (69).30,31,33,44,56
Benzosuberene 56 (1.43 g, 3.04 mmol) was dissolved in THF (10 mL), TBAF (3.65 mL, 3.6 mmol) was added, and the reaction mixture was stirred at room temperature for 12 h. The solution was washed with water and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure affording benzosuberene 69 (0.66 g, 1.9 mmol, 61%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ 6.72 (1H, d, J = 8.5 Hz), 6.58 (1H, d, J = 8 Hz), 6.51 (2H, s), 6.35 (1H, t, J = 7 Hz), 5.74 (1H, s), 3.93 (3H, s), 3.87 (3H, s), 3.81 (6H, s), 2.77 (2H, t, J = 7 Hz), 2.15 (2H, p, J = 7 Hz), 1.97 (2H, q, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 152.8, 145.0, 142.8, 142.3, 138.5, 137.3, 134.2, 127.7, 127.2, 120.8, 107.6, 105.3, 60.9, 56.1, 55.9, 33.6, 25.7, 23.5.
4.1.25. 2-Methoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-1-ol (72)
To benzosuberene 69 (0.66 g, 1.9 mmol) was added methanol (50 mL) and 10% Pd/C (0.42 g). Hydrogen gas was added, and the reaction mixture was stirrred at room temperature for 12 h. The reaction mixture was filtered through Celite®, and the Celite® was washed with EtOAc (3 x 30 mL). The organic phase (MeOH and EtOAc) was evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberane analogue 72 (0.16 g, 0.45 mmol, 24%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 6.54 (1H, d, J = 8.5 Hz), 6.47 (2H, s), 6.17 (1H, d, J = 8 Hz), 5.84 (1H, s), 4.17 (1H, d, J = 9.5 Hz), 3.89 (3H, s), 3.84 (9H, s), 3.28 (1H, q, J = 7.5 Hz), 2.73 (1H, t, J = 12 Hz), 2.17(1H, m), 2.02 (2H, m), 1.86 (2H, m), 1.46 (1H, q, J = 12 Hz). 13C NMR (125 MHz, CDCl3) δ 153.1, 144.7, 142.6, 141.2, 139.6, 136.2, 128.4, 118.6, 108.1, 105.7, 60.9, 56.1, 55.9, 49.5, 34.7, 30.6, 27.2, 25.4. HRMS: Obsvd 381.1764 [M + Na+], Calcd for C21H26O5Na+: 381.1672. HPLC (Method B): 15.30 min.
4.1.26. 2-Methoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-1-yl disodium phosphate (73)
To benzosuberane analogue 72 dissolved in CH2Cl2 (25 mL) was added POCl3 (0.11 mL, 1.1 mmol) and pyridine (0.1 mL, 1 mmol), and the reaction mixture was stirred at room temperature for 12 h. 2 M NaOH (1.69 mL, 3.38 mmol) was added, and the reaction was stirred for 5 min and extracted with CH2Cl2, and the organic layer was evaporated under reduced pressure. NaOH (2 mL, 2 M) was added to the resulting oil, and the solution was refluxed at 60 °C for 15 min. Water was removed under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 12 g C-18 column [solvent A: water; solvent B: acetonitrile; gradient: 0%A / 100%B (1 CV), 0%A / 100%B → 100%A / 0%B (10 CV), 0%A / 100%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford the benzosuberane phosphate salt 73 (0.024 g, 0.050 mmol, 17%) as a white solid. 1H NMR (500 MHz, D2O) δ 6.43 (2H, s), 6.36 (1H, d, J = 8.5 Hz), 6.07 (1H, d, J = 8.5 Hz), 4.04 (1H, d, J = 9.5 Hz), 3.61 (6H, s) 3.60 (3H, s), 3.56 (3H, s), 3.26 (1H, m), 2.55 (1H, t, J = 12 Hz), 1.90 (1H, m), 1.65 (4H, m), 1.16 (1H, q, J = 10.5 Hz). 13C NMR (125 MHz, D2O) δ 152.2, 150.3, 142.8, 140.5, 140.4, 139.0, 137.3, 134.6, 121.8, 108.6, 105.9, 60.8, 55.4, 48.6, 33.7, 30.0, 33.7, 29.7, 26.6. 31P NMR (200 MHz, D2O, 85% phosphoric acid reference) δ 0.97. HRMS: Obsvd 483.1190 [M + H+], Calcd for C21H26O5Na2P+: 483.1155. HPLC (Method A): 14.23 min.
4.1.27. 1,2-Dimethoxy-5-(4′-methoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (27)
To a solution of 4-methoxyphenyl bromide (0.719 g, 3.84 mmol) in THF (50 mL) at −78 °C was added n-BuLi (2.6 mL, 2.5 M in hexanes), and the reaction mixture was stirred for 30 min. Benzosuberone 13 (0.58 g, 2.6 mmol) in THF (15 mL) was added dropwise over 15 min. The reaction mixture was warmed to room temperature over 12 h. Upon completion, water was added, and the mixture was extracted with EtOAc (3 x 20 mL). The organic layer was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 80 g silica column [solvent A, EtOAc, solvent B, hexanes; gradient 0%A/ 100%B →100%A/ 0%B over 20.3 min; flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford alcohol 27 (0.45 g, 1.4 mmol, 54%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.39 (1H, d, J = 8.7 Hz), 7.15 (2H, d, J = 8.8 Hz), 6.83 (2H, d, J = 8.8 Hz), 6.79 (1H, d, J = 8.7 Hz), 3.89 (3H, s), 3.79 (3H, s), 3.75 (3H, s), 3.25 – 3.18 (1H, m), 2.67 – 2.59 (1H, m), 2.15 – 2.08 (2H, m), 1.93 – 1.86 (1H, m), 1.80 – 1.69 (2H, m), 1.43 – 1.35 (1H, m). 13C NMR (CDCl3, 126 MHz) δ 158.8, 151.7, 146.3, 139.1, 137.5, 135.4, 128.2, 122.31, 113.7, 108.8, 79.4, 61.0, 55.6, 55.2, 41.2, 27.3, 26.6, 25.2.
4.1.28. 3,4-Dimethoxy-9-(4′-methoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (36)
Tertiary alcohol analogue 27 (0.401 g, 1.22 mmol) was dissolved in acetic acid (10 mL), and the mixture was stirred for 12 h at ambient temperature. Water (30 mL) was added, and the reaction mixture was stirred for 2 h. The reaction mixture was extracted with EtOAc (3 x 20 mL). The organic solvent was evaporated under reduced pressure and the residue extracted with EtOAc (3 x 20 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberene analogue 36 (0.31 g, 1.0 mmol, 82%) as a clear oil. 1H NMR (CD3OD, 500 MHz) δ 7.17 (2H, d, J = 8.8 Hz), 6.87 (1H, d, J = 8.5 Hz), 6.86 (2H, d, J = 8.8 Hz), 6.71 (1H, d, J = 8.5 Hz), 6.30 (1H, t, J = 7.4 Hz), 3.89 (3H, s), 3.85 (3H, s), 3.81 (3H, s), 2.74 (2H, t, J = 7.0 Hz), 2.15 (2H, p, J = 7.1 Hz), 1.94 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz) δ 158.8, 151.3, 146.1, 142.2, 135.9, 135.2, 134.3, 129.0, 125.9, 125.0, 113.5, 109.2, 61.2, 55.6, 55.3, 34.6, 25.5, 24.0. HRMS: Obsvd 281.1597 [M + H+], Calcd for C19H23O2: 281.1536. HPLC (Method B): 19.08 min.
4.1.29. 1,2-Dimethoxy-5-phenyl-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (19)
To an oven dried flask, THF (50 mL) and phenylbromide (0.25 mL, 2.4 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (1.0 mL, 2.5 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 13 (0.39 g, 1.8 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 19 (0.29 g, 0.97 mmol, 55%) as a pale yellow oil. NMR characterization was performed after the next step.
4.1.30. 3,4-Dimethoxy-9-phenyl-6,7-dihydro-5H-benzo[7]annulene (28)
Acetic acid (15 mL) was added to tertiary alcohol 19 (0.29 g, 0.97 mmol), and the reaction mixture was stirred for 12 h at room temperature. The mixture was washed with water (50 mL) and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 50%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene analogue 28 (0.10 g, 0.36 mmol, 37%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.30 (5H, m), 6.78 (2H, s), 6.40 (1H, t, J = 7.5 Hz), 3.91 (3H, s), 3.90 (3H, s), 2.81 (2H, t, J = 7 Hz), 2.19 (2H, p, J = 7Hz), 2.01 (2H, q, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 151.5, 146.1, 142.9, 142.6, 135.9, 134.2, 128.1, 128.0, 127.5, 127.0, 125.1, 109.4, 61.2, 55.6, 34.5, 25.6, 24.1. HRMS: Obsvd 303.1363 [M + Na+], Calcd for C19H20O2Na: 303.1356. HPLC (Method B): 19.93 min.
4.1.31. 1,2-Dimethoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (26).31
To a solution of 3,4,5-trimethoxyphenyl bromide (0.67 g, 2.7 mmol) in THF (100 mL) at −78 °C was added n-BuLi (1.1 mL, 2.5 M in hexanes), and the reaction mixture was stirred for 30 min. Benzosuberone 13 (0.60 g, 2.7 mmol) in THF (15 mL) was added dropwise over 15 min. The reaction mixture was warmed to room temperature over 12 h. Water was added, and the mixture was extracted with EtOAc (100 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 56%A / 44%B (9.2 CV), 56%A / 44%B (1 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford alcohol 26 (0.39 g, 0.99 mmol, 34%) as a light yellow oil. 1H NMR (CDCl3, 500 MHz) δ 7.26 (1H, d, J = 8.7 Hz), 6.75 (1H, d, J = 8.8 Hz), 6.49 (2H, s), 3.87 (3H, s), 3.83 (3H, s), 3.74 (3H, s), 3.73 (6H, s), 3.26 – 3.20 (1H, m), 2.56 (1H, ddd, J = 14.1, 6.9, 3.0 Hz), 2.38 – 2.30 (1H, m), 2.21 – 2.21 (1H, m), 2.11 (1H, ddd, J = 14.0, 10.7, 3.1 Hz), 1.93 (1H, ddd, J = 15.3, 7.4, 3.8 Hz), 1.81-1.71 (2H, m), 1.50-1.42 (1H, m). 13C NMR (CDCl3, 126 MHz) δ 153.0, 151.8, 146.2, 141.6, 138.6, 137.2, 135.5, 123.0, 108.8, 104.2, 79.9, 61.0, 60.8, 56.1, 55.5, 41.3, 27.1, 26.3, 25.1.
4.1.32. 3,4-Dimethoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (35).31
Tertiary alcohol analogue 26 (0.5 g, 1 mmol) was dissolved in acetic acid (10 mL) and refluxed for 2 h. Water (30 mL) was added, and the reaction was refluxed for 2 h. The white precipitate thus obtained was filtered and washed with hexanes. On drying, it afforded benzosuberene analogue 35 (0.444 g, 1.2 mmol, 93%) as a colorless solid, which was not further purified. 1H NMR (CDCl3, 500 MHz) δ 6.79 (1H, d, J = 8.5 Hz), 6.77 (1H, d, J = 8.5 Hz), 6.51 (2H, s), 6.35 (1H, t, J = 7.3 Hz), 3.90 (3H, s), 3.89 (3H, s), 3.88 (3H, s), 3.82 (6H, s), 2.77 (2H, t, J = 7.0 Hz), 2.17 (2H, p, J = 7.1 Hz), 1.98 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 126 MHz) δ 152.9, 151.5, 146.1, 142.9, 138.4, 137.4 135.9, 133.8, 127.1, 125.3, 109.3, 105.3, 61.3, 60.9, 56.17, 55.6, 34.6, 25.6, 24.2. HRMS: Obsvd 393.1682 [M + Na+], Calcd for C22H26O5Na: 393.1672. HPLC (Method B): 17.52 min.
4.1.33. 5-(3′,5′-Bis(trifluoromethyl)phenyl)-1,2-dimethoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (25)
To a solution of 1-bromo-3,5-bis(trifluoromethyl)benzene (0.36 g, 1.2 mmol) in THF (13 mL) at −78 °C was added n-BuLi (0.55 mL, 2.5 M), and the reaction mixture was stirred for 1 h. Benzosuberone 13 (0.20 g, 0.91 mmol) in THF (5 mL) was added dropwise over 15 min. The reaction mixture was stirred for 20 h and was warmed to room temperature. The reaction mixture was diluted with H2O (25 mL) and extracted with EtOAc (2 × 25 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (15 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford alcohol 25 (0.45 g, 1.0 mmol, 84%) as a yellow solid. 1H NMR (CDCl3, 500 MHz) δ 7.70 (1H, s), 7.67 (2H, s), 6.92 (1H, d, J = 8.7 Hz), 6.63 (1H, d, J = 8.8 Hz), 3.76 (3H, s), 3.67 (3H, s), 3.16 (1H, ddd, J = 14.6, 7.8, 1.9 Hz), 2.50 (1H, s), 2.46 (1H, ddd, J = 14.3, 8.2, 2.8 Hz), 2.37 – 2.24 (1H, m), 2.06 (1H, ddd, J = 14.2, 9.6, 3.0 Hz), 1.91 – 1.85 (1H, m), 1.73 – 1.61 (1H, m), 1.59 – 1.50 (2H, m). 13C NMR (CDCl3, 125 MHz) δ 152.4, 149.8, 146.6, 137.2, 135.6, 131.6 (q, J = 33.1 Hz), 127.1 (q, J = 3.8 Hz), 123.5 (q, J = 272.7 Hz), 123.7, 121.3 (hept, J = 4.0 Hz), 109.5, 79.8, 61.1, 55.7, 41.6, 27.0, 25.2, 24.8.
4.1.44. 9-(3′,5′-Bis(trifluoromethyl)phenyl)-3,4-dimethoxy-6,7-dihydro-5H-benzo[7]annulene (34)
To a solution of tertiary alcohol 25 (0.45 g, 1.0 mmol) in EtOH (10 mL) was added 2 M HCl (10 mL, 20 mmol), and the reaction mixture was stirred overnight. The reaction mixture was then extracted using EtOAc (4 × 15 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 34 (0.22 g, 0.54 mmol, 54%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.75 (1H, s), 7.71 (2H, s), 6.77 (1H, d, J = 8.5 Hz), 6.63 (1H, d, J = 8.5 Hz), 6.49 (1H, t, J = 7.3 Hz), 3.90 (3H, s), 3.88 (3H, s), 2.76 (2H, t, J = 6.9 Hz), 2.18 (2H, p, J = 7.1 Hz), 2.03 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz) δ 152.2, 146.6, 144.7, 140.8, 136.1, 132.4, 131.6 (q, J = 33 Hz), 131.1, 128.0 (q, J = 3.8 Hz), 124.8, 123.6 (q, J = 272.7 Hz), 120.7 (hept, J = 4.0 Hz), 109.9, 61.4, 55.8, 34.3, 26.0, 24.3. 19F NMR (CDCl3, 470 MHz) δ −62.81. HRMS: Obsvd 401.0965 [M − CH3]−, alcd for C20H15F6O2: 401.0976. HPLC (Method B): 21.57 min.
4.1.45. 1,2-Dihydroxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (40).31,57
Ketone 13 (0.88 g, 4.0 mmol) was added to the ionic liquid [TMAH][Al2Cl7](20.0 mL, 0.497 M). The reaction mixture was subjected to microwave irradiation at 80 °C and 1 atm for 1 h. H2O (20 mL) was added to the mixture, and the resulting brown liquid was extracted with dichloromethane (3 x 20 mL). The organic extracts were dried over Na2SO4, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (13 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 40 (0.50 g, 2.6 mmol, 65% yield) as a brown solid. 1H NMR ((CD3)2CO, 500 MHz) δ 7.14 (1H, d, J = 8.3 Hz), 6.78 (1H, d, J = 8.8 Hz), 3.07 – 2.99 (2H, m), 2.67 – 2.59 (2H, m), 1.84 – 1.77 (2H, m), 1.77 – 1.71 (2H, m). 13C NMR ((CD3)2CO, 126 MHz) δ 205.4, 148.1, 142.0, 132.3, 128.9, 120.6, 112.3, 40.3, 24.4, 22.8, 21.0.
4.1.46. 1,2-Bis((tert-butyldimethylsilyl)oxy)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (43).31
To a solution of catechol 40 (0.68 g, 3.5 mmol) and DIPEA (2.7 mL, 16 mmol) in DMF (5 mL) at 0 °C was added TBSCl (1.60 g, 10.6 mmol) in portions. The reaction mixture was stirred for 18 h, diluted with H2O (5 mL), and extracted with Et2O (2 × 20 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 0%A / 100%B (1 CV), 0%A / 100%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford ketone 43 (1.51 g, 3.59 mmol, 99%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.29 (1H, d, J = 8.5 Hz), 6.75 (1H, d, J = 8.5 Hz), 2.97 – 2.94 (2H, m), 2.70 (2H, t, J = 6.2 Hz), 1.89 – 1.70 (4H, m), 1.02 (9H, s), 0.96 (9H, s), 0.24 (6H, s), 0.15 (6H, s). 13C NMR (CDCl3, 125 MHz) δ 205.3, 151.2, 143.9, 135.5, 134.0, 122.6, 118.6, 41.0, 26.5, 26.5, 25.3, 25.1, 21.9, 19.2, 18.9, −3.15, −3.19.
4.1.47. 1,2-Bis((tert-butyldimethylsilyl)oxy)-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (50).31
To a solution of 3,4,5-trimethoxyphenyl bromide (0.458 g, 1.85 mmol) in THF (20 mL) at −78 °C was added n-BuLi (0.96 mL, 2.5 M in hexanes), and the reaction mixture was stirred for 1 h. Benzosuberone 43 (0.639 g, 1.51 mmol) in THF (20 mL) was added dropwise over 15 min. The reaction mixture was stirred while warming to room temperature over 12 h. Water was added, and the mixture was extracted with EtOAc (4 x 20 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 42%A / 58%B (6 CV), 42%A / 58%B→70%A / 30%B (1 CV), 70%A / 30%B → 100%A/0%B, 100%A/0% B (1.1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford alcohol 50 (0.480 g, 0.81 mmol, 54%) as a clear oil. 1H NMR (CDCl3, 500 MHz) δ 7.10 (1H, d, J = 8.7 Hz), 6.72 (1H, d, J = 8.6 Hz), 6.46 (2H, s), 3.83 (3H, s), 3.74 (6H, s), 3.23 – 3.15 (1H, m), 2.57 (1H, ddd, J = 14.0, 6.2, 3.0 Hz), 2.23 – 2.07 (3H, m), 1.94 – 1.84 (1H, m), 1.81 – 1.66 (2H, m), 1.46 – 1.33 (1H, m), 1.00 (9H, s), 0.95 (9H, s), 0.24 (3H, s), 0.23 (3H, s), 0.15 (3H, s), 0.10 (3H, s). 13C NMR (CDCl3, 126 MHz) δ 152.9, 146.4, 143.7, 141.8, 139.1, 137.1, 133.9, 120.0, 117.5, 104.1, 80.0, 60.8, 56.0, 40.9, 26.8, 26.3, 26.2, 26.1, 25.9, 18.9, 18.6, −3.4, −3.6.
4.1.48. ((9-(3′,4′,5′-Trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene-3,4-diyl)bis(oxy))bis(tert-butyldimethylsilane) (57).31
Tertiary alcohol 50 (0.44 g, 0.75 mmol) was dissolved in acetic acid (5 mL) and stirred for 12 h at room temperature. The reaction was quenched with water (10 mL) and extracted with Et2O (3 x 10 mL). The combined organic extract was washed with sat. NaHCO3, brine, dried over Na2SO4, filtered, and evaporated under reduced pressure to afford the clear oil of TBS-protected benzosuberene analogue 57 (0.38 g, 0.66 mmol, 89%), which was used without further purification. 1H NMR (CDCl3, 500 MHz) δ 6.69 (1H, d, J = 8.4 Hz), 6.52 (1H, d, J = 8.4 Hz), 6.45 (2H, s), 6.33 (1H, t, J = 7.3 Hz), 3.85 (3H, s), 3.78 (6H, s), 2.70 (2H, t, J = 6.9 Hz), 2.10 (2H, q, J = 7.0 Hz), 1.95 (2H, q, J = 7.0 Hz), 1.04 (9H, s), 0.95 (9H, s), 0.24 (6H, s), 0.20 (6H, s). 13C NMR (CDCl3, 125 MHz) δ 152.7, 145.9, 143.4, 143.0, 138.6, 137.1, 134.4, 134.2, 126.8, 122.7, 117.9, 105.0, 60.9, 56.0, 33.9, 26.27, 26.25, 25.7, 24.5, 18.9, 18.7, −3.3, −3.4.
4.1.49. 9-(3′,4′,5′-Trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene-3,4-diol (70)
The di-TBS-protected analogue 57 (0.32 g, 0.56 mmol) was dissolved in THF (5 mL). To the solution, TBAF-3 H2O (1.4 mmol) was added and stirred for 3 h at room temperature. The reaction was quenched with water (15 mL), and the organic solvent was evaporated under reduced pressure. The resulting aqueous phase was then extracted with EtOAc (3 x 20 mL). The combined organic extract was washed with brine, dried over Na2SO4, filtered, evaporated under reduced pressure and purified by flash chromatography using a pre-packed 25 g silica gel column [solvent A, EtOAc, solvent B, hexanes; gradient 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (5.2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford catechol analogue 70 (0.17 g, 0.49 mmol, 88%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 6.68 (1H, d, J = 8.2 Hz), 6.52 (1H, d, J = 8.2 Hz), 6.49 (2H, s), 6.33 (1H, t, J =7.4 Hz), 5.29 (1H, s), 5.28 (1H, s), 3.86 (3H, s), 3.79 (6H, s), 2.71 (2H, t, J = 7.0 Hz), 2.15 (2H, p, J = 7.1 Hz), 1.96 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 126 MHz) δ 152.8, 142.9, 141.8, 140.8, 138.3, 137.3, 134.1, 128.5, 127.0, 121.7, 112.3, 105.3, 60.9, 56.1, 33.8, 25.6, 23.8. HRMS: Obsvd 365.1444 [M + H+], Calcd for C12H23O5: 365.1359. HPLC (Method B): 16.18 min.
4.1.50. 5-(3′,5′-Bis(trifluoromethyl)phenyl)-1,2-bis((tert-butyldimethylsilyl)oxy)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (46)
1-Bromo-3,5-bis(trifluoromethyl)benzene (0.36 g, 1.2 mmol) was dissolved in THF (13 mL) at −78 °C and n-BuLi (0.55 mL, 2.5 M) was then added. The reaction mixture was stirred for 1 h, and then ketone 43 (0.38 g, 0.91 mmol) in THF (5 mL) was added dropwise over 15 min. The reaction mixture was stirred for 20 h, warming from −78 °C to room temperature, and then diluted with H2O (25 mL) and extracted with EtOAc (3 × 25 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 100%A / 0%B (10 CV), 100%A / 0%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford alcohol 46 (0.43 g, 0.68 mmol, 74%) as a yellow solid. 1H NMR (CDCl3, 500 MHz) δ 7.77 (1H, s), 7.72 (2H, s), 6.92 (1H, d, J = 8.6 Hz), 6.73 (1H, d, J = 8.7 Hz), 3.25 (1H, ddd, J = 14.7, 7.6, 1.9 Hz), 2.57 (1H, ddd, J = 14.2, 7.3, 3.1 Hz), 2.31 (1H, s), 2.22 – 2.14 (2H, m), 1.96 – 190 (1H, m), 1.74 – 170 (1H, m), 1.64 – 1.58 (1H, m), 1.54 – 148 (1H, m), 1.01 (9H, s), 0.97 (9H, s), 0.26 (3H, s), 0.23 (3H, s), 0.19 (3H, s), 0.09 (3H, s). 13C NMR (CDCl3, 125 MHz) δ 149.7, 147.2, 144.3, 137.6, 134.0, 131.6 (q, J = 33.1 Hz), 127.2 (q, J = 3.2 Hz), 123.5 (q, J = 273.0 Hz), 121.3 (hept, J = 4.0 Hz), 120.6, 118.3, 79.8, 41.2, 26.8, 26.39, 26.35, 25.9, 25.2, 19.0, 18.7, −3.1, −3.2, −3.5, −3.9.
4.1.51((9-(3′,5′-Bis(trifluoromethyl)phenyl)-6,7-dihydro-5H-benzo[7]annulene-3,4-diyl)bis(oxy))bis(tert-butyldimethylsilane) (53)
Tertiary alcohol 46 (0.43 g, 0.67 mmol) was dissolved in EtOH (5 mL) and 2 M HCl (10 mL, 20 mmol) was then added. The reaction mixture was stirred overnight and then extracted using EtOAc (4 × 15 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 15%A / 85%B (10 CV), 15%A / 85%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 53 (0.22 g, 0.35 mmol, 53%) as a colorless oil. 1H NMR (CDCl3, 500 MHz) δ 7.75 (1H, s), 7.69 (2H, s), 6.72 (1H, d, J = 8.4 Hz), 6.49 (1H, t, J = 7.2 Hz), 6.40 (1H, d, J = 8.4 Hz), 2.73 (2H, t, J = 6.9 Hz), 2.15 (2H, p, J = 7.0 Hz), 2.03 (2H, q, J = 7.1 Hz), 1.07 (9H, s), 0.98 (9H, s), 0.26 (6H, s), 0.23 (6H, s). 13C NMR (CDCl3, 125 MHz) δ 146.8, 145.0, 144.0, 141.1, 134.7, 133.0, 131.5 (q, J = 33.0 Hz), 130.8, 127.9 (q, J = 3.1 Hz), 123.6 (q, J = 272.7 Hz), 122.3, 120.6 (hept, J = 3.9 Hz), 118.5, 33.8, 26.43, 26.39, 26.1, 24.8, 19.1, 18.9, −3.1, −3.2.
4.1.52. 9-(3′,5′-Bis(trifluoromethyl)phenyl)-6,7-dihydro-5H-benzo[7]annulene-3,4-diol (66)
TBS-protected analogue 53 (0.43 g, 0.84 mmol) was dissolved in THF (5 mL), and TBAF (1.00 mL, 1 M in THF) was added to the reaction flask. The reaction mixture was stirred for 18 h at ambient temperature, concentrated under reduced pressure, and H2O (5 mL) was then added. The reaction mixture was extracted with EtOAc (3 x 10 mL). The organic extract was dried over Na2SO4, filtered, concentrated under reduced pressure and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford catechol 66 (0.077 g, 0.20 mmol, 57 %) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.75 (1H, s), 7.71 (2H, s), 6.71 (1H, d, J = 8.2 Hz), 6.50 (1H, t, J = 7.3 Hz), 6.38 (1H, d, J = 8.2 Hz), 5.36 (1H, s), 5.25 (1H, s), 2.73 (2H, t, J = 6.9 Hz), 2.19 (2H, p, J = 7.0 Hz), 2.04 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz) δ 144.7, 142.4, 141.43, 140.9, 132.9, 131.6 (q, J = 32.9 Hz), 131.1, 128.8, 128.0 (q, J = 2.9 Hz), 123.6 (q, J = 272.7 Hz), 121.2, 120.7 (hept, J = 3.9 Hz), 112.9, 33.7, 26.0, 23.9. 19F NMR (CDCl3, 470 MHz) δ −62.83. HRMS: Obsvd 387.0805 [M − H]−, Calcd for C19H13F6O2: 387.0820. HPLC (Method B): 18.11 min.
4.1.53. 5-(3′-Methoxyphenyl)pent-4-enoic acid (2).31,33
To dissolved 3-(carboxypropyl)triphenyl phosphonium bromide (15.92 g, 37.09 mmol) in THF (500 mL) was added potassium tert-butoxide (8.20 g, 73.4 mmol), and the reaction mixture was stirred at room temperature for 1 h. 3-Methoxybenzaldehyde (4.5 mL, 37 mmol) was added, and the reaction mixture was stirred at room temperature for 12 h. The THF was evaporated, and the resulting material was quenched with 2 M HCl (75 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over sodium sulfate, filtered, and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 70%A / 30%B (10 CV), 70%A / 30%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 2 (5.63 g, 27.3 mmol, 74%) as a yellow solid. NMR characterization was performed after the next step.
4.1.54. 5-(3′-Methoxyphenyl)pentanoic acid (8).31,33,53
To dissolved carboxylic acid 2 (5.63 g, 27.3 mmol) in MeOH (100 mL) was added 10% palladium on carbon (0.44 g) and hydrogen gas. The reaction was stirred at room temperature for 12 h. The reaction mixture was then filtered through Celite®, and the Celite® was washed with EtOAc (3 x 50 mL). The organic phase (MeOH and EtOAc) was evaporated under reduced pressure to afford carboxylic acid 8 (4.27 g, 20.5 mmol, 75%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 11.2 (1H, s), 7.03 (1H, d, J = 8 Hz), 6.62 (2H, d, J = 5 Hz), 6.59 (1H, d, J = 8.5 Hz), 3.59 (3H, s), 2.44 (2H, t, J = 7.5 Hz), 2.21 (2H, d, J = 7.5 Hz), 1.52 (4H, m). 13C NMR (125 MHz, CDCl3) δ 180.2, 159.7, 143.7, 129.4, 120.9, 114.3, 111.1, 55.0, 35.6, 34.1, 30.7, 24.5.
4.1.55. 2-Methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (14)
To carboxylic acid 8 (4.43 g, 21.3 mmol) was added Eaton’s reagent (43 mL, 3 g per mmol of compound 8), and the mixture was stirred at room temperature for 12 h. The mixture was then poured over ice and neutralized with sodium bicarbonate. The aqueous layer was extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over sodium sulfate, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 14 (2.80 g, 14.7 mmol, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.59 (1H, d, J = 8.5 Hz), 6.61 (1H, dd, J = 8.5, 2.5 Hz), 6.51 (1H, d, J = 2.5 Hz), 3.63 (3H, s), 2.70 (2H, t, J = 6 Hz), 2.51 (2H, t, J = 6 Hz), 1.67 (2H, p, J = 7.5 Hz), 1.59 (2H, p, J = 5.5 Hz). 13C NMR (125 MHz, CDCl3) δ 203.5, 162.5, 144.1, 131.3, 131.0, 114.7, 111.6, 55.1, 40.5, 32.6, 24.9, 20.5.
4.1.56. 2-Hydroxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (39).31,55
To benzosuberone 14 (0.89 g, 4.7 mmol) in a 20 mL microwave vial was added [TMAH][Al2Cl7] (22 mL, 0.53 M), and the reaction mixture was subjected to microwave irradiation for 1 h at 80 °C. The reaction mixture was poured into water (50 mL) and extracted with EtOAc (3 x 25 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7% / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford alcohol 39 (0.66 g, 3.8 mmol, 80%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.73 (1H, d, J = 8.5 Hz), 6.75 (1H, dd, J = 8.5, 2.5 Hz), 6.67 (1H, d, J = 2 Hz), 3.84 (1H, s), 2.87 (2H, t, J = 6 Hz), 2.71 (2H, t, J = 6 Hz), 1.82 (4H, m). 13C NMR (125 MHz, CDCl3) δ 204.9, 159.9, 144.8, 131.6, 131.3, 116.3, 113.7, 40.7, 32.7, 25.0, 20.7.
4.1.57. 2-((tert-Butyldimethylsilyl)oxy)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (42).31
Phenol 39 (0.42 g, 2.4 mmol) was dissolved in dimethylformamide (50 mL). TBSCl (0.72 g, 4.8 mmol) and DIPEA (1.24 mL, 7.14 mmol) were added, and the solution was stirred for 12 h at room temperature. The reaction mixture was washed with water (50 mL) and extracted with EtOAc (5 x 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford TBS-protected 42 (0.53 g, 1.82 mmol, 77%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 7.62 (1H, d, J = 8.5 Hz), 6.64 (1H, dd, J = 8.5, 2 Hz), 6.56 (1H, d, J = 2 Hz), 2.77 (2H, t, J = 6 Hz), 2.59 (2H, t, J = 6 Hz), 1.75 (2H, p, J = 6.5 Hz), 1.68 (2H, p, J = 6 Hz), 0.90 (9H, s), 0.14 (6H, s). 13C NMR (125 MHz, CDCl3) δ 203.7, 159.0, 143.9, 132.0, 130.9, 120.8, 117.8, 40.5, 32.5, 25.5, 25.0, 20.6, 18.0, −4.5.
4.1.58. 2-((tert-Butyldimethylsilyl)oxy)-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (48).31
To an oven dried flask, THF (50 mL) and 3,4,5-trimethoxyphenyl bromide (1.01 g, 4.04 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (1.71 mL, 4.27 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 42 (0.53 g, 3.0 mmol) in THF (25 mL) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 48 (0.470 g, 1.02 mmol, 34%) as a clear oil. NMR characterization was performed after the next step.
4.1.59. tert-Butyldimethyl((9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-3-yl)oxy)silane (55).31
Acetic acid (10 mL) was added to tertiary alcohol 48 (0.47 g, 1.0 mmol), and the reaction mixture was stirred for 12 h at room temperature. The mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure to afford protected benzosuberene 55 (0.39 g, 0.89 mmol, 87%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.90 (1H, d, J = 8 Hz), 6.77 (1H, d, J = 2.5 Hz), 6.67 (1H, dd, J = 8.5, 2.5 Hz), 6.49 (2H, s), 6.34 (1H, t, J = 7 Hz), 3.86 (3H, s), 3.79 (6H, s), 2.60 (2H, t, J = 7 Hz), 2.15 (2H, p, J = 7.5Hz), 1.96 (2H, q, J = 7 Hz), 1.00 (9H, s), 0.23 (6H, s). 13C NMR (125 MHz, CDCl3) δ 154.4, 152.8, 143.7, 142.8, 138.4, 137.3, 133.0, 130.5, 127.0, 120.0, 117.3, 105.2, 60.9, 56.1, 35.0, 32.6, 25.7, 25.5, 18.2, −4.3.
4.1.60. 9-(3′,4′,5′-Trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-3-ol (68).31
TBS-protected 55 (0.39 g, 0.89 mmol) was dissolved in THF (25 mL), TBAF (1.06 mL, 1.06 mmol) was added, and the reaction mixture was stirred at room temperature for 12 h. The solution was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 68 (0.13 g, 0.40 mmol, 45%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.89 (1H, d, J = 8 Hz), 6.78 (1H, d, J = 3 Hz), 6.67 (1H, dd, J = 8.5 Hz, 2.5 Hz), 6.50 (2H, s), 6.33 (1H, t, J = 7.5 Hz), 6.21 (1H, s), 3.88 (3H, s), 3.79 (6H, s), 2.59 (2H, t, J = 7 Hz), 2.14 (2H, p, J = 7 Hz), 1.95 (2H, q, J = 7 Hz). 13C NMR (125 MHz, CDCl3) δ 154.9, 152.8, 144.0, 142.7, 138.7, 137.0, 132.1, 130.7, 127.1, 115.4, 112.9, 105.3, 61.0, 56.1, 35.0, 32.6, 25.5. HRMS: Obsvd 349.1417 [M + Na+], Calcd for C20H22O4Na: 349.1410. HPLC (Method B): 14.30 min.
4.1.61. 2-Methoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (20).30–31,56
To an oven dried flask, THF (50 mL) and 3,4,5-trimethoxyphenyl bromide (2.82 g, 11.4 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (4.9 mL,12 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 14 (1.60 g, 8.41 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78° C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 20 (2.29 g, 6.48 mmol, 76%) as a light yellow oil. NMR characterization was performed after the next step.
4.1.62. 3-Methoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (29).30–31,56
Acetic acid (15 mL) was added to tertiary alcohol 20 (2.29 g, 6.38 mmol), and the reaction mixture was stirred for 12 h at room temperature. The mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 29 (0.362 g, 1.06 mmol, 17%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 6.99 (1H, d, J = 8.5 Hz), 6.84 (1H, d, J = 2.5 Hz), 6.74 (1H, dd, J = 8.5, 2.5 Hz), 6.53 (2H, s), 6.37 (1H, t, J = 7.5 Hz), 3.78 (3H, s), 3.81 (3H, s), 3.79 (6H, s) 2.65 (2H, t, J = 7 Hz), 2.18 (2H, p, J = 7 Hz), 1.98 (2H, q, J = 7 Hz). 13C NMR (125 MHz, CDCl3) δ 158.5, 152.8, 143.7, 142.7, 138.5, 137.3, 132.3, 130.5, 126.8, 113.8, 111.1, 105.1, 60.7, 55.9, 54.9, 35.0, 32.7, 25.4. HRMS: Obsvd 363.1574 [M + Na+], Calcd for C21H24O4Na: 363.1567. HPLC (Method B): 18.33 min.
4.1.63. 3-Methoxy-6,7-dihydro-5H-benzo[7]annulen-9-yl trifluoromethanesulfonate (59)
To an oven dried flask, diisopropylamine (1.74 mL, 12.4 mmol) dissolved in THF (50 mL) was added and cooled to −78 °C. Then n-BuLi (5.0 mL, 12 mmol) was added, and the reaction was stirred for 15 min. Benzosuberone 14 (2.14 g, 11.3 mmol) dissolved in THF was added dropwise and stirred for 2 h at −78 °C. N-Phenyl-bis(trifluoromethanesulfonimide) (4.42 g, 12.4 mmol) dissolved in THF was then added dropwise, and the reaction mixture was stirred for 12 h while warming from −78 °C to room temperature. After 12 h, the THF was evaporated under reduced pressure, and the resulting solid was washed with water and extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford triflate 59 (2.28 g, 7.07 mmol, 68%) as a yellow solid. 1H NMR (500 MHz, CDCl3) 7.51(1H, d, J = 8.5 Hz), 6.86 (1H, dd, J = 8.5, 2.5 Hz), 6.81 (1H, d, J = 3 Hz), 6.15 (1H, t, J = 6 Hz), 3.79 (3H, s), 2.77 (2H, t, J = 6.5 Hz), 2.20 (2H, q, J = 7 Hz), 2.04 (2H, q, J = 7 Hz).
4.1.64. 3-Methoxy-9-(2′,3′,4′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (62)
Triflate 59 (2.28 g, 7.07 mmol) was dissolved in THF, and 2,3,4-trimethoxyphenyl boronic acid (1.65 g, 7.78 mmol), barium hydroxide octahydrate (3.35 g, 10.6 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.24 g, 0.21 mmol) were added to the solution and refluxed at 80 °C for 2 h. The solution was then filtered through Celite®, and the Celite® was washed with dichloromethane (3 × 30 mL). The combined organic solution (THF and dichloromethane) was evaporated under reduced pressure. The crude reaction mixture was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 45%A / 55%B (10 CV), 45%A / 55%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 62 (1.05 g, 3.08 mmol, 44%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.02 (1H, d, J = 8.5 Hz), 6.88 (1H, d, J = 2.5 Hz), 6.77 (1H, dd, J = 8.5, 2.5 Hz), 6.58 (2H, s), 6.41 (1H, t, J = 7.5 Hz), 3.91 (3H, s), 3.83 (3H s), 3.82 (6H, s), 2.69 (2H, t, J = 7 Hz), 2.21 (2H, p, J = 7 Hz), 2.00 (2H, q, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 158.5, 153.4, 152.8, 143.6, 142.7, 138.3, 137.4, 132.3, 130.4, 126.7, 123.5, 113.8, 111.1, 105.2, 60.6, 60.1, 55.8, 54.8, 35.0, 32.7, 25.4. HRMS: Obsvd 363.1573 [M + Na+], Calcd for C21H24O4Na: 363.1567. HPLC (Method B): 18.23 min.
4.1.65. 3-Methoxy-9-(4′-methoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (37)
To a solution of 4-methoxyphenyl bromide (0.886 g, 4.73 mmol) in THF (50 mL) at −78 °C was added n-BuLi (3.4 mL, 2.5 M in hexanes), and the reaction mixture was stirred for 30 min. Benzosuberone 14 (0.601 g, 3.15 mmol) in THF (25 mL) was added dropwise over a period of 15 min. The reaction mixture was stirred while warming to room temperature over 12 h. Water was added, and the mixture was extracted with EtOAc (100 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a pre-packed 40 g silica column [solvent A, EtOAc, solvent B, hexanes; gradient 0%A/100%B →100%A/0%B over 22.9 min; flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberene analogue 37 (0.32 g, 1.1 mmol, 36%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.20 (2H, d, J = 8.7 Hz), 6.94 (1H, d, J = 8.4 Hz), 6.83 (2H, d, J = 8.5 Hz), 6.82 (1H, d, J = 2.5 Hz), 6.73 (1H, dd, J = 8.5, 2.7 Hz), 6.29 (1H, t, J = 7.5 Hz) 3.83 (3H, s), 3.81 (3H, s), 2.63 (2H, t, J = 7.0 Hz), 2.16 (2H, p, J = 7.1 Hz), 1.96 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 126 MHz) δ 158.8, 158.3, 143.8, 142.1, 135.3, 133.0, 130.4, 125.9, 113.9, 113.50, 113.49, 111.1, 55.3, 55.2, 35.2, 32.8, 25.4. HRMS: Obsvd 311.1713 [M + H+], Calcd for C20H21O3 : 311.1642. HPLC (Method B): 17.65 min.
4.1.66. 3-Methoxy-2-methylbenzaldehyde.58
N′,N′,N′-trimethylethylene diamine (1.84 mL, 14.3 mmol) was dissolved in benzene (25 mL) and cooled to 0° C. n-BuLi (5.5 mL, 13 mmol) was added dropwise, and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was again cooled to 0° C, and m-anisaldehyde (1.54 mL, 13.3 mmol) was added. The reaction mixture was stirred for 15 min at room temperature and then cooled to 0° C. Phenyllithium (22.0 mL, 40.0 mmol) was added dropwise. The reaction mixture was stirred for 12 h at room temperature. THF (20 mL) was then added, and the reaction mixture was cooled to −78 °C. Methyl iodide (5.0 mL, 80 mmol) was added dropwise, and the reaction mixture was stirred for 30 min at room temperature. The reaction mixture was poured into cold 10% HCl (50 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford 3-methoxy-2-methylbenzaldehyde (2.01 g, 14.3 mmol, 98%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 9.97 (1H, s), 7.10 (1H, d, J = 8 Hz), 6.98 (1H, t, J = 8 Hz), 6.74 (1H, d, J = 8 Hz), 3.54 (3H, s), 2.23 (3H, s). 13C NMR (125 MHz, CDCl3) δ 192.0, 157.8, 134.9, 128.9, 126.3, 122.7, 114.8, 55.3, 10.0.
4.1.67. 5-(3′-Methoxy-2′-methylphenyl)pent-4-enoic acid (3)
To dissolved 3-(carboxypropyl)triphenyl phosphonium bromide (6.55 g, 15.3 mmol) in THF was added potassium tert-butoxide (3.30 g, 29.0 mmol), and the reaction mixture was stirred at room temperature for 1 h. 3-Methoxy-2-methylbenzaldehyde (2.01 g, 13.4 mmol) dissolved in THF was added to the original reaction mixture, and the reaction mixture was stirred at room temperature for 12 h. The THF was evaporated, and the resulting material was quenched with 2 M HCl (75 mL) and extracted with EtOAc (3 × 100 mL). The combined organic phase was evaporated under reduced pressure and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 75%A / 25%B (10 CV), 75%A / 25%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 3 (2.12 g, 9.62 mmol, 69%) as a yellow oil. NMR characterization was performed after the next step.
4.1.68. 5-(3′-Methoxy-2′-methylphenyl)pentanoic acid (9)
To dissolved carboxylic acid 3 (2.12 g, 9.62 mmol) in MeOH (100 mL) was added 10% palladium on carbon (0.44 g) and hydrogen gas. The reaction mixture was stirred at room temperature for 12 h. The mixture was then filtered through Celite®, and the Celite® was washed with EtOAc (3 × 50 mL). The combined organic phase (MeOH and EtOAc) was evaporated under reduced pressure. The residue was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 9 (1.20 g, 5.40 mmol, 59%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 11.94 (1H, s), 7.23 (1H, t, J = 8 Hz), 6.91 (1H, d, J = 7.5 Hz), 6.84 (1H, d, J = 8.5 Hz), 3.93 (3H, s), 2.77 (2H, t, J = 8 Hz), 2.51 (2H, t, J = 7 Hz), 2.34 (3H, s), 1.86 (2H, p, J = 7.5 Hz), 1.75 (2H, m). 13C NMR (125 MHz, CDCl3) δ 180.4, 157.9, 141.6, 126.1, 124.5, 121.6, 108.0, 55.5, 34.1, 33.4, 30.0, 24.7, 11.3.
4.1.69. 2-Methoxy-1-methyl-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (15)
To carboxylic acid 9 (1.20 g, 5.40 mmol) was added Eaton’s reagent (10.8 mL, 3 g per mmol of compound 9), and the mixture was stirred at room temperature for 12 h. It was then poured over ice and neutralized with sodium bicarbonate. The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 15 (2.43 g, 11.0 mmol, 74%) as a light yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.42 (1H, d, J = 8.5 Hz), 6.64 (1H, d, J = 8.5 Hz), 3.72 (3H, s), 2.76 (2H, t, J = 6 Hz), 2.53 (2H, t, J = 6 Hz), 2.09 (3H, s), 1.69 (2H, p, J = 7.5 Hz), 1.61 (2H, p, J = 6.5 Hz). 13C NMR (125 MHz, CDCl3) δ 205.9, 160.4, 140.4, 132.6, 127.4, 123.7, 107.6, 55.4, 40.4, 27.0, 24.1, 20.6, 11.0.
4.1.70. 2-Methoxy-1-methyl-5-(3,′4′,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (21)
To an oven dried flask, THF (50 mL) and 3,4,5-trimethoxyphenyl bromide (0.85 g, 3.4 mmol) were added, and the solution was cooled to −78 °C. n-Buli (1.5 mL, 3.6 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 15 (0.52 g, 2.6 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 60%A / 50%B (10 CV), 60%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 21 (2.29 g, 6.39 mmol, 76%) as a light yellow oil. NMR characterization was performed after the next step.
4.1.71. 3-Methoxy-4-methyl-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (30)
Acetic acid (15 mL) was added to tertiary alcohol 21 (0.48 g, 1.3 mmol), and the reaction mixture was stirred for 12 h at room temperature. The mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 30 (0.16. g, 0.45 mmol, 36%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 6.86 (1H, d, J = 8.5 Hz), 6.69 (1H, d, J = 8.5 Hz), 6.53 (2H, s), 6.33 (1H, t, J = 7 Hz), 3.87 (3H, s), 3.82 (3H s,) 3.80 (6H, s), 2.69 (2H, t, J = 6.5 Hz), 2.29 (3H, s), 2.12 (2H, p, J = 7 Hz), 1.91 (2H, q, J = 7 Hz). 13C NMR (125 MHz, CDCl3) δ 156.5, 152.8, 143.5, 141.6, 138.5, 137.3, 132.9, 127.4, 126.4, 123.1, 107.4, 105.3, 60.8, 56.1, 55.4, 34.0, 27.7, 25.5, 11.8. HRMS: Obsvd 377.1731 [M + Na+], Calcd for C22H26O5Na: 377.1723. HPLC (Method B): 19.56 min.
4.1.72. 2-Ethyl-3-methoxybenzaldehyde.59
N′,N′N′-trimethylethlene diamine (1.36 mL, 10.5 mmol) was dissolved in benzene (25 mL) and cooled to 0° C. n-BuLi (4.05 mL, 10.1 mmol) was added dropwise, and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was again cooled to 0 °C, m-anisaldehyde (1.34 mL, 9.84 mmol) was added, and the reaction mixture was stirred for 15 min at room temperature. The reaction mixture was then cooled to 0 °C, and phenyllithium (16.4 mL, 29.5 mmol) was added dropwise. The reaction mixture was stirred overnight at room temperature. THF (20 mL) was then added, and the reaction mixture was cooled to −78 °C. Ethyl iodide (3.7 mL, 59 mmol) was added dropwise, and the reaction mixture was stirred for 30 min at room temperature. The reaction mixture was poured into cold 10% HCl (50 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford 2-ethyl-3-methoxybenzaldehyde (0.91 g, 5.6 mmol, 57%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 10.24 (1H, s), 7.37 (1H, dd, J = 7.5, 1 Hz), 7.23 (1H, t, J = 8 Hz), 7.01 (1H, dd, J = 8.5, 1 Hz), 3.79 (3H, s), 3.01 (2H, q, J = 7.5 Hz), 1.14 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 192.1, 157.6, 135.9, 134.4., 126.7, 122.6, 115.5, 55.7, 17.5, 15.3.
4.1.73. 5-(2′-Ethyl-3′-methoxyphenyl)pent-4-enoic acid (4)
To dissolved 3-(carboxypropyl)triphenyl phosphonium bromide (3.56 g, 8.30 mmol) in THF (500 mL) was added potassium tert-butoxide (2.03 g, 18.1 mmol), and the reaction mixture was stirred at room temperature for 1 h. 2-Ethyl-3-methoxybenzaldehyde (1.35 g, 8.22 mmol) dissolved in THF (100 mL) was added to the original reaction mixture and stirred at room temperature for 12 h. The THF was evaporated, and the resulting material was quenched with 2 M HCl (75 mL) and extracted with EtOAc (3 × 100 mL). The combined organic layer was evaporated under reduced pressure and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 4 (1.78 g, 7.60 mmol, 92%) as an orange-yellow oil. NMR characterization was performed after the next step.
4.1.74. 5-(2′-Ethyl-3′-methoxyphenyl)pentanoic acid (10)
To dissolved carboxylic acid 4 (1.78 g, 7.60 mmol) in MeOH (50 mL) was added 10% palladium on carbon (0.58 g) and hydrogen gas. The reaction was stirred at room temperature for 12 h. The reaction mixture was then filtered through Celite®, and the Celite® was washed with EtOAc (3 × 50 mL). The combined organic phase (MeOH and EtOAc) was evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 10 (0.79 g, 3.3 mmol, 44%) as a clear oil. It is likely in this case that the carboxylic acid became methylated. 1H NMR (500 MHz, CDCl3) δ 7.14 (1H, t, J = 8 Hz), 6.82 (1H, d, J = 7.5 Hz), 6.75 (1H, d, J = 8.5 Hz), 3.83 (3H, s), 3.70 (3H, s), 2.76 (2H, q, J = 7.5 Hz), 2.70 (2H, t, J = 8 Hz), 2.39 (2H, J = 7.5 Hz), 1.79 (2H, p, J = 7 Hz), 1.68 (2H, p, J = 8 Hz), 1.22, (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 173.7, 157.6, 140.9, 130.6, 126.2, 121.7, 108.1, 55.2, 21.2, 33.9, 32.6, 21.1, 25.1, 19.2, 14.5.
4.1.75. 1-Ethyl-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (16)
To carboxylic acid 10 (0.79 g, 3.3 mmol) was added Eaton’s reagent (6.7 mL, 3 g per mmol of compound 10), and the reaction mixture was stirred at room temperature for 12 h. The mixture was then poured over ice and neutralized with sodium bicarbonate. The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 16 (0.32 g, 1.6 mmol, 52%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.46 (1H, d, J = 8.5 Hz), 6.70 (1H, d, J = 8.5 Hz), 3.77 (3H, s), 2.82 (2H, t, J = 6 Hz), 2.65 (2H, q, J = 7.5 Hz), 2.57 (2H, t, J = 6 Hz), 1.75 (2H, p, J = 7 Hz), 1.66 (2H, p, J = 6.5 Hz), 1.03 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 206.3, 160.3, 139.5, 132.9, 130.0, 127.7, 108.0, 55.4, 40.4, 25.3, 24.9, 20.4, 18.9, 14.5.
4.1.76. 1-Ethyl-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (22)
To an oven dried flask, THF (50 mL) and 3,4,5-trimethoxyphenyl bromide (0.52 g, 2.1 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (0.88 mL, 2.2 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 16 (0.32 g, 1.54 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78° C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 22 (0.20 g, 0.52 mmol, 33%) as a light yellow oil. NMR characterization was performed after the next step.
4.1.77. 4-Ethyl-3-methoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (31)
Acetic acid (15 mL) was added to tertiary alcohol 22 (0.20 g, 0.52 mmol), and the reaction mixture was stirred for 12 h at room temperature. The reaction mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 50%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 31 (0.085 g, 0.32 mmol, 45%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 6.85 (1H, d, J = 8.5 Hz), 6.69 (1H, d, J = 8.5 Hz), 6.52 (2H, s), 6.32 (1H, t, J = 7.5 Hz), 3.86 (3H, s), 3.83 (3H, s), 3.81 (6H, s), 2.78 (2H, q, J = 7.5 Hz), 2.69 (2H, t, J = 6.5 Hz), 2.14 (2H, p, J = 7 Hz), 1.92 (2H, q, J = 7 Hz), 1.18 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 156.5, 153.1, 143.8, 141.3, 139.0, 137.6, 133.4, 129.9, 127.9, 126.7, 107.8, 105.6, 61.2, 56.4, 55.7, 35.2, 27.5, 25.7, 20.0, 15.2. HRMS: Obsvd 391.1891 [M + Na+], Calcd for C23H28O4Na: 391.1880. HPLC (Method B): 20.66 min.
4.1.78. 3-Methoxy-2-propylbenzaldehyde.60
N′,N′,N′-trimethylethlene diamine (1.55 mL, 12.0 mmol) was dissolved in benzene (25 mL) and cooled to 0° C. n-BuLi (4.6 mL, 11 mmol) was added dropwise, and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was again cooled to 0 °C, and m-anisaldehyde (1.30 mL, 11.2 mmol) was added. The reaction mixture was stirred for 15 min at room temperature and cooled to 0° C. Phenyllithium (18.7 mL, 33.7 mmol) was added dropwise. The reaction mixture was stirred overnight at room temperature. THF (20 mL) was then added, and the reaction mixture was cooled to −78° C. Propyl iodide (6.7 mL, 67.32 mmol) was added dropwise, and the reaction mixture was stirred for 30 min at room temperature. The reaction mixture was poured into cold 10% HCl (50 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford 3-methoxy-2-propylbenzaldehyde (1.82 g, 10.2 mmol, 91%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 10.19 (1H, s), 7.32 (1H, d, J = 8 Hz), 7.16 (1H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8 Hz), 3.71 (3H, s), 2.94 (2H, t, J = 7 Hz), 1.48 (2H, sext, J = 7.5 Hz), 0.88 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 191.9, 157.8, 134.8, 134.3, 126.7, 122.3, 115.3, 55.5, 25.8, 24.2, 14.0.
4.1.79. 5-(3′-Methoxy-2′-propylphenyl)pent-4-enoic acid (5)
To dissolved 3-(carboxypropyl)triphenyl phosphonium bromide (4.43 g, 10.3 mmol) in THF (500 mL) was added potassium tert-butoxide (2.52 g, 22.5 mmol), and the reaction mixture was stirred at room temperature for 1 h. 3-Methoxy-2-propylbenzaldehyde (1.82 g, 10.2 mmol) dissolved in THF (100 mL) was added to the original reaction mixture and stirred at room temperature for 12 h. The THF was evaporated, and the residue was quenched with 2 M HCl (75 mL) and extracted with EtOAc (3 × 100 mL). The combined organic phase was evaporated under reduced pressure and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford compound 5 (2.25 g, 9.06 mmol, 89%) as a yellow oil. NMR characterization was performed after the next step.
4.1.80. 5-(3′-Methoxy-2′-propylphenyl)pentanoic acid (11)
To dissolved compound 5 (2.25 g, 9.06 mmol) in MeOH (50 mL) was added 10% palladium on carbon (0.47 g) and hydrogen gas. The reaction mixture was stirred at room temperature for 12 h. The mixture was then filtered through Celite®, and the Celite® was washed with EtOAc (3 × 50 mL). The combined organic phase (MeOH and EtOAc) was evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford compound 11 (0.87 g, 3.5 mmol, 38%) as a clear oil. It is likely that the carboxylic acid was methylated during this reaction. 1H NMR (500 MHz, CDCl3) δ 7.14 (1H, t, J = 8 Hz), 6.83 (1H, d, J = 7.5 Hz), 6.75 (1H, d, J = 8 Hz), 3.82 (3H, s), 3.71 (3H, s), 2.71 (4H, m), 2.40 (2H, t, J = 7.5 Hz), 1.79 (2H, p, J = 7.5 Hz), 1.66 (4H, m), 1.09 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 173.4, 157.5, 140.9, 128.8, 125.9, 121.2, 107.6, 54.8, 50.9, 33.5, 32.3, 30.7, 27.8, 24.7, 23.1, 14.2.
4.1.81. 2-Methoxy-1-propyl-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (17)
To carboxylic acid 11 (0.87 g, 3.5 mmol) was added Eaton’s reagent (7.0 mL, 3 g per mmol of compound 11), and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was then poured over ice and neutralized with sodium bicarbonate. The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 17 (0.56 g, 2.41 mmol, 69%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.45 (1H, d, J = 8.5 Hz), 6.68 (1H, d, J = 8.5 Hz), 3.75 (3H, s), 2.81 (2H, t, J = 6.5 Hz), 2.59 (4H, m), 1.74 (2H, p, J = 7 Hz), 1.65 (2H, p, J = 6 Hz), 1.42 (2H, sext, J = 7.5 Hz), 0.91 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 206.2, 160.4, 139.7, 132.9, 128.5, 127.6, 107.8, 55.2, 40.3, 27.6, 25.3, 24.8, 23.2, 20.3, 14.2.
4.1.82. 2-Methoxy-1-propyl-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (23)
To an oven dried flask, THF (50 mL) and 3,4,5-trimethoxyphenyl bromide (0.80 g, 3.3 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (1.4 mL, 3.4 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 1 h. Benzosuberone 17 (0.56 g, 2.4 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78° C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 23 (0.53g, 1.3 mmol, 52%) as a colorless oil. NMR characterization was performed after the next step.
4.1.83. 3-Methoxy-4-propyl-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (32)
Acetic acid (10 mL) was added to tertiary alcohol 23 (0.53 g, 1.3 mmol), and the reaction mixture was stirred for 12 h at room temperature. The reaction mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction mixture was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 32 (0.13 g, 0.34 mmol, 25%) as a cream-colored solid. 1H NMR (500 MHz, CDCl3) δ 6.86 (1H, d, J = 8.5 Hz), 6.70 (1H, d, J = 8.5 Hz), 6.53 (2H, s), 6.33 (1H, t, J = 7.5 Hz), 3.88 (3H, s), 3.84 (3H, s), 3.82 (6H, s), 2.72 (4H, m), 2.15 (2H, p, J = 7 Hz), 1.93 (2H, q, J = 7 Hz), 1.59 (2H, sext, J = 7 Hz), 1.04 (3H, t, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3) δ 156.4, 152.8, 143.5, 141.3, 138.7, 137.3, 133.1, 128.3, 127.6, 126.4, 107.5, 105.3, 60.9, 56.2, 55.4, 35.0, 28.6, 27.3, 25.5, 23.8, 14.5. HRMS: Obsvd 405.2043 [M + Na+], Calcd for C24H30O4Na+: 405.2036. HPLC (Method B): 21.53 min.
4.1.84. 2-(2′,3′-Dimethoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole.61–62
A mixture of 2,3-dimethoxybenzoic acid (5.00 g, 27.5 mmol) in SOCl2 (10 mL) was stirred at room temperature for 24 h. The excess of SOCl2 was evaporated under reduced pressure, and the residue was diluted with CH2Cl2. (80 mL). This solution was added dropwise to a solution of 2-amino-2-methylpropan-1-ol (4.89 g, 55 mmol) in CH2Cl2 (150 mL) at −10 °C and stirred for 20 h while warming to room temperature. The resulting suspension was filtered, and the filtrate was evaporated to give the crystalline amide. The latter was treated dropwise with SOCl2 (10 mL) and stirred at room temperature for another 7 h. The mixture was then poured into diethyl ether (400 mL) to form a suspension, which was then cooled to 0° C, and aqueous NaOH solution (20%, 100 mL) was added. The aqueous layer was extracted with diethyl ether (3 × 100 mL). The combined organic phase was dried with Na2SO4, filtered, and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford 2-(2,3-dimethoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole (5.22 g, 22.2 mmol, 81%) as a yellow solid. 1H NMR (CDCl3, 500 MHz) δ 7.31-7.00 (3H, m), 4.11 (2H, s), 3.86 (6H, m), 1.39 (6H, s). 13C NMR (CDCl3, 125 MHz) δ 161.3, 153.3, 148.7, 123.8, 123.4, 122.6, 115.0, 79.2, 67.3, 61.4, 56.1, 28.3.
4.1.85. 2-(2′-Butyl-3′-methoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole.63
A solution of 2-(2,3-dimethoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole (5.02 g, 21.4 mmol) in THF (80 mL) was stirred and cooled to −40 °C in a cyclohexanone/dry ice bath. n-BuLi (13 mL, 2.5 M) was added dropwise to the reaction flask. The reaction mixture was stirred for 5 h while warming to 0° C. The reaction was quenched with saturated aqueous NH4Cl and extracted with diethyl ether (3 × 30 mL). The combined organic phase was dried with Na2SO4 and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford 2-(2-butyl-3-methoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole as yellow liquid (4.99 g, 19.1 mmol, 89%). 1H NMR (CDCl3, 500 MHz) δ 7.23 (1H, dd, J = 7.5 Hz, 1.2 Hz), 7.15 (1H, t, J = 8.0 Hz), 6.91 (1H, dd, J = 8.1, 1.1 Hz), 4.07 (2H, s), 3.82 (3H, s), 1.53-1.47 (2H, m), 1.38 (6H, s), 1.41-1.33 (4H, m), 0.93 (3H, t, J = 7.4Hz). 13C NMR (CDCl3, 125 MHz) δ 163.0, 157.7, 132.0, 129.2, 126.1, 121.9, 112.3, 78.8, 67.7, 55.7, 32.4, 28.4, 26.5, 23.1, 14.0.
4.1.86. 2-Butyl-3-methoxybenzoic acid.63–64
A solution of 2-(2-butyl-3-methoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole (4.69 g, 17.95 mmol) in 4.5 M HCl (100 mL) was refluxed for 21 h. After cooling to room temperature, the reaction mixture was extracted with diethyl ether (3 × 30 mL), and the combined organic phase was washed with brine, dried with Na2SO4, and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford 2-butyl-3-methoxybenzoic acid as a colorless liquid (2.45 g, 11.8 mmol, 65%). 1H NMR (CDCl3, 500 MHz) δ 7.55 (1H, dd, J = 7.9 Hz, 1.1 Hz), 7.24 (1H, t, J = 8.0 Hz), 7.05 (1H, dd, J = 8.3 Hz, 0.9 Hz), 3.87 (3H, s), 3.01 (2H, m), 1.54 (2H, m), 1.43 (2H, m), 0.95 (3H, t, J = 7.3 Hz). 13C NMR (CDCl3, 125 MHz) δ 173.1, 158.0, 134.5, 129.9, 126.2, 122.9, 114.4, 55.8, 32.4, 26.3, 23.1, 13.9.
4.1.87. (2-Butyl-3-methoxyphenyl)methanol
2-butyl-3-methoxybenzoic acid (2.45 g, 11.8 mmol) was dissolved in THF (40 mL) and stirred for 10 min. LiAlH4 (7.6 mL, 2.0 M) was then added dropwise, and the reaction mixture was stirred for 10 h while warming to room temperature. The reaction was carefully quenched with a H2O/THF (1:4) solution, followed by aqueous NaOH (15%, 20 mL), and a precipitate formed. The unwanted precipitate was was removed by filtration through Celite®. The Celite® and unwanted precipitate were washed with CH2Cl2. The H2O/THF/CH2Cl2 filtrate was extracted with EtOAc (3 × 30 mL), and the organic layer was rinsed with brine and dried with Na2SO4. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford (2-butyl-3-methoxyphenyl)methanol as a colorless liquid (1.94 g, 9.97 mmol, 85%). 1H NMR (CDCl3, 500 MHz) δ 7.19 (1H, t, J = 8.5 Hz), 7.01 (1H, d, J = 7.6 Hz), 6.83 (1H, d, J = 8.3 Hz), 4.72 (2H, d, J = 5.3 Hz), 3.83 (3H, s), 2.69 (2H, t, J = 7.4 Hz), 1.49 (4H, m), 0.95 (3H, t, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz) δ 157.7,139.6, 129.8, 126.6, 120.4, 110.0, 63.2, 55.5, 32.4, 25.5, 23.1, 14.0.
4.1.88. 2-Butyl-3-methoxybenzaldehyde
To a well stirred solution of pyridinium chlorochromate (2.586 g, 12 mmol) in CH2Cl2 (20 mL) at room temperature, (2-butyl-3-methoxyphenyl)methanol (1.936 g, 9.97 mmol) dissolved in CH2Cl2 (15 mL) was slowly added via syringe. The reaction mixture was stirred at room temperature for 12 h. The reaction mixture was filtered through Celite®, and the Celite® was washed thoroughly with CH2Cl2. The filtrate was concentrated under reduced pressure and further purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford 2-butyl-3-methoxybenzaldehyde as a dark yellow liquid (1.91 g, 9.93 mmol, 99%). 1H NMR (CDCl3, 500 MHz) δ 10.34 (1H, s), 7.46 (1H, dd, J = 7.8 Hz, 1.1 Hz), 7.30 (1H, t, J = 8.0 Hz), 7.09 (1H, dd, J = 8.1 Hz, 0.75 Hz), 3.87 (3H, s), 3.06 (2H, m), 1.53 (2H, m), 1.42 (2H, m), 0.95 (3H, t, J = 7.3 Hz). 13C NMR (CDCl3, 125 MHz) δ 192.3, 157.8, 135.0, 134.7, 126.7, 122.1, 115.5, 55.8, 33.5, 23.8, 22.8, 13.9.
4.1.89. 5-(2′-Butyl-3′-methoxyphenyl)pent-4-enoic acid (6)
A mixture of 3-(carboxypropyl) triphenylphosphonium bromide (4.52 g, 10.6 mmol) and potassium tert-butoxide (2.62 g, 23.2 mmol) in THF (100 mL) was stirred for 1 h at room temperature. 2-Butyl-3-methoxybenzaldehyde (2.03 g, 10.6 mmol) in THF (20 mL) was added dropwise to the reaction mixture and stirred for 12 h at room temperature. The reaction was quenched with 2 M HCl (15 mL), then extracted with EtOAc (3 × 50 mL). The combined organic phase was washed with brine, dried with Na2SO4, and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 3%B → 60%A/40% B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 6 (2.38 g, 9.07 mmol, 86%). NMR characterization was performed after the next step.
4.1.90. 5-(2′-Butyl-3′-methoxyphenyl)pentanoic acid (12)
Carboxylic acid 6 (2.38 g, 9.07 mmol) was mixed with 10% Pd/C (0.15 g). Methanol (25 mL) was slowly added, and the reaction mixture was stirred for 12 h at room temperature under a H2 atmosphere. The suspension was filtered through Celite®, and the Celite® was rinsed with EtOAc. The filtrate (MeOH and EtOAc) was concentrated under reduced pressure, and the crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 20%A/ 80%B (3 CV), 20%A/ 80%B → 100%A/ 0%B (10 CV), 100%A/ 0%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 12 (1.48 g, 56.0 mmol, 61%) as a yellow-brown liquid. 1H NMR (CDCl3, 500 MHz) δ 7.09 (1H, t, J = 7.9 Hz), 6.77 (1H, d, J = 7.6 Hz), 6.72 (1H, d, J = 8.1 Hz), 3.81 (3H, s), 2.63 (4H, m), 2.40 (2H, t, J = 7.3 Hz), 1.75 (2H, m), 1.64 (2H, m), 1.44 (4H, m), 0.96 (3H, t, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz) δ 179.4, 157.7, 141.1, 129.5, 126.1, 121.5, 108.1, 55.4, 34.0, 32.5, 32.3, 30.8, 25.7, 24.8, 23.2, 14.0.
4.1.91. 1-Butyl-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (18)
Eaton’s reagent (28 mL) was added to carboxylic acid 12 (1.48 g, 5.6 mmol) and sonicated until the 12 dissolved. The reaction mixture was then stirred at room temperature for 12 h. Ice was poured into the reaction flask, and then the reaction mixture was neutralized with a sat. NaHCO3 solution and extracted with EtOAc (3 × 50 mL). The combined organic phase was dried with Na2SO4 and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford ketone 18 (1.13 g, 4.59 mmol, 82%) as a yellow liquid. 1H NMR (CDCl3, 500 MHz) δ 7.53 (1H, d, J = 8.6 Hz), 6.77 (1H, d, J = 8.6 Hz), 3.86 (3H, s), 2.89 (2H, m), 2.67 (4H, m), 1.83 (2H, p, J = 6.5 Hz), 1.75 (2H, m), 1.42 (4H, m), 0.96 (3H, t, J = 7.1 Hz). 13C NMR (CDCl3, 125 MHz) δ 206.8, 160.6, 139.8, 133.0, 128.9, 127.7, 108.0, 55.5, 40.5, 32.5, 26.5, 25.5, 25.5, 23.0, 20.0, 20.5, 14.0.
4.1.92. 1-Butyl-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (24)
5-Bromo-1,2,3-trimethoxybenzene (1.70 g, 6.9 mmol) was dissolved in THF (20 mL) and cooled to −78 °C. n-BuLi (4.06 mL, 2.5 M) was added dropwise, and the reaction mixture was stirred at −78° C. After 1 h, ketone 18 (1.13 g, 4.6 mmol) in THF (15 mL) was added dropwise to the reaction flask. The reaction mixture was stirred for 12 h while warming to room temperature. The reaction was quenched with water (40 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried with Na2SO4 and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 24 (0.76 g, 1.8 mmol, 40%). 1H NMR (CDCl3, 500 MHz) δ 7.39 (1H, d, J = 8.7 Hz), 6.72 (1H, d, J = 8.8 Hz), 6.51 (2H, s), 3.84 (3H, s), 3.82 (3H, s), 3.74 (6H, s), 2.96 (2H, m), 2.67 (4H, m), 1.82 (4H, m), 1.39 (4H, m), 0.92 (3H, t, J = 6.9 Hz).
4.1.93. 4-Butyl-3-methoxy-9-(3′,4′,5′-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (33)
The tertiary alcohol 24 (0.76 g, 1.8 mmol) was dissolved in acetic acid (10 mL), and the reaction mixture was stirred for 6 h. The reaction was quenched with water (50 mL) and extracted with EtOAc (3 × 20 mL). The combined organic phase was washed with brine, dried with Na2SO4 and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford benzosuberene 33 (0.76 g, 1.8 mmol, quantitative) as a yellowish oil. 1H NMR (CDCl3, 500 MHz) δ 6.84 (1H, d, J = 8.5 Hz), 6.69 (1H, d, J = 8.5 Hz), 6.51 (2H, s), 6.32 (1H, t, J = 7.4 Hz), 3.86 (3H, s), 3.83 (3H, s), 3.81 (6H, s), 2.74 (2H, m), 2.68 (2H, t, J = 6.9 Hz), 2.13 (2H, p, J = 7.0 Hz), 1.91 (2H, q, J = 7.3 Hz), 1.53 (2H, m), 1.46 (2H, m), 0.98 (3H, t, J = 7.3 Hz). 13C NMR (CDCl3, 125 MHz) δ 156.4, 152.8, 143.5, 141.2, 138.7, 137.3, 133.1, 128.5, 127.5, 126.4, 107.5, 105.3, 60.9, 56.2, 55.4, 34.9, 32.9, 27.3, 26.3, 25.5, 23.2, 14.1. HRMS: Obsvd 397.2374 [M+H]+, calcd for C25H33O4: 397.2373. HPLC (Method B): 22.30 min.
4.2. Biological Evaluations
4.2.1. SRB Assay65–66
Inhibition of human cancer cell growth was assessed using the sulforhodamine B assay, as previously described.65 Cancer cell lines were plated at 9000 cells/well into 96-well plates using DMEM supplemented with 5% fetal bovine serum/ 1% gentamicin sulfate and incubated for 24 h. Serial dilutions of the compounds were then added. After 48 h, the cells were fixed with trichloroacetic acid, washed, dried, stained with sulforhodamine B dye (Acid red 52), solubilized, and read at 540 nm and normalized to 630 nm with an automated Biotek plate reader. A growth inhibition of 50% (GI50 or the drug concentration causing 50% reduction in the net protein increase) was calculated from the absorbance data.
4.2.2. Colchicine Binding Assay
Inhibition of [3H]colchicine binding to tubulin was determined using 0.1 mL reaction mixtures. Each reaction mixture contained 1.0 μM tubulin, 5.0 μM [3H]colchicine (from Perkin-Elmer), 5% (v/v) dimethyl sulfoxide, potential inhibitors at 1.0, 5.0 or 50 μM and components that were previously demonstrated to stabilize the colchicine binding activity of tubulin67 (1.0 M monosodium glutamate [adjusted to pH 6.6 with HCl in a 2.0 M stock solution], 0.5 mg/mL bovine serum albumin, 0.1 M glucose-1-phosphate, 1.0 mM MgCl2, and 1.0 mM GTP). Incubation was for 10 min at 37 °C, a time point at which the binding reaction in the control is 40–60% complete. Reactions were stopped by adding 2.0 mL of ice-cold water and placing the samples on ice. Each sample was poured onto a stack of two DEAE-cellulose filters, followed immediately by 6 mL of ice-cold water, and the water was aspirated under reduced vacuum. The filters were washed with 2 mL water x 3 and, following removal of excess water under a strong vacuum, placed into vials containing 5 mL of Biosafe II scintillation cocktail. Samples were counted the next day in a Beckman scintillation counter. Samples with potential inhibitors were compared to controls with no inhibitor to determine percent inhibition. All samples were corrected for the amount of colchicine that bound to the filters in the absence of tubulin.
4.2.3. Inhibition of Tubulin Polymerization
Tubulin assembly experiments were performed using 0.25 mL reaction mixtures (final volume).68 The mixtures contained 1 mg/mL (10 μM) purified bovine brain tubulin, 0.8 M monosodium glutamate (pH 6.6, as above), 4% (v/v) dimethyl sulfoxide, 0.4 mM GTP, and varying compound concentrations. Initially, all components except GTP were preincubated for 15 min at 30 °C in 0.24 mL. After chilling the mixtures on ice, 10 μL of 10 mM GTP was added. The reaction mixtures were then transferred to cuvettes held at 0 °C in Beckman DU-7400 and DU-7500 spectrophotometers equipped with electronic temperature controllers. The temperature was jumped to 30 °C over about 30 s, and polymerization was followed turbidimetrically at 350 nm for 20 min. Each reaction set included a reaction mixture without compound, and the IC50 was defined as compound concentration that inhibited extent of assembly by 50% after 20 min at 30 °C.
4.2.4. In Vivo Tumor Model
Human breast cancer cells, MDA-MB-231(ATCC), were transfected with a lentivirus containing a firefly luciferase reporter. Highly expressing stable clones were isolated to create the cell line, MDA-MB-231-Luc, which was kindly provided by Dr. Edward Graves, Stanford University.51 Induction of tumors was carried out by injecting 106 cells mixed with 30% Matrigel™ (BD Biosciences, San Jose, CA) into the mammary fat pads of female SCID mice (UTSW breeding colony). Tumors were allowed to grow to a size of approximately 5 mm in diameter, determined by calipers, before selection for BLI or histological analysis. All animal procedures were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
4.2.5. In Vivo Bioluminescence Imaging (BLI)
Bioluminescence imaging was carried out as described previously.69 Briefly, anesthetized, tumor bearing mice (O2, 2% isoflurane, Henry Schein Inc., Melville, NY) were injected subcutaneously in the fore-back neck region with 80 μL of a solution of luciferase substrate, D-luciferin (sodium salt, 120 mg/kg, in saline, Gold Biotechnology, St. Louis, MO). Mice were maintained under anesthesia (2% isoflurane in oxygen, 1 dm3/min), while baseline bioluminescence imaging was performed using a Xenogen IVIS® Spectrum (Perkin-Elmer, Alameda, CA). A series of BLI images was collected over 35 min using the following settings: auto exposure time, f-stop = 2, Field of view = D, binning = 4 (medium). Light intensity-time curves obtained from these images were analyzed using Living Image® software. Mice were injected intraperitoneally with either 120 μL of saline (vehicle), CA4P (provided by OXiGENE 120 mg/kg in saline as used previously7 or analogue 73 (20, 30 or 40 mg/kg) in saline immediately after baseline BLI. Bioluminescence imaging was repeated, with new luciferin injections 4 and 24 h later. Dosing with 20 and 30 mg/kg was repeated in a separate cohort of mice.
Supplementary Material
Acknowledgments
The authors are grateful to the National Cancer Institute of the National Institutes of Health (Grant No. 5R01CA140674 to K.G.P, M.L.T, and R.P.M), the Cancer Prevention and Research Institute of Texas (CPRIT, Grant No. RP140399 to K.G.P, M.L.T, and R.P.M), and OXiGENE, Inc. (grant to K.G.P. and M.L.T.) for their financial support of this project, and to the NSF for funding the Varian 500 MHz NMR spectrometer (Grant no. CHE-0420802). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. The authors would also thank Dr. James Karban and Dr. Michelle Nemec (Director) for the use of the shared Molecular Biosciences Center at Baylor University, Dr. Alejandro Ramirez (Mass Spectrometry Core Facility, Baylor University) and Dr. Kevin Klausmeyer and Marissa Penney (X-ray analysis). The authors are grateful to Mr. Tyler Goddard (Baylor University) for his contributions to the synthesis of certain analogues, and to Jeni Gerberich (UTSW) and Dr. Li Li (UTSW) for valuable technical assistance. Imaging was facilitated with the assistance of Resources of the Harold C. Simmons Cancer Center supported through an National Institutes of Health National Cancer Institute Cancer Center Support Grant [Grant 1P30 CA142543], specifically, the Southwestern Small Animal Imaging Resource, and Live Cell Imaging Resource. The IVIS Spectrum was purchased with support of 1S10RR024757.
Footnotes
Supplementary data including 1H NMR, 13C NMR, 19F NMR, 31P NMR, HPLC, HRMS for final target compounds and intermediates (1H NMR, 13C NMR, 19F NMR only), X-ray crystallography for compound 72, an alternative synthetic procedure for compound 30, and molecular docking for compounds 29, 62, and 72 associated with this article can be found in the online Supplementary data file.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siemann DW, Chaplin DJ, Horsman MR. Cancer. 2004;10:2491. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]
- 2.Siemann DW. Cancer Treat Rev. 2011;37:63. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tozer GM, Kanthou C, Baguley BC. Nat Rev Cancer. 2005;5:423. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- 4.Kanthou C, Tozer GM. Expert Opin Ther Targets. 2007;11:1443. doi: 10.1517/14728222.11.11.1443. [DOI] [PubMed] [Google Scholar]
- 5.Horsman MR, Bohn AB, Busk M. Exp Oncol. 2010;32:143. [PubMed] [Google Scholar]
- 6.Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR, Marme D, Lorusso PM. Clin Cancer Res. 2005;11:416. [PubMed] [Google Scholar]
- 7.Mason RP, Zhao D, Liu L, Trawick ML, Pinney KG. Integrat Biol. 2011;3:375–387. doi: 10.1039/c0ib00135j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dougherty GJ, Chaplin DJ. In: Vascular Disruptive Agents for the Treatment of Cancer. Meyer T, editor. Chaper 1 Springer; New York: 2010. pp. 1–27. [Google Scholar]
- 9.Siemann DW. In: Vascular-Targeted Therapies in Oncology. Siemann D, editor. John Wiley & Sons; London, UK: 2006. pp. 1–8. [Google Scholar]
- 10.Pettit GR, Singh SB, Boyd MR. J Med Chem. 1995;38:1666. doi: 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- 11.Oshumi K, Nakagawa R, Fukuda Y. J Med Chem. 1998;41:3022. doi: 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- 12.McGowan AT, Fox BW. Cancer Chemother Pharmacol. 1990;26:79. doi: 10.1007/BF02940301. [DOI] [PubMed] [Google Scholar]
- 13.El-Zayat AAE, Degan D, Drabek S. Anticancer Drug Des. 1993;4:19. [Google Scholar]
- 14.Pettit GR, Minardi MD, Boyd MR, Pettit RK. Anticancer Drugs Des. 2000;15:397. [PubMed] [Google Scholar]
- 15.Pettit GR, Lippert JW., III Anticancer Drug Des. 2000;15:203. [PubMed] [Google Scholar]
- 16.Boehle AS, Sipos B, Kliche U, Kalthoff H, Dohrmann P. Ann Thorac Surg. 2001;71:1657. doi: 10.1016/s0003-4975(01)02408-0. [DOI] [PubMed] [Google Scholar]
- 17.Pettit GR, Moser BR, Boyd MR. Anticancer Drug Des. 2001;16:185. [PubMed] [Google Scholar]
- 18.Ohno T, Kawano K, Tahara K. Gastroenterology. 2001;120:2831. [Google Scholar]
- 19.Oshumi K, Hatanaka R, Nakagawa R. Anticancer Drugs. 1999;14:539. [Google Scholar]
- 20.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experentia. 1989;45:209. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 21.Pettit GR, Singh SB, Niven ML, Hamel E, Schmidt JM. J Nat Prod. 1987;50:119. doi: 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- 22.Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Cancer Res. 1997;57:1829. [PubMed] [Google Scholar]
- 23.Pettit GR, Rhodes MR. Anticancer Drug Des. 1998;13:183. [PubMed] [Google Scholar]
- 24.Cooney MM, Ortiz J, Bukowski RM, Remick SC. Curr Onocol Rep. 2005;7:90–95. doi: 10.1007/s11912-005-0033-x. [DOI] [PubMed] [Google Scholar]
- 25.Kirwan IG, Liadman PM, Swaine DJ, Anthoney DA, Pettit GR, Lippert JW, III, Shnyder SD, Cooper PA, Bibby MC. Clin Cancer Res. 2004;10:1446–1453. doi: 10.1158/1078-0432.ccr-0518-03. [DOI] [PubMed] [Google Scholar]
- 26.Kretzschmann VK, Fürst R. Phytochem Rev. 2014;13:191–206. [Google Scholar]
- 27.Baguley BC, McKeage MJC. Clin Invest. 2012;2:985–993. [Google Scholar]
- 28.Liu P, Qin Y, Wu L, Yang S, Li N, Wang H, Xu H, Sun K, Zhang S, Han X, Sun Y, Shi Y. Anti-Cancer Drugs. 2014;25:462–471. doi: 10.1097/CAD.0000000000000070. [DOI] [PubMed] [Google Scholar]
- 29.Chaplin DJ, Horsman MR, Siemann DW. Curr Opin Invest Drugs. 2006;7:522–528. [PubMed] [Google Scholar]
- 30.Sriram M, Hall JJ, Grohmann NC, Strecker TE, Wootton T, Franken A, Trawick ML, Pinney KG. Bioorg Med Chem. 2008;16:8161. doi: 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- 31.Pinney KG, Sriram M, George C, Tanpure RP. WO 2012068284 A2 20120524. PCT Int Appl. 2012
- 32.Pinney KG, Mocharla VP, Chen Z, Garner CM, Ghatak A, Hadimani M, Kessler J, Dorsey JM, Edvardsen K, Chaplin DJ, Prezioso J, Ghatak UR. 20040043969 A1. US Patent Appl Publ. 2004
- 33.Tanpure RP, George CS, Strecker TE, Devkota L, Tidmore JK, Lin CM, Herdman CA, MacDonough MT, Sriram M, Chaplin DJ, Trawick ML, Pinney KG. Bioorg Med Chem. 2013;21:8019. doi: 10.1016/j.bmc.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanpure RP, George CS, Sriram M, Strecker TE, Tidmore JT, Hamel E, Charlton-Sevcik AK, Chaplin DJ, Trawick ML, Pinney KG. Med Chem Comm. 2012;3:720. doi: 10.1039/C2MD00318J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadimani MB, MacDonough MT, Ghatak A, Strecker TE, Lopez R, Sriram M, Nguyen BL, Hall JJ, Kessler RJ, Shirali AR, Liu L, Garner CM, Pettit GR, Hamel E, Chaplin DJ, Mason RP, Trawick ML, Pinney KG. J Nat Prod. 2013;76:1668. doi: 10.1021/np400374w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flynn BL, Gill GS, Grobelny DW, Chaplin JH, Paul D, Leske AF, Lavranos TC, Chalmers DK, Charman SA, Kostewicz E, Shackleford DM, Morizzi J, Hamel E, Jung MK, Kremmidiotis G. J Med Chem. 2011;54:6014. doi: 10.1021/jm200454y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanpure RP, Harkrider AR, Strecker TE, Hamel E, Trawick ML, Pinney KG. Bioorg Med Chem. 2009;17:6993. doi: 10.1016/j.bmc.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinney KG, Pettit GR, Trawick ML, Jelinek C, Chaplin DJ. In: Antitumor Agents from Natural Products. 2. Kingston D, Newman D, Cragg G, editors. CRC Press, Taylor and Francis Group; Boca Raton, FL: 2012. pp. 27–63. [Google Scholar]
- 39.Pinney KG. In: Vascular-targeted Therapies in Oncology. Siemann DW, editor. John Wiley & Sons; London, UK: 2006. pp. 95–121. [Google Scholar]
- 40.Chen Z, Mocharla VP, Farmer JM, Pettit GR, Hamel E, Pinney KG. J Org Chem. 2000;65:8811. doi: 10.1021/jo0004761. [DOI] [PubMed] [Google Scholar]
- 41.Pinney KG, Bounds AD, Dingeman KM, Mocharla VP, Pettit GR, Bai R, Hamel E. Bioorg Med Chem Lett. 1999;9:1081. doi: 10.1016/s0960-894x(99)00143-2. [DOI] [PubMed] [Google Scholar]
- 42.Pinney KG, Pettit GR, Mocharla VP, Del PMM, Shirali A. WO9839323 A1 19980911. PCT Int Appl. 1998
- 43.Kessler RJ. Masters Thesis. Baylor University; Waco, TX: Dec, 2002. Synthesis and Evaluation of New Inhibitors of Tubulin Polymerization and Their Corresponding Prodrugs as Potential Vascular Targeting Agents. [Google Scholar]
- 44.Chen Z, O’Donnell CJ, Maderna A. Tetrahedron Lett. 2012;53:64. [Google Scholar]
- 45.Chen Z, Maderna A, Sukuru SCK, Wagenaar M, O’Donnell CJ, Lam MH, Musto S, Loganzo F. Biorg Med Chem Letters. 2013;23:6688. doi: 10.1016/j.bmcl.2013.10.039. [DOI] [PubMed] [Google Scholar]
- 46.Eaton PE, Carlson GR, Lee JT. J Org Chem. 1973;38:4071. [Google Scholar]
- 47.Tanis VM, Moya C, Jacobs RS, Little RD. Tetrahedron. 2008;64:10649–10663. [Google Scholar]
- 48.Kemperman G, Roeters T, Hilberink P. Eur J Org Chem. 2003:1681–1686. [Google Scholar]
- 49.Pascal VP, Kepler JA, Pettit GR, Hamel E. Mol Pharm. 2000;57:180. [PubMed] [Google Scholar]
- 50.Pettit GR, Grealish MP, Hearld DL, Boyd MR, Hamel E, Pettit RK. J Med Chem. 2000;43:2731. doi: 10.1021/jm000045a. [DOI] [PubMed] [Google Scholar]
- 51.Zhao D, Richer E, Antich PP, Mason RP. FASEB J. 2008;22:2445–2451. doi: 10.1096/fj.07-103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alhasan MK, Liu L, Lewis MA, Magnusson J, Mason RP. PLoS ONE. 2012;7:e46106. doi: 10.1371/journal.pone.0046106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardner PD, Horton WJ, Thompson G, Twelves RR. J Am Chem Soc. 1952;74:5527. [Google Scholar]
- 54.Caunt D, Crow WD, Haworth RD, Vodoz CA. J Chem Soc. 1950:1631. [Google Scholar]
- 55.Horton WJ, Pitchforth LL. J Org Chem. 1960;25:131. [Google Scholar]
- 56.Pinney KG, Sriram M. WO 2006138427 A2 20061228. PCT Int Appl. 2006
- 57.Carpenter PD, Humphreys DJ, Proctor GR, Rees LG. J Chem Soc, Perkins Trans 1: Org Bio-Org Chem. 1974;13:1527. [Google Scholar]
- 58.Comins DL, Brown JD. J Org Chem. 1989;54:3730. [Google Scholar]
- 59.Richtzenhain H, Meyer-Delius M. Chem Ber. 1948;81:81. [Google Scholar]
- 60.Gapinksi DM, Roman CR, Rinkema LE. J Med Chem. 1988;31:172. doi: 10.1021/jm00396a027. [DOI] [PubMed] [Google Scholar]
- 61.Meyers AI, Mihelich ED. J Am Chem Soc. 1975;97:7383. [Google Scholar]
- 62.Meyers AI, Gabel R, Mihelich ED. J Org Chem. 1978;43:1372. [Google Scholar]
- 63.Flippin LA, Berger J, Parnes JS, Gudiksen MS. J Org Chem. 1996;61:4812. doi: 10.1021/jo9602175. [DOI] [PubMed] [Google Scholar]
- 64.Flippin LA, Carter DS, Dubree NJP. Tetrahedron Lett. 1993;34:3255. [Google Scholar]
- 65.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A, Gray-Goodrich M, Campbell M, Mayo J, Boyd M. J Natl Cancer Inst. 1991;83:767. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 66.Vichai V, Kirtikara K. Nat Protoc. 2006;1:1112. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 67.Hamel E, Lin CM. Biochim Biophys Acta. 1981;675:226. doi: 10.1016/0304-4165(81)90231-2. [DOI] [PubMed] [Google Scholar]
- 68.Hamel E. Cell Biochem Biophys. 2003;38:1. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 69.Liu L, Beck H, Wang X, Hsieh H, Mason RP, Liu X. PLoS ONE. 2012;7:e43314. doi: 10.1371/journal.pone.0043314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.